Intermolecular C–H bond activation of benzene and pyridines by a vanadium(III) alkylidene including a stepwise conversion of benzene to a vanadium-benzyne complex†
José G.
Andino
a,
Uriah J.
Kilgore
a,
Maren
Pink
a,
Andrew
Ozarowski
b,
J.
Krzystek
b,
Joshua
Telser
c,
Mu-Hyun
Baik
a and
Daniel J.
Mindiola
*a
aDepartment of Chemistry and the Molecular Structure Center, Indiana University, Bloomington, IN 47405. E-mail: mindiola@indiana.edu; Fax: (+1) 812-855-8300
bNational High Magnetic Field Laboratory, Florida State University, Tallahassee, Florida 32310, USA
cDepartment of Biological, Chemical and Physical Sciences, Roosevelt University, Chicago, Illinois 60605, USA
First published on 18th June 2010
Abstract
Breaking of the carbon–hydrogen bond of benzene and pyridine is observed with (PNP)V(CH2tBu)2 (1), and in the case of benzene, the formation of an intermediate benzyne complex (C) is proposed, and indirect proof of its intermediacy is provided by identification of (PNP)V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O(η2-C6H4) in combination with DFT calculations.
O(η2-C6H4) in combination with DFT calculations.
Activating C–H bonds of hydrocarbons constitutes an active area of continued development, since such feedstocks could be converted into commodity products by subsequent functionalization.1 One particular reaction that we investigated is the conversion of benzene to benzyne by the mere activation of two adjacent C–H bonds by a combination of 1,2-CH addition and σ-bond metathesis steps. Converting benzene to benzyne constitutes an attractive goal given the wide array of reactivity observed with the latter, especially in the realm of selective transformations in organic chemistry.2 Because of our continuing interest in 3d transition metal systems engaging in metal–carbon multiple bonds, we have explored low-valent vanadium alkyl complexes of the type (PNP)V(CH2tBu)2 (1), (PNP = N[2-P(CHMe2)2-4-methylphenyl]2−).3 The inherent reactivity of these complexes stems from the formation of a transient alkylidene (PNP)V
CHtBu (A), as well as the reactivity of 1 or A towards two-electron oxidants including the activation of atmospheric nitrogen to afford a dinuclear compound with a terminal alkylidene and bridging N2 moieties, [(PNP)VCHtBu]2(μ2;η1,η1-N2).3 In this work, we demonstrate through a series of deuteration and oxidation reactions that 1 can activate two arene ortho-C–H bonds in benzene, to ultimately furnish a rare example of a 3d transition metal benzyne complex.4,5 Labeling studies in combination with theoretical analysis are provided in order to understand the mechanism of conversion of benzene to benzyne. Analogously, we also demonstrate that 1, by route of the unsaturated alkylidene species, A, can perform C–H activation of 2-picolines to yield the corresponding pyridyl complexes.
We found that compound 1 is remarkably stable in the solid state, but gradually transforms to a transient species in solution phase that we propose to be the unsaturated V(III) alkylidene complex A.3 This hypothesis has been probed with a variety of two-electron oxidants, or other reagents such as NCtBu, N2, and 2,2′-bpy. In the latter case, the coordinatively saturated V(III) alkylidene could be trapped in the form of (PNP)VCHtBu(2,2′-bpy).3 Complex 1 is paramagnetic and the μeff value of 2.74(7)μB collected at 25 °C in C6D6 is in accord with the expected spin-only value of 2.83μB for a high-spin d2 configuration.3 To address if complex 1 is indeed a high-spin system with a metal centric V(III) oxidation state supported by a redox non-active PNP ligand we carried out high-frequency and -field EPR (HFEPR‡) measurements of polycrystalline samples from 5–60 K at frequencies ranging from 50 to 300 GHz. A representative spectrum recorded at 10 K and 224 GHz is shown in Fig. 1, and can be recognized readily as originating from a spin triplet (S = 1) state of nearly axial symmetry, with a strong “half-field” transition corresponding to an off-axis turning point of the ΔMS = ±2 transition and a set of ΔMS = ±1 turning points. Simulations have been carried out assuming a perfect powder distribution at each frequency to accompany the experimental spectra. The resonant fields provide the spin Hamiltonian (see ESI†)6 parameters and the relative intensities of the two branches further allows determination of the sign of D, which is positive in this case (Fig. 1). The parameter E is given the same sign by convention, and parameters are as follows: S = 1; D = +3.93 cm−1, E = +0.145 cm−1, gx = gy = 1.955, gz = 1.99 (Fig. 1). These parameters do not change upon raising the temperature up to 60 K. A signal from a V(IV) (3d1, S = 1/2) impurity is also observed at a g value less than 2. The integrated intensity of this impurity relative to that of the V(III) species of interest is very low, as the latter covers the entire field range from 3–12 T, while the former is essentially a “spike” at 8 T.6 Qualitatively, our spectroscopic results demonstrate that use of sufficiently high frequencies combined with high resonant magnetic fields allows observation of EPR resonances from systems traditionally regarded as “EPR-silent”, especially in the context of an organometallic precursor leading to metal–carbon multiple bonds. The relatively large magnitude of D for (PNP)V(CH2tBu)2 (∼4 cm−1) is consistent with a system that is best described as V(III) rather than as an organic (ligand-centered) diradical, which would exhibit zfs much below 1 cm−1. The D value is larger than for five-coordinate V(III), but lower than for classical octahedral S = 1 complexes.7,8 Having understood the role of the unpaired electrons in 1, we investigated its reactivity with benzene.
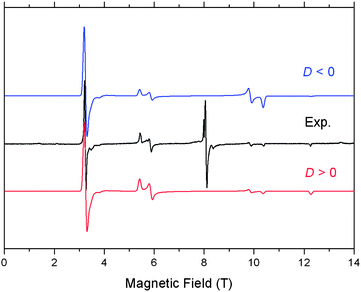 | ||
| Fig. 1 HFEPR spectrum of polycrystalline 1 at 224 GHz and 10 K (black trace) accompanied by two powder pattern simulations, both using: S = 1, |D| = 3.93, |E| = 0.145 cm−1; gx = gy = 1.955; gz = 1.99. Single-crystal linewidth used: 40 mT, isotropic. The upper, blue trace uses D < 0 while the lower, red trace uses D > 0. The group of resonances at ∼8 T likely originates from minor V(IV) impurities. | ||
In C6D6 at 110 °C in the absence of N2, complex 1 slowly undergoes thermal activation of the arene C–H bond within 3 h of reaction time, presumably via 1,2-CH bond addition across the VCHtBu ligand in transient A to furnish the intermediate we speculate to be (PNP)V(CHDtBu)(C6D5) (B-D6) (Scheme 1).6 CH3tBu is generated in the reaction mixture when the solution is assayed by 1H NMR and GC-MS. We found that extended thermolysis of the mixture at 110 °C for >3 h resulted in gradual formation of a new compound, when the solution was monitored by 1H NMR and UV-vis spectroscopy. Unfortunately, the paramagnetic and lipophilic nature of the mixture prevented more detailed characterization of species present in solution. To examine the final product, presumably formed from C–H activation of benzene over an extended period of time, we resorted to two-electron redox chemistry with N2O (110 °C, 4 h) of the solution mixture which had been heated for 6 h at 110 °C to ensure complete consumption of 1 in C6D6, which produced the d0 complex (PNP)VO(η2-C6D4) (2-D4), as a brown colored powder (Scheme 1).6 The isotopomer, 2-D4, can be analogously prepared from thermolysis of 1 in C6H6, in the presence of N2O. No exchange is observed between 2-D4 and C6H6 under identical thermolytic conditions, although the conditions used to generate the metal benzyne complex is not surprising, since Teuben and co-workers have estimated relatively high barriers for formation of these species types.5
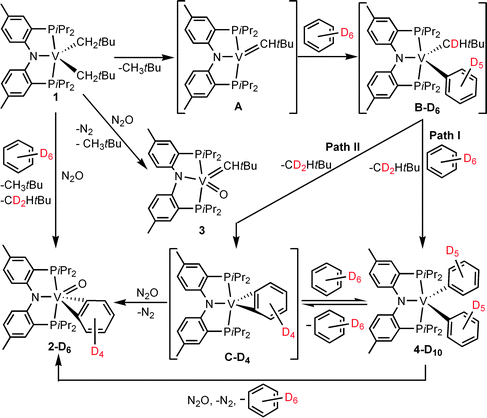 | ||
| Scheme 1 C–H bond activation of benzene by the alkylidene intermediate A to ultimately produce complex 2. The two proposed pathways are depicted along with the synthesis of 3 from 1 and N2O. | ||
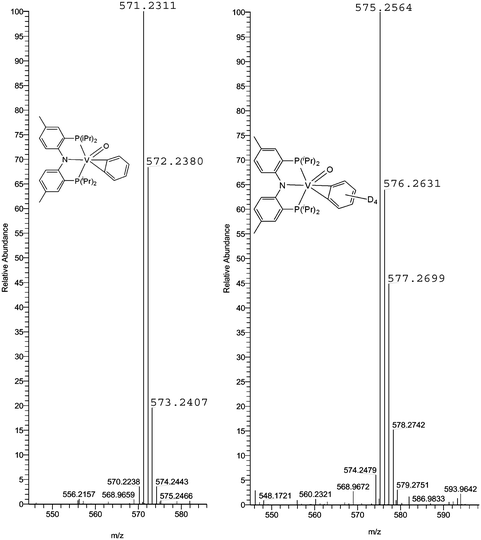
Even though we were unable to obtain single crystals of complex 2, 13C NMR spectroscopic data are consistent with an η2-benzyne ligand having inequivalent α-carbon resonances (157.6 and 156.6 ppm). Because of the skewing of the PNP phosphine arms, complex 2 displays also two broad resonances at 65.2 and 60.2 ppm in the 31P NMR spectrum. Both 13C and 31P NMR resonances are broad presumably due to scalar coupling of the 51V to the 31P and/or 13C nuclei. Also observed are the inequivalent benzyne resonances at 8.06 (d, 1H, 3JH–H = 5.4 Hz), 8.02 (d, 1H, 3JH–H = 5.7 Hz), and 7.64 (overlapping m, 2H) ppm in the 1H NMR spectrum of 2 (Fig. 2). In order to resolve the multiplet at 7.64 ppm a deconvolution routine9 was applied to the spectrum which revealed a pattern reminiscent of unsymmetrically 1,2-substituted halobenzenes.9 As shown in Fig. 2, the α-protons display only the strong coupling to the β-protons while the latter display a more complicated pattern due to long range coupling inasmuch as a roofing effect due to the approaching resonances of each proton.9 Not surprisingly, a similar pattern for the benzyne resonances in complex Cp2Zr(η2-C6H4)(PMe3) was observed by 1H and 13C NMR spectroscopy.2 We unambiguously distinguish these resonances from those of the PNP aryl groups, since isotopomer, 2-D4, does not display such resonances. Formation of an oxo-benzyne complex was further established by high-resolution CI-MS of 2 (theoretical [M]+ 571.2338; exp. 571.2311) and that of its isotopomer, 2-D4 (theoretical [M]+, 575.2589; exp. 575.2564), both of which are shown in Fig. 3. We propose the oxo ligand in 2 to be terminal on the basis of CI-MS and that the precursor, 1, can also be oxidized with N2O to furnish the mononuclear and thus terminally coordinated oxo product (PNP)VO(CHtBu) (3) and CH3tBu, quantitatively (Scheme 1).3 In fact, we have prepared mononuclear vanadium tellurides having one or two of these ligands in a terminal mode,10 therefore hinting that a bridging oxo ligand would be rather rare for such ligand scaffold, especially in a coordinatively saturated environment. Based on our data, we cannot refute the possibility of the O-atom inserting into the benzyne ligand to furnish a chelating oxometallacyclobutene vanadium(III) species,11 but we would expect this type of complex to be paramagnetic due to the lack of suitable π-acceptor ligands. To unambiguously ascertain if complex 3 has indeed a benzyne and terminal oxo ligand, a DFT optimized structure was computed for both full and truncated models.6 In both cases, the preferred geometry for 3 displays the terminal oxo ligand orthogonal to the benzyne plane and this complex is lower in energy by >15 kcal mol−1 than a hypothetical O-inserted product such as an oxometallacyclobutene vanadium(III) species (Fig. 4).6 The terminal vanadium-oxo distance of 1.592 Å is analogous to that found for the structure of 3 (1.595(4) Å),3 while the benzyne C1–C2 closely resembles the distance observed in molecular structure of Cp2Zr(η2-C6H4)(PMe3) (1.364(8) Å).2 In addition, calculation of the chemical shifts using the optimized geometry confirmed the assignment of benzyne resonances from 1H NMR spectrum of 2. In both cases, the benzyne protons display a unique pattern where a pair of doublets at 8.06 and 8.02 ppm correspond to the α-protons that will only couple with the β-protons. As shown in Fig. 2, the inequivalent β-protons display what appears to be a quintet at 7.64 ppm as a result of near superimposition of their coupling to each other and the α-protons (vide supra).
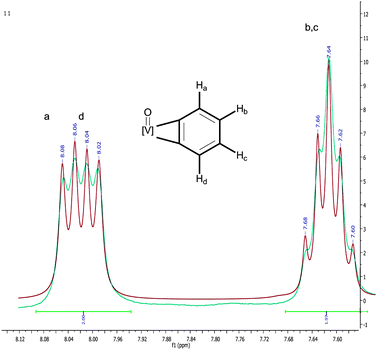 | ||
| Fig. 2 Expansion of benzyne region of 1H NMR spectrum of complex 2. Blue trace represents the original 1H NMR spectrum and the red trace is the deconvoluted spectrum. | ||
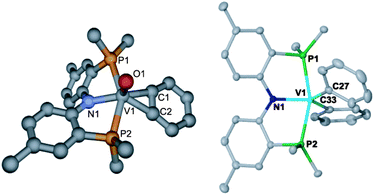 | ||
| Fig. 4 Left figure is an optimized structure predicted by DFT for complex 2, with methyls replacing iPr and hydrogen atoms omitted for clarity. Right figure is the molecular structure of complex 4 omitting H-atoms, and isopropyl methyls on P for the purpose of clarity and with thermal ellipsoids displayed at the 50% probability level. Selected metrical parameters (distances in Å, angles in °), for 3: V1–C1, 2.033; V1–C2, 2.031; V1–P1, 2.451; V1–P2, 2.458; V1–N, 2.100; V1–O, 1.592; C1–C2, 1.338; N1–V1–C1, 131.41; P1–V1–P2, 149.78. For 4: V1–C27, 2.0869(12); V1–C33, 2.0628(12); V1–N1, 1.9750(10); V1–P1, 2.4875(3); V1–P2, 2.4944(3); P1–V1–P2, 159.451(13); N1–V1–C27, 118.38(4); N1–V1–C33, 119.10(5). | ||
Due to the paramagnetic nature of 1 and the likely intermediates ultimately leading to 2, we conducted further isotopic labeling studies with C6D6. Treatment of 1 with C6D6 (110 °C for 6 h) revealed formation of CH3tBu and CD2HtBu as volatile by-products (GCMS and 1H NMR spectroscopy) formed in the reaction mixture prior to oxidation with N2O, thus suggestive of α-hydrogen abstraction preceding the C–H bond activation of the arene, and not supporting the plausible alternative of σ-bond metathesis playing the main role. Therefore, we speculate complex B to be a likely intermediate generated from C–H bond activation by the alkylidene ligand in transient A. From the putative complex B two likely pathways can be proposed: (Path I) a stepwise mechanism via a σ-bond metathesis of benzene to yield diphenyl, (PNP)V(C6H5)2 (4), followed by β-hydrogen abstraction to form C and benzene, or (Path II) a concerted β-hydrogen abstraction step to yield the vanadium(III) benzyne intermediate, (PNP)V(η2-C6H4) (C) (Scheme 1). Under the premise that 1 does not undergo σ-bond metathesis with benzene, and the precedence that Legzdins and co-workers12 have observed benzene C–H bond activation via an isolable Mo-benzyne complex, our Path II proposed in Scheme 1 is therefore favored.
To establish if Path I was plausibly operating in this reaction, we focused our attention on complex 4, since this species could be a precursor to 2 as well as an intermediate formed along the C–H activation of benzene. We prepared compound 4 independently in order to compare the 1H NMR spectra with the crude reaction mixture obtained from thermolysis at 110 °C (>6 h, without subjecting the mixture to N2O). The treatment of (PNP)VCl23 with two equiv. of PhMgBr rapidly produced 4 as orange colored crystals (79% isolated yield) subsequent to workup of the reaction mixture. Complex 4 has been characterized by 1H NMR spectroscopy, solution Evans magnetization measurement (μeff = 2.78μB), CI-MS and single crystal X-ray diffraction analysis (Fig. 4).6 The molecular structure of 4, collected on a small crystal using synchrotron radiation,‡6 features a vanadium complex confined to a pseudo trigonal bipyramid with standard V–C distances (2.0628(12) and 2.0869(12) Å) in accord with other vanadium(III) bis-alkyl systems reported in the literature.3,13 The overall gross structural geometry of 4 resembles the structure of the neopentylidene precursor 1. As suspected from our previous experiments, the 1H NMR spectrum of complex 4 displays similarly shifted resonances to those observed for crude reaction mixtures of 1 with benzene, while thermolysis of isolated samples of 4 with N2O produce 2 cleanly.
Having prepared 4 independently, and to address if the two-electron oxidation step promotes elimination of benzene via this species, complex 4 was first thermolyzed in C6H6 at 110 °C for 3 h, and the volatiles then replaced with THF-D8. Oxidation of the reaction mixture with N2O in THF-D8 (110 °C, 6 h) does indeed confirm C6H6 to be formed, therefore implying that the two-electron redox step promotes β-hydrogen abstraction to form the benzyne ligand in 2. We cannot, however, infer from this study if 4 is present with some of the vanadium(III) benzyne intermediate, C, or if this species is ever formed prior to the two-electron oxidation.
Due to the paramagnetic nature of intermediates A and B, we also do not have compelling kinetic data to decipher whether formation of A or 1,2-CH bond addition is the overall rate-determining step along the intermolecular benzene C–H activation process. Previous kinetic studies by Girolami,4 Gibson,14 Hessen,15 Legzdins,16 and us17 have clearly established that formation of the M–C multiple bond is generally the slow step in the intermolecular C–H activation sequence. To probe for whether or not the proposed intermediates and their corresponding barriers en route to formation of 4 are electronically and thermodynamically feasible, we computed reaction Path II in addition to other plausible pathways.6 As we began to model the reaction using DFT, we resorted to a truncated model in which the iPr substituents were replaced by Me groups.6 We noted that the activation of the first benzene molecule was competitive with respect to loss of neopentane (using a truncated model), which means the latter could be the rate-limiting step based on precedence in the literature (vide supra).4,14–17 However, when we included the full model in our calculations, the activation of benzene is the rate-determining step. We attribute the differences between the two models to be the stabilization gained by loss of bulky alkane such as neopentane, as well as the difficulty in accessing the vanadium center when sterically encumbering iPr groups are placed in the phosphorus sites. One important difference to take into account when comparing our system with a similar d0 Ti system17 is the presence of two unpaired electrons on vanadium, which results in an overall increase in the repulsion of the metal center with the incoming substrate as well as the leaving group. In accord with the HFEPR data for 1, the potential energy surface (PES) shown in Fig. 5 includes all models in a triplet ground state. As observed in Fig. 5, formation of B represents a concerted and slow process with intermediate A having a thermodynamic driving force of 18.28 kcal mol−1. Without a doubt, the formation of B represents the most difficult step since it must traverse a barrier of ∼33 kcal mol−1. From our calculations, the barriers associated with subsequent steps should be readily accessed with the high temperatures used in these experiments (i.e. >100 °C). With the exception of TS-3, all other activated complexes are 4-centered involving the linear transfer of a hydrogen atom to minimize distortion (Fig. 5). From our calculation, conversion of C to 4 represents a fast step, presumably due to the reactive and unsaturated nature of the benzyne ligand. Based on our barriers leading to TS-3 and TS-4 from the common intermediate, C, we can clearly establish the benzyne ligand to be more selective for aromatic vs. an aliphatic C–H bond activation.6 Therefore, this PES together with our isotopic labeling studies and independent synthesis of 3 from intermediate 4 tantalizingly suggest Path II to be the most likely route to C–H activation of benzene. In contrast to previous work involving 1,2-CH addition of an arene across the M–C multiple bond4,14-17 our computational studies suggest C–H bond activation of the arene (A → B) to be the slowest step along formation of 4.
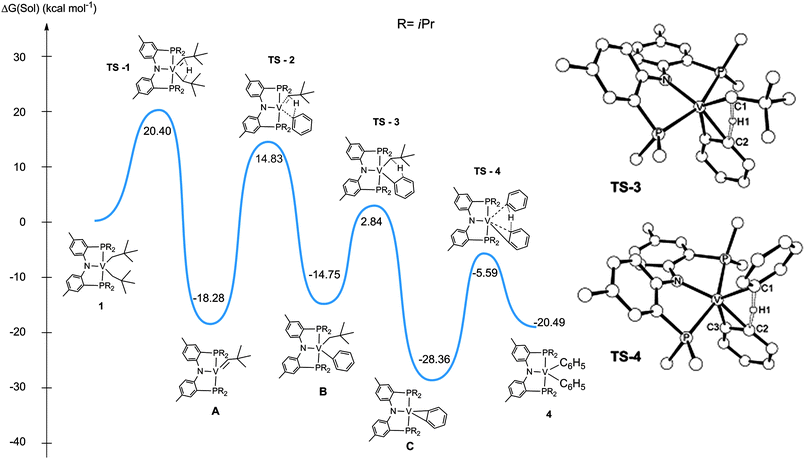 | ||
| Fig. 5 Reaction profile for the most plausible reaction pathway from 1 to 4 (B3LYP/6-31G**/LACVP). | ||
Benzene is not the only substrate amenable to C–H activation since 2-picoline and 2-phenyl picoline react slowly (>4 days, N2 excluded) at 25 °C with 1 to afford the pyridyl complexes (PNP)V(CH2tBu)(η2-NC5RH4) (R = CH3 (5) or Ph (6), respectively, Scheme 2).6 Unfortunately, we were unable to isolate the product resulting from pyridine activation by precursor 1. Unlike the benzene C–H activation intermediate, B, compounds 5 and 6 are crystalline, isolable products obtained in 20 and 22% yield, respectively.6 Solution Evans magnetic susceptibility measurements are in accord with both systems having two unpaired electrons (μeff = 2.7μB for 5, and 2.8μB for 6), while high-resolution CI-MS reveals the corresponding ions [M]+, 642.3386 (theoretical for 5 is 642.3437) and [M]+ 704.3587 (theoretical for 6 is 704.3593), respectively. Most notably, Fig. 6 depicts the two solid state structures 5 and 6, thus unambiguously confirming the presence of both neopentyl and pyridyl moieties.6 The two crystal structures are akin with distances V–Npyridyl (2.1200(15) and 2.1680(12) Å), V–Cpyridyl (2.0514(18) and 2.0414(14) Å), and V–Cneopentyl (2.1262(18) and 2.1191(14) Å). Other salient metrical parameters for each are shown with Fig. 6. It was found that oxidation of 5 or 6 under a bed of N2O results in quantitative formation of 3 along with liberation of the corresponding substituted pyridine (Scheme 2).6
 | ||
| Scheme 2 C–H activation of 2-picolines to form the pyridyl complexes 5 and 6 and treatment with N2O to form the oxo alkylidene 3 and free picoline | ||
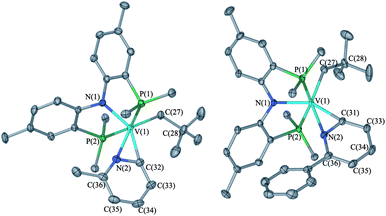 | ||
| Fig. 6 Molecular structure of complexes 5 (left) and 6 omitting H-atoms, solvent, and isopropyl methyls on P for the purpose of clarity. For 5, a co-crystallized HPNP was also omitted. Thermal ellipsoids are displayed at the 50% probability level. Selected metrical parameters (distances in Å, angles in °): For 5: V1–C32, 2.0514(18); V1–N2, 2.1200(15); V1–C27, 2.1262(18); V1–N1, 2.0369(14); V1–P1, 2.5462(6); V1–P2, 2.5442(6); P1–V1–P2, 153.503(19); V1–C27–C28, 127.31(13); N2–V1–C32, 37.70(6). | ||
Conclusions
We have demonstrated that a transient, four-coordinate vanadium alkylidene generated from a high-spin metal-centric diradical can engage in intermolecular 1,2-CH addition reactions of benzene and pyridines. We have also discovered that two-electron oxidation chemistry of a V(III) diphenyl complex accessed by use of N2O can promote formation of a rare example of a V(V)-benzyne complex which is trapped as an oxo. Although complex 2 is in limited company with another vanadium benzyne complex, CpV(C6H4)(PMe3)2,5 our work has established that benzyne can be derived from double deprotonation of benzene and by using vanadium–carbon multiple bonds. Future work will explore the chemistry of complexes such as 2, since the benzyne motif, derived by double deprotonation of benzene, could be further functionalized or transferred. Use of DFT has been essential to confirm the identity of the benzyne-oxo complex, 3. Combining spectroscopic data with the computed DFT model of this diamagnetic species leaves little room for ambiguity both theoretically and experimentally. The energy profile provides a clear picture of the intermediacy of benzyne complex C. This knowledge enables us to plan a strategy to systematically activate and functionalize arenes and pyridines, and we are conducting studies to model the feasibility of certain products.Notes and references
- R. H. Crabtree, Chem. Rev., 1985, 85, 245 CrossRef CAS
; R. G. Bergman, Science, 1984, 223, 902 CAS
- S. L. Buchwald, B. T. Watson and J. C. Huffman, J. Am. Chem. Soc., 1986, 108, 7411 CrossRef CAS
- U. J. Kilgore, C. A. Sengelaub, M. Pink, A. R. Fout and D. J. Mindiola, Angew. Chem., Int. Ed., 2008, 47, 3769 (
Angew. Chem.
, 2008
, 120
, 3829
) CrossRef CAS
- A transient titanium benzene derived from benzene activation has been proposed on the basis of isotopic labelling studies. J. Cheon, D. M. Rogers and G. S. Girolami, J. Am. Chem. Soc., 1997, 119, 6804 Search PubMed
- J. K. F. Buijink, K. R. Kloetstra, A. Meetsma, J. H. Teuben, W. J. J. Smeets and A. L. Spek, Organometallics, 1996, 15, 2523 CrossRef CAS
- See supporting information for details†.
- S. Ye, F. Neese, A. Ozarowski, D. Smirnov, J. Krzystek, J. Telser, J.-H. Liao, C.-H. Hung, W.-C. Chu, Y.-F. Tsai, R.-C. Wang, K.-Y. Chen and H.-F. Hsu, Inorg. Chem., 2010, 49, 977 CrossRef CAS
- J. Krzystek, A. T. Fiedler, J. J. Sokol, A. Ozarowski, S. A. Zvyagin, T. C. Brunold, J. R. Long, L.-C. Brunel and J. Telser, Inorg. Chem., 2004, 43, 5645 CrossRef CAS
- W. B. Smith and G. M. Cole, J. Phys. Chem., 1965, 69, 4413 CrossRef CAS
- U. J. Kilgore, J. A. Karty, M. Pink, X. Gao and D. J. Mindiola, Angew. Chem., Int. Ed., 2009, 48, 2394 CAS
- Insertion of O-atom into strained metallacyclopropenes, using a thermodynamically powerful oxidant such as N2O have been documented. A. K. List, K. Koo, A. L. Rheingold and G. L. Hillhouse, Inorg. Chim. Acta, 1998, 270, 399 Search PubMed
- K. Wada, C. B. Pamplin, P. Legzdins, B. O. Patrick, I. Tsyba and R. Bau, J. Am. Chem. Soc., 2003, 125, 7035 CrossRef CAS
- P. H. M. Budzelaar, A. B. von Oort and A. G. Orpen, Eur. J. Inorg. Chem., 1998, 1485 CrossRef CAS
- M. P. Coles, V. C. Gibson, W. Clegg, M. R. J. Elsegood and P. A. Porrelli, Chem. Commun., 1996, 1963 RSC
- H. van der Heijden and B. Hessen, J. Chem. Soc., Chem. Commun., 1995, 145 RSC
- C. B. Pamplin and P. Legzdins, Acc. Chem. Res., 2003, 36, 223 CrossRef CAS
- A. R. Fout, J. L. Scott, D. L. Miller, B. C. Bailey, J. C. Huffman, M. Pink and D. J. Mindiola, Organometallics, 2009, 28, 331 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available: Complete structural, synthetic and spectroscopic data as well as all information regarding HFEPR data collection and simulation. CCDC reference numbers 730602–730604. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c0sc00201a |
| ‡ ChemMatCARS Sector 15 is principally supported by the National Science Foundation/Department of Energy under grant number CHE-0535644. Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357. HFEPR studies were supported by the National High Magnetic Field Laboratory, which is funded by the NSF through Cooperative Agreement DMR 0654118, the State of Florida, and the DOE. J.T. and J.K. acknowledge NHMFL UCGP grant 5062. |
| This journal is © The Royal Society of Chemistry 2010 |