 Open Access Article
Open Access ArticleEnd-functionalized glycopolymers as mimetics of chondroitin sulfate proteoglycans†
Song-Gil
Lee‡
,
Joshua M.
Brown‡
,
Claude J.
Rogers
,
John B.
Matson
,
Chithra
Krishnamurthy
,
Manish
Rawat
and
Linda C.
Hsieh-Wilson
*
Division of Chemistry and Chemical Engineering and Howard Hughes Medical Institute, California Institute of Technology, Pasadena, California 91125, USA. E-mail: lhw@caltech.edu; Fax: +1 626 564 9297; Tel: +1 626 395 6101
First published on 23rd July 2010
Abstract
Glycosaminoglycans are sulfated polysaccharides that play important roles in fundamental biological processes, such as cell division, viral invasion, cancer and neuroregeneration. The multivalent presentation of multiple glycosaminoglycan chains on proteoglycan scaffolds may profoundly influence their interactions with proteins and subsequent biological activity. However, the importance of this multivalent architecture remains largely unexplored, and few synthetic mimics exist for probing and manipulating glycosaminoglycan activity. Here, we describe a new class of end-functionalized ring-opening metathesis polymerization (ROMP) polymers that mimic the native-like, multivalent architecture found on chondroitin sulfate (CS) proteoglycans. We demonstrate that these glycopolymers can be readily integrated with microarray and surface plasmon resonance technology platforms, where they retain the ability to interact selectively with proteins. ROMP-based glycopolymers are part of a growing arsenal of chemical tools for probing the functions of glycosaminoglycans and for studying their interactions with proteins.
Introduction
Carbohydrates possess greater structural diversity than either nucleic acids or proteins. Although they participate in a wide range of critical processes and alterations in their structure have been linked to a number of human diseases, they remain under-explored targets for chemical biology and pharmaceutical chemistry. We have embarked on a program to study a large class of sulfated polysaccharides known as glycosaminoglycans, with the goals of understanding their structure–function relationships and gaining insight into the molecular mechanisms underlying their biological activity.Glycosaminoglycans (GAGs) are polymers, composed of 10–200 repeating sulfated disaccharide units (Fig. 1A).1 GAGs contain regions of high and low sulfation,2 with highly sulfated regions serving as binding sites for proteins.3 These protein interactions endow GAGs with the ability to regulate essential processes such as cell division, viral invasion, blood coagulation and neuronal regeneration.1a,b,3a,4 GAGs are covalently attached to various proteoglycan proteins, with some proteoglycans bearing as many as 100 sugar chains (Fig. 1B).5 It has been established using synthetic glycopolymers and oligosaccharides for other systems that the relative position and density of sugars can impact the avidity and specificity of glycan–protein interactions.6 However, despite these important advances, the role of the multivalent architecture found in native GAG structures has remained largely unexplored.
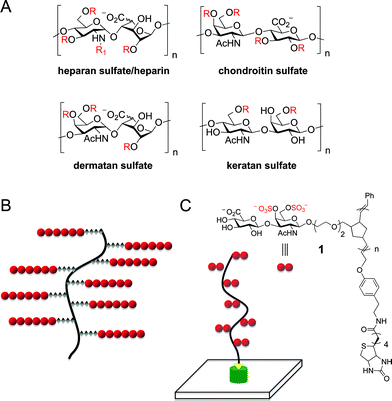 | ||
| Fig. 1 (A) Structures of representative GAG classes. R = SO3− or H; R1 = SO3−, H, or Ac; n = ∼10–200. (B) Schematic representation of a proteoglycan, which typically consists of multiple GAG chains attached to a protein core. (C) Biotin end-functionalized ROMP polymers as mimetics of CS proteoglycans. n = ∼80–280. | ||
Here, we describe a new class of end-functionalized GAG mimetic glycopolymers that are designed to mimic the multivalent presentation of chondroitin sulfate (CS) on proteoglycans. We demonstrate that these glycopolymers can be integrated with microarray as well as surface plasmon resonance (SPR) technology platforms to probe GAG–protein interactions and study the activity of specific sulfation motifs.
Results and discussion
To mimic the orientation of the sugar chains on proteoglycans, we designed glycopolymer 1, which has an end-functionalized biotin moiety to achieve the desired orientation of the pendant sugars and to facilitate attachment of the polymer to surfaces (Fig. 1C). We chose a norbornene-based backbone to allow for multivalent display of the sugar chains at defined, chemically controlled intervals and to confer a degree of rigidity to the structure. Previous studies in our laboratory have demonstrated that glycopolymers containing complex, highly anionic di- and tetrasaccharides can be generated using ring-opening metathesis polymerization (ROMP) chemistry, although more flexible cis-cyclooctene monomers were employed.7 In addition to increasing the structural rigidity of the resultant polymer, norbornene-based monomers would have the advantage of enabling access to block copolymers for controlling the sulfation motifs between GAG chains.Glycopolymer 2 was synthesized from monomer 3, which contains the biologically active CS-E disaccharide unit (Scheme 1).8 Briefly, trichloroacetimidate donor 4 was coupled to norbornene acceptor 5 to provide fully protected disaccharide 6 in good yield and with excellent β-stereoselectivity. Radical–mediated conversion of the N-trichloroacetyl group to an N-acetyl group and DDQ oxidation of the p-methoxybenzylidene acetal afforded diol 7. Sulfation using sulfur trioxide·trimethylamine complex afforded the desired norbornene monomer 3 in 83% yield. A major challenge for the polymerization reaction was the low solubility of sulfated oligosaccharides in the non-coordinating, aprotic solvents typically used for ROMP. Fortunately, the fully protected, sulfated monomer 3 was soluble in MeOH/(CH2Cl)2 co-solvent mixtures, and polymerization with 1.0 mol% of Grubbs' catalyst (H2IMes)(Py)2(Cl)2Ru = CHPh (8)9 led to complete conversion to the desired glycopolymer 9 within 5 min (Table 1, degree of polymerization (DP) = 97; polydispersity index (PDI) = 1.17). Lowering the catalyst concentration to 0.5 mol% produced glycopolymer 10 with exceptionally long chain lengths (DP = 281; PDI = 1.07). Longer polymers with narrower polydispersities were attainable with norbornene relative to cis-cyclooctene monomers,7,10 which may be advantageous for biological activity. The CS glycomimetics 2 and 11 were obtained after desilylation and sequential LiOOH–NaOH treatment in 71–80% yield over two steps.
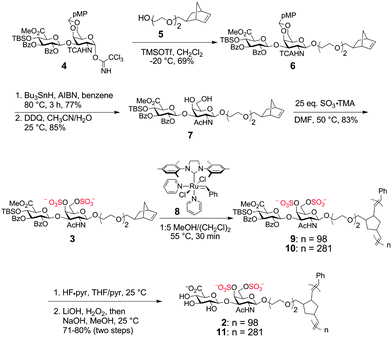 | ||
| Scheme 1 Synthesis of Norbornene-based CS Glycopolymers | ||
| entry | monomer | mol % catalyst | polymer | n (DP) | % yield | Mna/g mol−1 | PDIa |
|---|---|---|---|---|---|---|---|
| a Number average molecular weight and polydispersity index were determined by GPC (0.2 M LiBr in DMF for entries 1–2; 100 mM NaNO3 and 200 ppm NaN3 in water for entries 3–4). b Yield for 3 steps (polymerization, desilylation, saponification). | |||||||
| 1 | 3 | 1.0 | 9 | 97 | 87 | 105,100 | 1.17 |
| 2 | 3 | 0.5 | 10 | 281 | 92 | 283,100 | 1.07 |
| 3 | 3 | 1.0 | 1 | 86 | 74b | 68,480 | 1.05 |
| 4 | 7 | 1.0 | 15 | 82 | 55b | 48,480 | 1.10 |
We next investigated end-functionalization of the glycopolymers with a biotin moiety. Previous studies have established that internal cis-olefin or enol ether terminating agents (TA) can be used for the direct, efficient capping of ROMP polymers.11 Addition of the biotin terminating agent 1211b to the reaction mixture after completion of the living polymerization, resulted in the desired end-capping of the glycopolymer to produce 13 (Scheme 2). Desilylation and saponification afforded glycopolymer 1 in 74% overall yield over the 3 steps (Table 1, DP = 86; PDI = 1.05). The end-capping efficiency of 20%, as determined using a colorimetric biotin quantification assay and 1H NMR, was modest due to the limited solubility of the unquenched glycopolymer and 12. For comparison, we also synthesized the biotin-functionalized unsulfated glycopolymer 15 (DP = 82; PDI = 1.10) from monomer 7 using a similar approach.
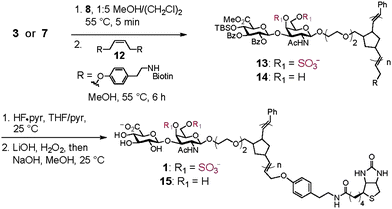 | ||
| Scheme 2 Synthesis of Biotin End-Functionalized CS Glycopolymers | ||
To explore the ability of the glycopolymers to interact with protein receptors, glycopolymers 1 and 15 were attached to microarray surfaces. A high-precision contact-printing robot was used to deliver nanoliter volumes of the biotin-labeled glycopolymers to streptavidin-coated slides, yielding spots approximately 200 μm in diameter. We examined the binding of monoclonal antibodies 2D11 and 2D5, which are selective for the CS-E and CS-C sulfation motifs, respectively.3b,8a The microarrays were incubated with each antibody (70 nM), and protein binding was visualized using a secondary Cy3-conjugated goat anti-mouse antibody. Antibody 2D11 bound selectively to CS-E sulfated glycopolymer 1 and showed no detectable binding to unsulfated glycopolymer 15 (Fig. 2A). Moreover, no binding of the CS-C antibody 2D5 to either glycopolymer was observed, consistent with the selective recognition of specific sulfated epitopes. We also examined the binding of several growth factors, including glial cell-derived neurotrophic factor (GDNF), a growth factor important for the survival and differentiation of dopaminergic neurons.12 Although the binding of GDNF to a highly sulfated mixture of chondroitin and dermatan sulfate chains has been studied,3c its ability to recognize homogeneous, well-defined CS structures has not been explored. Significant binding of GDNF to the CS-E glycopolymer, but not the unsulfated glycopolymer, was observed (Fig. 2B), indicating a clear preference of GDNF for the sulfated sugar epitope.
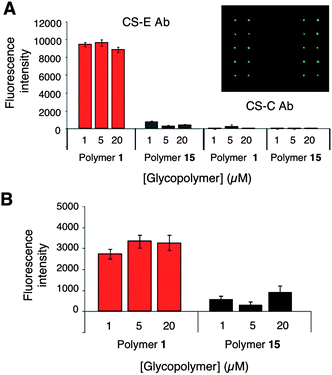 | ||
| Fig. 2 (A) Binding of the CS-E Ab (left) and CS-C Ab (right) to CS-E glycopolymer 1 and unsulfated glycopolymer 15 immobilized on microarrays. (B) Binding of GDNF to glycopolymers 1 and 15. Each microarray contained 640 spots, and the bar graphs represent the quantification for selected concentrations of glycopolymer (1280 data points per protein). The inset shows binding of the CS-E Ab to 1 on a representative portion of the array. See Supplementary Information† for details. | ||
Finally, we investigated whether the end-functionalized glycopolymers could be used to facilitate quantitative, real-time analysis of GAG–protein interactions using surface plasmon resonance (SPR). Glycopolymers 1 and 15 were immobilized on streptavidin–conjugated CM5 sensor chips at low density (RL ≈ 25 RU) to prevent mass transfer-limited kinetics. Binding of GDNF to the glycopolymers was assessed by flowing GDNF over the chip at various concentrations (2, 1, 0.5 nM) and recording the SPR sensorgrams (50 μL min−1, 25 °C). As shown in Fig. 3, GDNF interacted with the CS-E sulfated glycopolymer, but not with the unsulfated glycopolymer, consistent with the microarray results. Binding of GDNF to glycopolymer 1 was characterized by a slow initial rate of association that rapidly reached equilibrium, followed by a slow rate of dissociation. By plotting the response at equilibrium for varying concentrations of GDNF (0.25–62 nM), we obtained a dissociation constant (KD) of 6 ± 1 nM for the interaction between GDNF and glycopolymer 1. It is well known that monovalent CS and heparan sulfate disaccharides exhibit weak binding affinity for proteins and minimal biological activity.7,8b,13 Indeed, binding of GDNF to a biotinylated CS-E disaccharide could not be detected under these conditions (Fig. S2†). Thus, the observation that our glycopolymers interact strongly with proteins suggests that the multivalent display of sulfated epitopes between GAG chains plays a critical role in enhancing their interactions with proteins. Together, our studies demonstrate that end-functionalized ROMP glycopolymers can effectively engage glycosaminoglycan-binding proteins and function as novel mimetics for CS glycosaminoglycans.
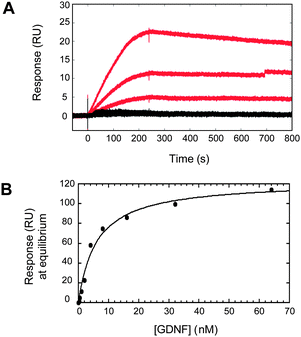 | ||
| Fig. 3 Surface plasmon resonance of GDNF binding to CS glycopolymers 1 and 15. GDNF at varying concentrations (2, 1, and 0.5 nM) binds to the CS-E sulfated glycopolymer (red), but not the unsulfated polymer (black). (B) The dissociation constant (KD) for the interaction between 1 and GDNF was measured by plotting the response at equilibrium for varying concentrations of GDNF. Nonlinear regression analysis gave a KD of 6 ± 1 nM. | ||
Conclusions
We have generated a new class of CS glycomimetic polymers that display defined sulfation motifs, while mimicking the multivalent architecture of native GAG chains. Our studies demonstrate that these glycopolymers can be efficiently attached to surfaces, where they approximate physiological cell–cell and cell–extracellular matrix interactions and retain the ability to engage proteins. ROMP-based glycopolymers are part of a growing arsenal of chemical tools for studying the structure–activity relationships of GAGs. We anticipate that they will prove valuable for understanding how multivalency, not only within but also between, GAG chains enhances the avidity, specificity and cooperativity of GAG–protein interactions. Future studies will focus on extending the methodology reported herein to polymers and block co-polymers with varied sulfation patterns and applying them as tools to manipulate CS activity in various biological contexts.References
- (a) I. Capila and R. J. Linhardt, Angew. Chem. Int. Ed. Engl., 2002, 41, 390 CrossRef CAS; (b) K. Sugahara, T. Mikami, T. Uyama, S. Mizuguchi, K. Nomura and H. Kitagawa, Curr. Opin. Struct. Biol., 2003, 13, 612 CrossRef CAS; (c) C. I. Gama and L. C. Hsieh-Wilson, Curr. Opin. Chem. Biol., 2005, 9, 609 CrossRef CAS.
- (a) H. Desaire, T. L. Sirich and J. A. Leary, Anal. Chem., 2001, 73, 3513 CrossRef CAS; (b) J. E. Turnbull and J. T. Gallagher, Biochem. J., 1991, 273(Pt 3), 553 CAS.
- (a) R. Raman, V. Sasisekharan and R. Sasisekharan, Chem. Biol., 2005, 12, 267 CrossRef CAS; (b) S. E. Tully, M. Rawat and L. C. Hsieh-Wilson, J. Am. Chem. Soc., 2006, 128, 7740 CrossRef CAS; (c) C. D. Nandini, N. Itoh and K. Sugahara, J. Biol. Chem., 2005, 280, 4058 CAS; (d) S. S. Deepa, Y. Umehara, S. Higashiyama, N. Itoh and K. Sugahara, J. Biol. Chem., 2002, 277, 43707 CrossRef CAS; (e) E. L. Shipp and L. C. Hsieh-Wilson, Chem. Biol., 2007, 14, 195 CrossRef CAS.
- (a) S. Mizuguchi, T. Uyama, H. Kitagawa, K. H. Nomura, K. Dejima, K. Gengyo-Ando, S. Mitani, K. Sugahara and K. Nomura, Nature, 2003, 423, 443 CrossRef CAS; (b) J. M. Trowbridge and R. L. Gallo, Glycobiology, 2002, 12, 117R CrossRef CAS; (c) Y. Shen, A. P. Tenney, S. A. Busch, K. P. Horn, F. X. Cuascut, K. Liu, Z. He, J. Silver and J. G. Flanagan, Science, 2009, 326, 592 CrossRef CAS.
- L. Kjellen and U. Lindahl, Annu. Rev. Biochem., 1991, 60, 443 CrossRef CAS.
- (a) D. M. Lewallen, D. Siler and S. S. Iyer, ChemBioChem, 2009, 10, 1486 CrossRef CAS; (b) J. E. Gestwicki, C. W. Cairo, L. E. Strong, K. A. Oetjen and L. L. Kiessling, J. Am. Chem. Soc., 2002, 124, 14922 CrossRef CAS; (c) N. Horan, L. Yan, H. Isobe, G. M. Whitesides and D. Kahne, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 11782 CrossRef CAS; (d) M. Dhayal and D. M. Ratner, Langmuir, 2009, 25, 2181 CrossRef CAS; (e) H. M. Branderhorst, R. Ruijtenbeek, R. M. Liskamp and R. J. Pieters, ChemBioChem, 2008, 9, 1836 CrossRef CAS; (f) K. Godula, D. Rabuka, K. T. Nam and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 4973 CrossRef CAS; (g) J. E. Gestwicki, C. W. Cairo, D. A. Mann, R. M. Owen and L. L. Kiessling, Anal. Biochem., 2002, 305, 149 CrossRef CAS; (h) J. Geng, G. Mantovani, L. Tao, J. Nicolas, G. Chen, R. Wallis, D. A. Mitchell, B. R. G. Johnson, S. D. Evans and D. M. Haddleton, J. Am. Chem. Soc., 2007, 129, 15156 CrossRef CAS; (i) K. Matsuura, H. Kitakouji, N. Sawada, H. Ishida, M. Kiso, K. Kitajima and K. Kobayashi, J. Am. Chem. Soc., 2000, 122, 7406 CrossRef CAS.
- M. Rawat, C. I. Gama, J. B. Matson and L. C. Hsieh-Wilson, J. Am. Chem. Soc., 2008, 130, 2959 CrossRef CAS.
- (a) C. I. Gama, S. E. Tully, N. Sotogaku, P. M. Clark, M. Rawat, N. Vaidehi, W. A. Goddard, 3rd, A. Nishi and L. C. Hsieh-Wilson, Nat. Chem. Biol., 2006, 2, 467 CrossRef CAS; (b) S. E. Tully, R. Mabon, C. I. Gama, S. M. Tsai, X. Liu and L. C. Hsieh-Wilson, J. Am. Chem. Soc., 2004, 126, 7736 CrossRef CAS.
- (a) K. D. Camm, N. M. Castro, Y. Liu, P. Czechura, J. L. Snelgrove and D. E. Fogg, J. Am. Chem. Soc., 2007, 129, 4168 CrossRef CAS; (b) J. A. Love, J. P. Morgan, T. M. Trnka and R. H. Grubbs, Angew. Chem., Int. Ed., 2002, 41, 4035 CrossRef CAS.
- R. Walker, R. M. Conrad and R. H. Grubbs, Macromolecules, 2009, 42, 599 CrossRef CAS.
- (a) J. B. Matson and R. H. Grubbs, Macromolecules, 2008, 41, 5626 CrossRef CAS; (b) J. B. Matson and R. H. Grubbs, Macromolecules, 2010, 43, 213 CrossRef CAS; (c) R. M. Owen, J. E. Gestwicki, T. Young and L. L. Kiessling, Org. Lett., 2002, 4, 2293 CrossRef CAS.
- (a) L. F. Lin, D. H. Doherty, J. D. Lile, S. Bektesh and F. Collins, Science, 1993, 260, 1130 CrossRef CAS; (b) A. Tomac, E. Lindqvist, L. F. Lin, S. O. Ogren, D. Young, B. J. Hoffer and L. Olson, Nature, 1995, 373, 335 CrossRef CAS.
- J. L. de Paz, C. Noti, F. Bohm, S. Werner and P. H. Seeberger, Chem. Biol., 2007, 14, 879 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures and compound characterizations. See DOI: 10.1039/c0sc00271b |
| ‡ These authors equally contributed to this work. |
| This journal is © The Royal Society of Chemistry 2010 |