Chiral pyridyl phosphinites with large aryl substituents as efficient ligands for the asymmetric iridium-catalyzed hydrogenation of difficult substrates†‡
David H.
Woodmansee
a,
Marc-André
Müller
a,
Marcus
Neuburger
b and
Andreas
Pfaltz
*a
aDepartment of Chemistry, University of Basel, St. Johanns-Ring 19, CH-4056, Basel, Switzerland. E-mail: andreas.pfaltz@unibas.ch; Fax: +41 61-267-1103
bM. Neuburger Laboratory for Chemical Crystallography, University of Basel, Spitalstrasse 51, 4056, Basel, Switzerland
First published on 12th May 2010
Abstract
Using a flexible synthesis, new chiral iridium N,P complexes with sterically demanding aryl substituents were synthesized and used in the asymmetric hydrogenation of difficult substrates. Unprecedented enantioselectivities were obtained in the asymmetric hydrogenation of α-substituted α,β-unsaturated esters and dihydronaphthalenes. The variety of aryl substituents incorporated into the catalyst has made it possible to obtain useful structure selectivity relationships for important classes of substrates.
Introduction
Catalytic asymmetric hydrogenation is one of the most widely used and reliable catalytic methods for the preparation of optically active compounds.1 High enantioselectivity, low catalyst loadings, essentially quantitative yields, perfect atom economy, and mild conditions are attractive features of this transformation. For these reasons asymmetric hydrogenation is the most utilized synthetic asymmetric methodology in industry and forms the critical transformation in several commercial scale processes.2 Logically, discovery of high performance chiral catalysts crucially depends on the development of appropriate ligands3 and this field is in constant development with new generations of ligands appearing every year.A plethora of chiral phosphine ligands are known which induce very high enantioselectivity in rhodium- and ruthenium-catalyzed hydrogenations.4–7 However, the range of olefins that can be hydrogenated with rhodium and ruthenium complexes with high enantiomeric excess is still limited as these catalysts require the presence of a coordinating group next to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond. With unfunctionalized olefins, they generally show low reactivity and unsatisfactory enantioselectivity. Iridium complexes such as the Crabtree catalyst on the other hand are more active in the reduction of CC bonds without adjacent coordinating functional groups and stronger coordinating functional groups are frequently observed as inhibitors of iridium based catalysts. In this sense, iridium complexes are complementary to rhodium and ruthenium complexes.
C bond. With unfunctionalized olefins, they generally show low reactivity and unsatisfactory enantioselectivity. Iridium complexes such as the Crabtree catalyst on the other hand are more active in the reduction of CC bonds without adjacent coordinating functional groups and stronger coordinating functional groups are frequently observed as inhibitors of iridium based catalysts. In this sense, iridium complexes are complementary to rhodium and ruthenium complexes.
Some years ago we discovered a new class of homogenous hydrogenation catalysts capable of the asymmetric reduction of unfunctionalized olefins: iridium complexes of the chiral N,P ligand progenitor, phosphino-oxazoline 1 (PHOX).8,9 These catalysts could achieve unprecedented levels of enantioselectivity with a wide range of unfunctionalized olefins, exhibiting impressive activity in the hydrogenation of tri- and even tetrasubstituted olefins. In this respect, they resembled the Crabtree catalyst, [Ir(Cy3P)(pyridine)(COD)]PF6 (Cy = cyclohexyl, COD = cyclooctadiene),10 which provided the impetus for our early work. In addition, promising results were also obtained with certain functionalized alkenes for which no suitable catalysts were available at that time. In our initial studies we found that the careful choice of solvent and anion was the key to successful catalysis as only in weakly coordinating solvents with a virtually non-coordinating anion such as BArF (tetrakis[bis-3,5-(trifluoromethyl)phenyl]borate) could high turnover numbers (>5000) be obtained.8,11 Further modifications of PHOX led to the creation of ThrePHOX 2, SimplePHOX 3a and the later developed NeoPHOX 3b. All of these ligands are highly effective, easily accessible from the chiral pool and in the case of 2 two iridium catalysts with different ThrePHOX derivatives have become commercially available.12–14 Work in our group and in other laboratories is now focusing on the extension of iridium catalyzed asymmetric hydrogenation to new substrate classes which give unsatisfactory results with known catalysts.15–17
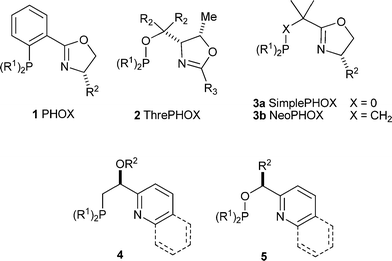
In an endeavor to synthesize catalysts that more closely match the Crabtree catalyst, ligands 4 and 5 were synthesized in our group.18,19 We were encouraged by the results to pursue catalysts with 5- and 6-membered ring backbones 6a, 6b, and 8 in the hope of reaching higher enantioselectivities with a more rigid system. Complexes 6a, 6b, and 8 proved to be remarkably active and selective in the enantioselective hydrogenation of purely alkyl substituted olefins,20,21 furans,22 and indoles23 as well as playing a featured role in a handful of total syntheses.12,24,25
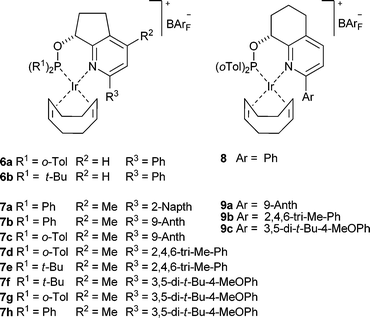
The current trend in catalyst development in this field is not towards more general catalysts capable of handling all possible substitution patterns around a CC bond but to customize a catalyst for improvement in a given class of substrates. It would therefore be highly advantageous to have an adaptable system that could be quickly modified to improve enantioselectivity. In this report we present a flexible synthesis of sterically encumbered N,P complexes based on the scaffolds 6 and 8 and the successful asymmetric hydrogenation of several examples of trisubstituted olefins that gave unsatisfactory results with previous catalysts.
Discussion and results
Catalyst synthesis
Data from earlier work have allowed us to develop some guidelines that have led to more selective and active catalysts in the reduction of weakly coordinating trisubstituted olefins. Catalysts with both bis(tert-butyl) and bis(ortho-tolyl)phosphinites in combination with a 5-membered ring backbone such as 6a,b gave superior enantioselectivity than the less bulky phosphinite analogues. However, catalysts with a 6-membered ring based scaffold such as 8 perform best with bis(ortho-tolyl)phosphinites and often give better results compared to the bis(tert-butyl)phosphinite analogues or complexes with a less sterically demanding phosphinite. We also found the substituent on the ortho position in the pyridine ring to be critical to both activity and selectivity with the optimal size and shape being phenyl. Larger groups such as tert-butyl were not tolerated in this position while smaller groups gave lower enantioselectivity. We saw this substitution dependence of the catalyst as an opportunity to extend the reactivity and selectivity profile of the catalyst.In order to explore further structural modifications we required a flexible synthesis that would allow for the incorporation of a variety of aromatic substituents on the pyridine ring at a later phase of the route. Shortly after our initial publications the group of Zhou reported a route to nearly the same complexes that utilized a Suzuki coupling to install the aromatic group at a late stage.26 All attempts to adapt this method to our system failed to produce appreciable amounts of product. Noting that there are very few reports of coupling hindered boronic acids with 2-pyridyl chlorides in the literature we decided to replace the chlorine for a bromine atom but our efforts led to low yield of product and complex tar-like mixtures. Realizing the hydroxyl group created a chelating structure with the adjacent pyridine ring that could be responsible for slow to nonexistent catalyst turnover we decided to investigate two possible strategies to circumvent this problem, one incorporating the Suzuki coupling before the hydroxylation and a second route via a silyl protected pyridyl alcohol (Scheme 1).
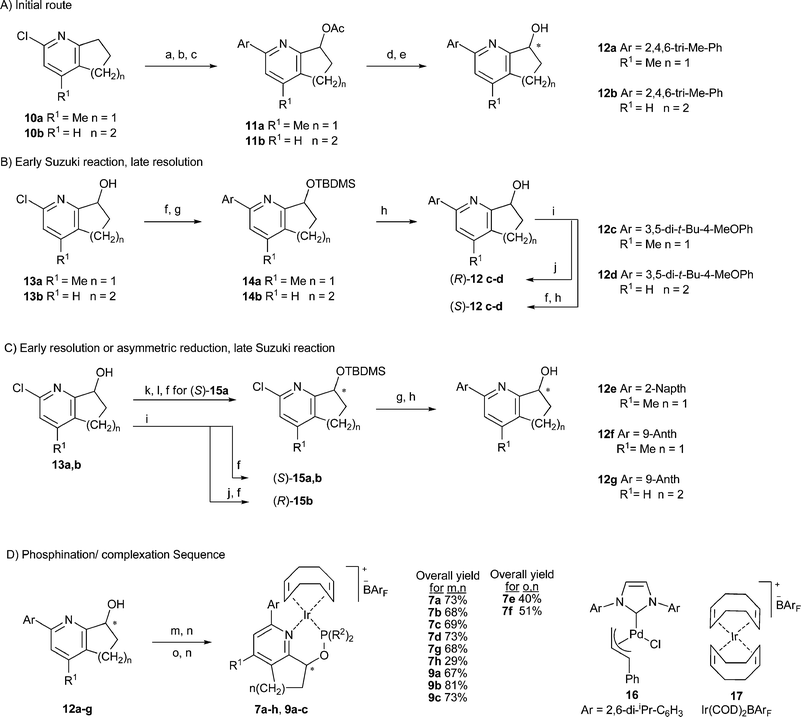 | ||
| Scheme 1 Synthesis of catalysts 7a–h and 9a–c. Reagents: (a) catalyst 16 (1–2 mol%), ArB(OR)2 (2 eq.), NaOH (3 eq.), 105–115 °C, 48 h, 86%, (b) MCPBA, DCM, rt, 6 h, 89–95%, (c) acetic anhydride, 130 °C, 8 h, 56–86%, (d) Chiralcel OD, (e) 4 M NaOH, THF, 65 °C, 89–94%, (f) TBDMSCl (2.5 eq.), imidazole (3 eq.), DMF, overnight, 86–98%, (g) catalyst 16 (4 mol%), ArB(OR)2 (1.5–2 eq.), NaOH (3 eq.), 50 °C, 18 h, 86%, (h) TBAF, 3H2O (3 eq.), THF, 50 °C, 79–84%, (i) Novozyme 435, DIPE, vinyl acetate, (j) K2CO3, MeOH, 90–95%, (k) Swern, 95%, (l) (−)-DIPCl (1.5 eq.), THF, −50 °C, 2 days, ethanolamine (1.5 eq.), 51% yield 99% ee, (m) DMAP (1 eq.), Ar2PCl (1 eq.), THF, 24 h, (n) complex 17 (1 eq.), (o) KH (1.5 eq.), DMF, ClP(tBu)2, 2 days. | ||
The starting materials 10a,b and 13a,b have been reported by several groups, the chemistry is well known and commercial sources exist for intermediates 10a,b, 13b and the desmethyl analogue of 13a.27–31 More importantly several methods for the preparation of enantioenriched alcohols have been reported.26,32–35 Our initial concern was to obtain first test samples of enantiopure catalyst quickly and in this regard chiral semipreparative HPLC can deliver perfect enantiomeric purity. We were able to successfully separate the mesityl functionalized pyridyl esters 11a,b followed by hydrolysis to give the desired enantiopure alcohols 12a,b. Although pyridines with anthracene substituents were easily resolved with large differences in the retention times of the enantiomers these crystalline polyaromatics were virtually insoluble in the mobile phases typically used. Other substitution patterns on the pyridine ring were found to also have solubility issues as well as inferior resolution in spite of extensive investigation of HPLC conditions. In light of these problems we decided to use the silyl ether route given in sections B and C of Scheme 1 for the formation of the other enantiopure alcohols 12b–g.
Uenishi et al.36,37 reported the kinetic resolution of simple tetrahydroquinoline-8-ols with lipase, which we later used and optimized in the preparation of enantiopure alcohols en route to catalysts 6a,b, and 8.38 Given our previous success with this method we thought it could also be used to provide reasonable quantities of the more elaborate pyridyl alcohols and we were able to resolve alcohols 12c,d with 3,5-disubstituted aryl groups in excellent enantioselectivity. However, all attempts to resolve pyridyl alcohols with sterically more demanding 2,6-disubstituted aryl groups failed in both the acylation of the racemic alcohol and the hydrolysis of the racemic ester with polymer immobilized Candida antarctica lipase B. We attribute the loss of reactivity in both the forward and reverse reactions to a locked perpendicular ring system that blocks the approach of the alcohol to the reactive site in the enzyme. X-Ray crystal structures of the R-alcohol 12g indicated a very sequestered hydroxyl group buried in a 6-membered ring formed from hydrogen bonding with a second pyridyl alcohol to form a dimer and a second interaction between two anthracene groups bound together by edge face C–H–π interactions that completed an encumbered tetramer.
Fortunately we were able to resolve enzymatically the chlorides 13a and 13b with some success. In small test resolutions we followed the enantioselectivity with time to gain a better understanding of the reaction parameters and optimal time (see ESI†). We found that the tetrahydroquinoline 13b gave excellent enantioselectivity in the production of the R-acetate with a shallow sloped linear behavior; the S-alcohol was produced in 99% ee within 28 h albeit with a small loss of enantioselectivity of the R-acetate. The R-acetate 13b could be easily recrystallized from cold pentane with 56% return of the product in 99% ee. The 5-membered bicyclic 13a proved to be much more difficult to resolve with a lower ee in the initial acyl ester formed and a far more dramatic loss of ee with the R-acetate 13a over time. However, the S-alcohol of 13a could be obtained with 99% ee with a small loss of yield.
We previously reported38 a simple procedure for the isolation of the S-pyridyl alcohol parents of catalysts 6a,b and 8 after enzymatic kinetic resolution by formation of a phthalic acid monoester. The phthalate could be separated from the reaction mixture by precipitation and filtration without need of chromatography. This approach was unsuccessful for 12c,d as hydrolysis of the phthalate was unfeasible with the 2-chloropyridine derivatives 13a,b with the harsh basic conditions used to cleave the phthalate ester. We therefore resorted to a silyl ether formation with the resolved reaction mixture to greatly simplify the chromatography.
Dissatisfied with this result we attempted a number of asymmetric reductions of the parent ketone with a commercial oxazaborolidine catalyst and found similar results to those of Zhou et al.26 and Hu et al.32 who reported enantioselectivities up to 93% ee. We were somewhat more successful using (−)-DIPCl in a stoichiometric reduction which generated the desired S-alcohol 13a in 99% ee and 51% yield. The reduction was extremely sensitive to temperature and addition technique with addition of the solid DIPCl to a −50 °C solution in THF and stirring for two days at that temperature providing the optimal yield and selectivity. Subsequent Suzuki coupling of the silyl ether followed by deprotection produced the naphthalene and anthracene analogues 12e,f and the known alcohol 12g39,40 in over 90% yield.
Of the commercially available palladium catalysts we tried we found the carbene based catalyst 1641 under modified conditions to provide very clean conversion of the pyridyl chlorides to hindered biaryls in high yield with a limited amount of by-product. Notably the simple pyridines reacted with lower catalyst loadings of 1–2 mol%, fewer equivalents of boronic acid or ester and could withstand higher temperatures. Silyl ether derivatives would undergo deprotection at higher temperatures so greater catalyst loadings up to 4 mol% and larger amounts of boronic acid were required. Yields were greater than 80% for all cases and solubility issues with the boronic acid could often be avoided by simply switching to a pinacol ester. The silyl protecting group also had the added benefit of reducing the polarity of the desired compound and facilitating purification by column chromatography. The resulting silyl ethers were sluggish in the deprotection step with TBAF but gentle heating furnished the desired alcohols in high yield. A reaction sequence of the chiral alcohol with diarylphosphine chlorides with DMAP as an activating base followed by a simple filtration through silica gel into a solution of Ir(COD)2BArF or [Ir(COD)Cl]2 and NaBArF generated the desired complexes in reasonable yields for most of the catalysts.
Formation of tert-butyl derived phosphinites was far more challenging; the reactions required highly concentrated conditions and potassium hydride in DMF with extended reaction times. Concentration of the reaction mixture was a critical parameter with the reaction proceeding within reasonable time at higher concentration and slowing down to abysmal levels (ca. 7–10 days) under more dilute conditions. X-Ray quality crystals of complexes 7a, 7e, 7f, and 9a were grown from mixtures of DCM and pentane. The solid state structures indicate a very congested environment about the metal center for all of the catalysts with both the ortho and meta substituents of the pendant biaryl moiety coming in close contact with substituents on the phosphorus atom (Fig. 1).
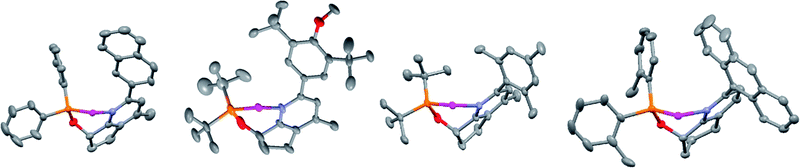 | ||
| Fig. 1 Crystal structures of 7a, 7f (mirror image), 7e, and 9a; COD and counterions are omitted for clarity. | ||
Asymmetric hydrogenation of trisubstituted olefins
Structure–activity relationships were immediately apparent in the initial screening of common substrates. In reductions of (E)-α-methylstilbene 18a at 1 mol% catalyst loading the reaction displayed a preference for catalysts with less encumbrance about the metal center to accommodate the sterically demanding substrate (Table 1). We observed some differences from our previous studies with catalyst 6a,b and 8 in that simple diphenylphosphino groups suffice for excellent enantioselectivity in combination with the sterically demanding 3,5-di-tert-butyl-4-methoxyphenyl group for catalyst 7h or even a simple β-naphthyl group as in 7a. Catalyst 9c reached only 73% conversion albeit with 99% enantioselectivity, similar to catalyst 8 which reduced the same substrate in 99% ee but only 20% conversion under identical conditions.|
|
|||
|---|---|---|---|
| Catalyst | ee (%) | Catalyst | ee (%) |
| a All reactions were run for 2 h at 1 mol% catalyst loading and 50 bar H2 pressure in 0.5 mL absolute DCM at room temperature. b 73% conversion. c Ref. 13. | |||
| (S)-7a | 98 (S) | (S)-7g | 99 (S) |
| (S)-7b | 95 (S) | (S)-7h | 99 (S) |
| (S)-7c | 93 (S) | (S)-9c | 99 (S)b |
| (R)-7f | 94 (R) | (R)-6a | 99 (R)c |
Allylic alcohol 19a was found to be more sensitive to steric hindrance than substrate 18a (Table 2). We attribute the loss of activity with the larger ligands to catalyst inhibition by the more strongly coordinating primary hydroxyl group competing for a limited amount of space with the olefin leading to a less reactive system. The best performing catalyst was 7a which also had the least sterically demanding groups, coming very close to the enantioselectivity of 6a and with better conversion. The di-tert-butyl-phosphine catalyst 7f gave very poor results with 12% ee and 62% conversion in comparison with the closely related di-o-tolyl-phosphine catalyst 7g which gave 93% ee and full conversion. A similar trend can be seen when changing from the bulky ortho-tolyl substituted 7c, which gives abysmal enantioselectivity of 29% and 25% yield, to the less demanding diphenylphosphino derivative 7b which gives 80% ee and 99% yield.
|
|
|||||
|---|---|---|---|---|---|
| Catalyst | ee (%) | Conversion (%) | Catalyst | ee (%) | Conversion (%) |
| a All reactions were run for 16 h at 1 mol% catalyst loading and 50 bar H2 pressure in 0.5 mL absolute DCM at room temperature. b Over 80% converted to the branched alkyl aldehyde. c Hydrogenation gives the S-enantiomer. d Ref. 20. | |||||
| (S)-7a | 95 | 99 | (S)-7g | 93 | 99 |
| (S)-7b | 80 | 99 | (S)-7h | 92 | 70 |
| (S)-7c | 29 | 25 | (S)-9a | 84 | 99 |
| (S)-7d | 88 | 99b | (S)-9b | 57 | 99 |
| (S)-7e | 72 | 99 | (S)-9c | 86 | 34 |
| (R)-7fc | 12 | 62 | (R)-6ac,d | 97 | 95 |
Steric parameters seem to have far greater consequences for this class of substrate, namely 2-methyl-3-phenyl-propanal, which is produced in trace quantities with catalyst 6a, became the predominant product with the highly congested 7d. This finding is consistent with the work of Mazet et al.42,43 who have optimized this 1,3 hydride shift into a viable asymmetric process by maximizing steric interactions with the allylic alcohol in a highly restricted pocket about the iridium metal center. Clearly the addition of very sterically demanding aromatic substituents about the metal center creates a more sensitive system and changes that are typically well tolerated with other substrates can have drastic effects under these circumstances.
We found that a number of our more encumbered catalysts were well suited toward the asymmetric reduction of cyclic dihydronaphthalenes, which are known as difficult substrates, and these catalysts have given the highest enantioselectivity reported to date with olefin 20a (Table 3). They gave good to excellent enantioselectivities at 1 mol% catalyst loading and a pressure of 50 bar of hydrogen. Lowering the catalyst loading below 1 mol% however gave lower enantioselectivity and conversion was greatly reduced.
|
|
|||
|---|---|---|---|
| Catalyst | ee (%) | Catalyst | ee (%) |
| a All reactions were run for 4 h at 1 mol% catalyst loading and 50 bar H2 pressure in 500 μL absolute DCM at room temperature. b Configuration was (S), opposite of what is depicted. c From our previous report ref. 20. | |||
| (S)-7a | 49 | (S)-7g | 97 |
| (S)-7b | 95 | (S)-7h | 96 |
| (S)-7c | 97 | (R)-9ab | 99 |
| (S)-7d | 99 | (S)-9b | 99 |
| (S)-7e | 97 | (S)-9c | 98 |
| (R)-7fb | 94 | (R)-6bb,c | 92 |
Encouraged by the performance with the simple dihydronaphthalene we turned our attention to the reduction of a more difficult analogue, 21a, which we reported in the asymmetric synthesis of desmethyl methoxycalamenene (Table 4).12 We were pleased to discover several of our catalysts performed with excellent enantioselectivity and conversion. Higher loading with catalyst 9a at higher concentrations and with higher pressures afforded the reduced product 21b in 96% conversion and 98% ee with only a small trace of the aromatized product 21c as well as the tetrasubstituted olefin 21d (Fig. 2).
|
|
||||
|---|---|---|---|---|
| Catalysta | ee (%) | Conversion (%) | 21b (%)b | 21c (%)b |
| a Reactions were run for 3 h with 0.1 M concentration of substrate, 1 mol% catalyst loading and 50 bar H2 pressure in absolute DCM at room temperature unless otherwise noted. b Determined by GC. c ca. 1.8% of 21d from the isomerization of 21a was present. d Reactions were run for 3 h with 0.25 M concentration of substrate and 75 bar H2 pressure in absolute DCM with 2 mol% catalyst loading. | ||||
| (S)-7c | 91 (S) | >99 | 96 | 4 |
| (R)-7f | 95 (R) | >99 | 98 | 2 |
| (S)-9a | 96 (S) | 84 | 80 | 2c |
| (S)-9a | 98d (S) | 99 | 96 | 0.8c |
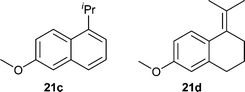 | ||
| Fig. 2 Side products 21c,d. | ||
To test the limits of what was possible with these catalysts we synthesized the dihydronaphthalene 22a. Reduction of the hindered cyclic olefin was highly sensitive to conditions, catalyst loading and concentration. The reaction was very sluggish and a pressure of 100 bar for 24 h with 2 mol% catalyst was required for complete conversion (Scheme 2). Formation of naphthalene 22c was reduced at higher concentrations and the enantioselectivity improved slightly but the ee was very dependent on catalyst loading and solvent purity.
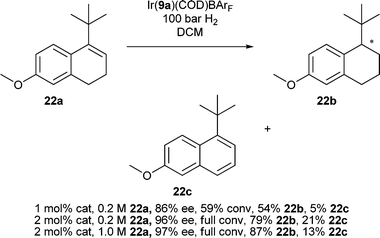 | ||
| Scheme 2 Asymmetric hydrogenation results for the reduction of 22a. | ||
Investigations of olefins 23 and 24 were very encouraging (Fig. 3). High enantioselectivity was obtained with an improvement over our previously reported results for (E)- and (Z)-24 with catalyst loadings as low as 0.25 mol%. This result encouraged us to reexamine the reduction of (E,E)-farnesol 25. We were pleased to see excellent enantioselectivity of greater than 99% ee and an increase in diastereoselectivity with catalyst 7c compared to the best results of our previous study with catalysts 8 and 6a.
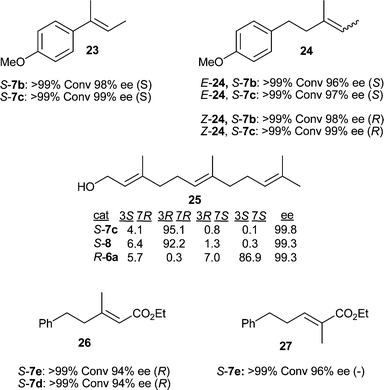 | ||
| Fig. 3 Asymmetric hydrogenation results for substrates 23–27. | ||
Having established these ligands as effective in the reduction of these challenging substrates we turned our attention to the conjugate reduction of ester 26. The mesityl derived catalysts 7d and 7e reduced the α,β-unsaturated ester in 94% ee and greater than 99% conversion. The reduction of α-substituted esters has been a long standing challenge with iridium N,P catalyzed hydrogenations.44 Andersson et al. have proposed a rationale for this with a computational model which indicates a steric and electronic mismatch effect for α-methyl cinnamic methyl ester with an iridium N,P hydride complex.45 We attempted the reduction of 27 and we were surprised to find higher enantioselectivity than with the β-methyl analogue 26. A thorough analysis on a series of esters easily prepared from commercially available α-methylcinnamic acid was quite revealing (Table 5).
|
|
|||
|---|---|---|---|
| ee (%)a | |||
| Catalyst | 29a | 29b | 29c |
| a All reactions were carried out at 50 bar for 18 h at room temperature; conversions less than full are indicated in brackets. b Configuration was (S), opposite of what is depicted. | |||
| (S)-7a | 7 (8) | 3 (97) | 20 (19) |
| (S)-7b | 83 | 80 (17) | 91 (81) |
| (S)-7c | 87 | 83 | 93 |
| (S)-7d | 82 (77) | 80 (88) | 92 (69) |
| (S)-7e | 97 | 97 | 99 |
| (R)-7f | 76b | 95b | 93b |
| (S)-7g | 29 | 8 | 46 |
| (R)-9a | 89b | 92b | 97b |
| (S)-9b | 86 | 81 | 91 (70) |
| (S)-9c | 15 | 6 | 0 (58) |
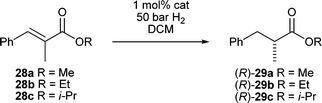
Hydrogenations of cinnamic esters 28a–c were quite slow and high pressure with longer reaction times were required to reach full conversion. Catalyst 7e clearly stood out with 97% ee for the methyl and ethyl ester and almost perfect enantioselectivity for the isopropyl ester. Larger ester groups also increased the enantioselectivity for other catalysts by a considerable measure. It also appears that the substituents on the phosphine atom play a crucial role in complexes 7a–g with a 5-membered ring backbone. The most striking example of this phosphine dependence is given by catalysts 7f and 7g. While 7f with a bis(tert-butyl)phosphino group induces 95% ee, the bis(ortho-tolyl)phosphine analogue 7g shows almost no selectivity.
These results stand in sharp contrast to those obtained with the allylic alcohol 19a which has a closely related geometry to the α-methyl cinnamic esters. Both systems contain a methyl group α to the coordinating oxygen function, E geometry about the CC bond and a conjugated phenyl group but demonstrate opposite trends with the range of tested catalysts. Apparently the allylic alcohols give the best performance with a sterically more accessible catalyst whereas the α-methyl cinnamic esters function better with large substituents surrounding both the metal center and the ester group. However, both substrates give the same configuration of product from the same catalyst enantiomer. Burgess and Zhao have also seen large differences in selectivity between allylic alcohols and methyl ester analogues in diastereoselective reductions, but in contrast to our system the alcohol directed the reduction to the opposite enantioface compared to the ester.46 These results suggest a change in binding modes between these two classes of substrates which implicates a change in the structural requirements for high enantioselectivity.
Conclusion
We have presented a flexible synthesis to hindered pyridine based N,P iridium complexes which expands the scope of both what catalysts can be made and the substrates that can be hydrogenated with high enantioselectivity. This report marks the highest enantioselectivities recorded to date for the asymmetric hydrogenation of dihydronaphthalenes and α-substituted α,β-unsaturated esters. The range of complexes we were able to synthesize has yielded valuable structure–selectivity relations that provide a useful basis for further studies.Acknowledgements
We are grateful for financial support from the Swiss National Science Foundation and the Federal Commission for Technology and Innovation (KTI). We are also grateful for the generous gift of the Nolan NHC catalyst 16 from Umicore AG & Co. We thank Lars Tröndlin for the preparation of substrates 27, 28a–c, and Felix Voss for the synthesis of catalyst starting materials.Notes and references
- J. M. Brown, Hydrogenation of Functionalized Carbon–Carbon bonds, Springer, Berlin, 1999 Search PubMed.
- N. B. Johnson, I. C. Lennon, P. H. Moran and J. A. Ramsden, Acc. Chem. Res., 2007, 40, 1291–1299 CrossRef CAS.
- R. Noyori, Angew. Chem., Int. Ed., 2002, 41, 2008–2022 CrossRef CAS.
- G. Erre, S. Enthaler, K. Junge, S. Gladiali and M. Beller, Coord. Chem. Rev., 2008, 252, 471–491 CrossRef CAS.
- A. J. Minnaard, B. L. Feringa, L. Lefort and J. G. de Vries, Acc. Chem. Res., 2007, 40, 1267–1277 CrossRef CAS.
- W. Tang and X. Zhang, Chem. Rev., 2003, 103, 3029–3070 CrossRef CAS.
- W. Zhang, Y. Chi and X. Zhang, Acc. Chem. Res., 2007, 40, 1278–1290 CrossRef CAS.
- A. Lightfoot, P. Schnider and A. Pfaltz, Angew. Chem., Int. Ed., 1998, 37, 2897–2899 CrossRef CAS.
- A. Pfaltz, J. Blankenstein, R. Hilgraf, E. Hormann, S. McIntyre, F. Menges, M. Schonleber, S. P. Smidt, B. Wustenberg and N. Zimmermann, Adv. Synth. Catal., 2003, 345, 33–43 CrossRef CAS.
- R. Crabtree, Acc. Chem. Res., 1979, 12, 331–337 CrossRef CAS.
- S. P. Smidt, N. Zimmermann, M. Studer and A. Pfaltz, Chem.–Eur. J., 2004, 10, 4685–4693 CrossRef CAS.
- M. G. Schrems and A. Pfaltz, Chem. Commun., 2009, 6210–6212 RSC.
- S. P. Smidt, F. Menges and A. Pfaltz, Org. Lett., 2004, 6, 2023–2026 CAS.
- F. Menges and A. Pfaltz, Adv. Synth. Catal., 2002, 344, 40–44 CrossRef CAS.
- T. L. Church and P. G. Andersson, Coord. Chem. Rev., 2008, 252, 513–531 CAS.
- S. J. Roseblade and A. Pfaltz, Acc. Chem. Res., 2007, 40, 1402–1411 CrossRef.
- X. Cui and K. Burgess, Chem. Rev., 2005, 105, 3272–3296 CrossRef CAS.
- A. Pfaltz and W. J. Drury, III, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5723–5726 CrossRef CAS.
- W. J. Drury, III, N. Zimmermann, M. Keenan, M. Hayashi, S. Kaiser, R. Goddard and A. Pfaltz, Angew. Chem., Int. Ed., 2004, 43, 70–74 CrossRef.
- S. Bell, B. Wustenberg, S. Kaiser, F. Menges, T. Netscher and A. Pfaltz, Science, 2006, 311, 642–644 CrossRef CAS.
- A. Wang, B. Wüstenberg and A. Pfaltz, Angew. Chem., Int. Ed., 2008, 47, 2298–2300 CrossRef CAS.
- S. Kaiser, S. P. Smidt and A. Pfaltz, Angew. Chem., Int. Ed., 2006, 45, 5194–5197 CrossRef.
- A. Baeza and A. Pfaltz, Chem.–Eur. J., 2010, 16, 2036–2039 CrossRef CAS.
- T. Yoshinari, K. Ohmori, M. G. Schrems, A. Pfaltz and K. Suzuki, Angew. Chem., Int. Ed., 2010, 49, 881–885 CAS.
- G. G. Bianco, H. M. C. Ferraz, A. M. Costa, L. C. V. Costa-Lotufo, C. U. Pessoa, M. O. de Moraes, M. G. Schrems, A. Pfaltz and L. F. Silva, J. Org. Chem., 2009, 74, 2561–2566 CrossRef CAS.
- Q.-B. Liu, C.-B. Yu and Y.-G. Zhou, Tetrahedron Lett., 2006, 47, 4733–4736 CrossRef CAS.
- M. P. A. Lyle, N. D. Draper and P. D. Wilson, Org. Biomol. Chem., 2006, 4, 877–885 RSC.
- M. P. A. Lyle and P. D. Wilson, Org. Lett., 2004, 6, 855–857 CrossRef CAS.
- M. P. A. Lyle, A. A. Narine and P. D. Wilson, J. Org. Chem., 2004, 69, 5060–5064 CrossRef CAS.
- K. Ito, M. Yoshitake and T. Katsuki, Tetrahedron, 1996, 52, 3905–3920 CrossRef CAS.
- K. Ito, S. Tabuchi and T. Katsuki, Synlett, 1992, 575–576 CrossRef CAS.
- Y. Xie, H. Huang, W. Mo, X. Fan, Z. Shen, Z. Shen, N. Sun, B. Hu and X. Hu, Tetrahedron: Asymmetry, 2009, 20, 1425–1432 CrossRef CAS.
- S. J. Roseblade and A. Pfaltz, Synthesis, 2007, 3751–3753 CAS.
- F. K. Cheung, C. Lin, F. Minissi, A. L. Criville, M. A. Graham, D. J. Fox and M. Wills, Org. Lett., 2007, 9, 4659–4662 CrossRef CAS.
- C. Mazet, S. Roseblade, V. Koehler and A. Pfaltz, Org. Lett., 2006, 8, 1879–1882 CrossRef CAS.
- J. Uenishi and M. Hamada, Synthesis, 2002, 625–630 CrossRef CAS.
- J. I. Uenishi, T. Hiraoka, S. Hata, K. Nishiwaki, O. Yonemitsu, K. Nakamura and H. Tsukube, J. Org. Chem., 1998, 63, 2481–2487 CrossRef CAS.
- M. Maywald and A. Pfaltz, Synthesis, 2009, 3654–3660 CAS.
- S. C. Zimmerman, Z. Zeng, W. Wu and D. E. Reichert, J. Am. Chem. Soc., 1991, 113, 183–196 CrossRef.
- S. C. Zimmerman and Z. Zeng, J. Org. Chem., 1990, 55, 4789–4791 CrossRef CAS.
- O. Navarro, N. Marion, J. Mei and S. P. Nolan, Chem.–Eur. J., 2006, 12, 5142–5148 CrossRef.
- L. Mantilli, D. Gerard, S. Torche, C. Besnard and C. Mazet, Angew. Chem., Int. Ed., 2009, 48, 5143–5147 CrossRef CAS , S5143/5141–S5143/5119.
- L. Mantilli and C. Mazet, Chem. Commun., 2010, 46, 445–447 RSC.
- M. Schönleber, R. Hilgraf and A. Pfaltz, Adv. Synth. Catal., 2008, 350, 2033–2038 CrossRef CAS.
- C. Hedberg, K. Kallstrom, P. Brandt, L. K. Hansen and P. G. Andersson, J. Am. Chem. Soc., 2006, 128, 2995–3001 CrossRef CAS.
- J. Zhao and K. Burgess, Org. Lett., 2009, 11, 2053–2056 CrossRef CAS.
Footnotes |
| † Dedicated to Professor Albert Eschenmoser on the occasion of his 85th birthday. |
| ‡ Electronic supplementary information (ESI) available: Full characterization of catalysts 7a–h and 9a–c, preparation of intermediates and 1H and 13C spectra. Representative chromatograms and details of the separation of uncommonly tested substrates. CCDC reference numbers 768973–768978. For ESI and crystallographic data for 7a, 7e, 7f, 9a, 12g, and 17 in CIF format see DOI: 10.1039/c0sc00223b |
| This journal is © The Royal Society of Chemistry 2010 |