(Imido)vanadium(V)-alkyl, -alkylidene complexes exhibiting unique reactivity towards olefins and alcohols
Kotohiro
Nomura
*a and
Wenjuan
Zhang
b
aDepartment of Chemistry, Tokyo Metropolitan University, 1-1 Minami Osawa, Hachioji, Tokyo 192-0397, Japan. E-mail: ktnomura@tmu.ac.jp
bKey Laboratory of Engineering Plastics, Institute of Chemistry, Chinese Academy of Sciences, Beijing, 100190, People's Republic of China. E-mail: zhangwj@iccas.ac.cn
First published on 28th May 2010
Abstract
This minireview introduces recent results in the synthesis of a series of (imido)vanadium(V)-alkyl complexes, and some reactions with alcohols (phenols) that are proposed to proceed via intermediates involving coordination of the alcohols (phenols). (Imido)vanadium(V)-alkylidene complexes, prepared by α-hydrogen elimination in the presence of a neutral donor ligand (PMe3, etc.), exhibited high activity for ring-opening metathesis polymerisation of cyclic olefins (norbornene) even at high temperature; these are promising for olefin metathesis by vanadium complex catalysts.
 Kotohiro Nomura | Kotohiro Nomura received his PhD in 1993 from Osaka University, before joining the group of Richard Schrock (MIT). He then moved to Sumitomo Chemical Company, Ltd. and then the NAIST as an associate professor in 1998. Recently he moved to Tokyo Metropolitan University as a full professor. His research focuses on the design of molecular catalysts. He received The CSJ Award for Young Chemists in Technical Development from The Chemical Society of Japan in 1996, and The Society Award (Chemical & Engineering) from the Catalysis Society of Japan in 2001. He has co-authored over 160 publications and is an editorial board member for J. Mol. Catal. A: Chem. |
 Wenjuan Zhang | Wenjuan Zhang joined the group of Wen-Hua Sun (ICCAS) for her PhD in olefin polymerisation. She then moved to the group of Kotohiro Nomura (NAIST) in 2006, as a JSPS postdoctoral fellow for two years. She focused on vanadium(V) chemistry for olefin coordination insertion and metathesis polymerisation and related organometallic chemistry. After finishing her fellowship, she returned to ICCAS as an associate professor in 2008. Her recent research has focused on the design of transition metal catalysts for olefin polymerisation, and related organometallic chemistry, and ring-opening polymerisation. She received the Award for Excellent Young Scientists from the Institute of Chemistry, Chinese Academy of Sciences, both in 2005 and 2009. |
Introduction
Efficient carbon–carbon bond formation is one of the most important reactions in organic synthesis as well as in polymer synthesis, and metal-alkyl and metal-alkylidene species are known to play essential roles. The synthesis and reaction chemistry of metal-alkyl complexes have thus been considered to be important not only for designing efficient catalysts, but also for a better understanding of the organic reactions, especially in terms of catalytic cycles or reaction pathways.1 Polyolefins produced by metal catalysed olefin coordination/insertion polymerisation are important synthetic polymers in our daily life, and the market capacity (even of the conventional polyethylene and polypropylene) still increases every year. Catalyst development is considered as core technology, therefore considerable attention has been devoted to the design of efficient catalysts that are highly effective for the synthesis of new polymers which cannot be prepared using conventional catalysts.2–5Classical Ziegler-type vanadium catalysts (e.g. VOCl3, VCl4, VCl3–AlBr3, AlCl3–AlPh3, AliBu3, SnPh4) are known to display unique characteristics in olefin polymerisation. For example, the catalyst system afforded (i) high molecular weight linear polyethylene with uniform molecular weight distribution,6 and (ii) high molecular weight amorphous polymers applied to the synthesis of ethylene/propylene/diene copolymers (called EPDM, synthetic rubbers)7,8 as well as ethylene/cyclic olefin copolymers (COC, as commercialised as ‘APEL™’ in Mitsui Chemicals, Inc., information and electronic materials). Moreover, (iii) the catalyst system [V(acac)3 (acac = acetylacetonato)–Et2AlCl] polymerises propylene to give not only a syndiotactic “living” polymer with narrow molecular weight distribution (Mw/Mn = 1.05–1.20),9 but also diblock copolymers of propylene and methyl methacrylate (MMA).10
Due to the promising characteristics demonstrated above, the design and synthesis of new vanadium complex catalysts directed toward controlled polymerisation has thus been an attractive target.3a,b,5 Although examples of the synthesis of vanadium complexes used as catalyst precursors for olefin polymerisation are known, successful examples which exhibit the above unique characteristics of vanadium were limited until recently.3a,b,5a,b Moreover, examples concerning the synthesis and reaction chemistry of vanadium-alkyls were limited until recently,11–13 probably due to these vanadium-alkyls tending to be reactive and/or thermally labile, and reductions to lower oxidation states often occurred in the reactions with organometallic reagents.13
Olefin metathesis [such as ring-opening metathesis polymerisation (ROMP), ring-closing metathesis and cross metathesis reactions etc.], introduces promising possibilities for the synthesis of functional polymers as well as valuable organic compounds,14 as demonstrated especially by molybdenum15 and ruthenium.16 Transition metal-alkylidene complexes, especially high-oxidation-state early transition metal alkylidene complexes, attract considerable attention15,17 because they play essential roles as catalysts in olefin metathesis and Wittig-type or group transfer reactions,14,15,17 as demonstrated especially by molybdenum (so called Schrock type complexes).14,15 However, as described below, no examples that promote the olefin metathesis reaction using vanadium(V)-alkylidene complexes were known until recently.
In this minireview, our recent efforts concerning (i) the synthesis of a series of (imido)vanadium(V)-alkyl, and alkylidene complexes, including recent examples by others, and (ii) some reactions of these vanadium(V)-alkyl complexes with alcohols (phenols, ROH) that proceed via coordination of ROH to the vanadium metal center are discussed.16
1. Synthesis of (imido)vanadium(V)-dichloro complexes as effective catalyst precursors for olefin polymerisation
A series of (imido)vanadium(V)-dichloride complexes, V(NAr)Cl2(X) [Ar = 2,6-Me2C6H3; X = aryloxo (1),18 ketimide (2),19 phenoxy-imine (3),20 (2-anilidomethyl)pyridine (4)21] or V(NAd)Cl2(X) [Ad = 1-adamantyl; X = aryloxo (5), ketimide (6)],22 could be prepared by treating (ArN)VCl3 with the corresponding lithium salts [such as LiN![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CtBu2, LiOAr etc.] in Et2O or toluene (Scheme 1). The crystallographic results (Fig. 1) revealed that the ketimide-dichloride analogue, V(NAr)Cl2(NCtBu2) (2a), folds in a distorted tetrahedral geometry around V, and the complex (2a) is a 14 electron species.19 As shown in Scheme 2, complex 2a reacted with one equivalent of PMe3 to afford V(NAr)Cl2(NCtBu2)(PMe3) that folds in a distorted trigonal bipyramidal structure around V; the reaction of 2a with bis(dimethylphosphino)ethane (dmpe) afforded V(NAr)Cl2(NCtBu2)(dmpe), which folds in a distorted octahedral geometry around V.19
CtBu2, LiOAr etc.] in Et2O or toluene (Scheme 1). The crystallographic results (Fig. 1) revealed that the ketimide-dichloride analogue, V(NAr)Cl2(NCtBu2) (2a), folds in a distorted tetrahedral geometry around V, and the complex (2a) is a 14 electron species.19 As shown in Scheme 2, complex 2a reacted with one equivalent of PMe3 to afford V(NAr)Cl2(NCtBu2)(PMe3) that folds in a distorted trigonal bipyramidal structure around V; the reaction of 2a with bis(dimethylphosphino)ethane (dmpe) afforded V(NAr)Cl2(NCtBu2)(dmpe), which folds in a distorted octahedral geometry around V.19
 | ||
| Scheme 1 Selected examples for (imido)vanadium(V) dichloride complexes. | ||
 | ||
| Fig. 1 Structures for V(NAr)Cl2(NCtBu2) (2a, top left), V(NAr)Cl2(NCtBu2)(PMe3) (top right) and V(NAr)Cl2(NCtBu2)(dmpe) (bottom).19 All hydrogen atoms are omitted for clarity. | ||
 | ||
| Scheme 2 Synthesis of (arylimido)vanadium(V)-dichloro complexes containing a ketimide ligand.19 | ||
As described in the introduction, these complexes were effective catalyst precursors for ethylene (co)polymerisation in the presence of Al cocatalysts.18,20–24 In particular, the arylimido-aryloxo analogues (1) showed remarkable catalytic activities not only for ethylene polymerisation,18,23 but also for ethylene/norbornene copolymerisation in the presence of halogenated Al alkyls (Et2AlCl, Me2AlCl, EtAlCl2etc.), affording high molecular weight polymers with uniform distributions.23 Selected results in ethylene polymerisation using V(NAr)Cl2(O-2,6-Me2C6H3)–Al cocatalyst systems are summarised in Table 1.23b
| Complex/μmol | Al cocatalyst | Solvent | Activityb kg-PE/mol-V h | TOFc/h−1 | M w × 10−5 | M w/Mnd | M η × 10−5 |
|---|---|---|---|---|---|---|---|
| a Selected data from ref. 23b. Conditions: solvent + cocatalyst solution = total 30 mL, ethylene 8 atm, 0 °C (25 °C with MAO), 10 min, Al cocatalyst 250 μmol (MAO 2.5 mmol). b Activity in kg-polymer/mol-V h. c TOF = (molar amount of ethylene consumed)/(mol-V h). d GPC data in o-dichlorobenzene vs. polystyrene standards. e Molecular weight by viscosity. f Insoluble in o-dichlorobenzene for GPC measurement. g Cp*TiCl2(O-2,6-iPr2C6H3) was used, ethylene 4 atm, 25 °C, 10 min (cited from ref. 3d). | |||||||
| 0.05 | Me2AlCl | Toluene | 27500 | 980000 | —f | — | 89.8 |
| 0.05 | Et2AlCl | Toluene | 11700 | 415000 | 36.5 | 1.42 | 98.7 |
| 0.05 | i Bu2AlCl | Toluene | 52000 | 1850000 | —f | — | 125 |
| 0.01 | i Bu2AlCl | Toluene | 64800 | 2310000 | —f | — | |
| 0.05 | EtAlCl2 | Toluene | 37400 | 1330000 | 6.02 | 3.04 | |
| 1.0 | MAO | Toluene | 2930 | 104000 | 28.7 | 1.64 | |
| 0.2 (Ti)g | MAO | Toluene | 8400 | 298000 | 12.4 | 1.90 | |
| 0.05 | Et2AlCl | C6H5Cl | 19000 | 677000 | —f | — | 38.3 |
| 0.05 | EtAlCl2 | C6H5Cl | 64400 | 2300000 | 24.4 | 3.14 | |
| 0.05 | Me2AlCl | CH2Cl2 | 19700 | 702000 | 24.2 | 3.38 | |
| 0.05 | Et2AlCl | CH2Cl2 | 13200 | 471000 | 13.4 | 3.93 | |
| 0.05 | i Bu2AlCl | CH2Cl2 | 45200 | 1610000 | —f | — | 58.5 |
| 0.01 | EtAlCl2 | CH2Cl2 | 584000 | 20800000 | —f | — |
The catalytic activity in the ethylene polymerisation was highly dependent upon the Al cocatalyst employed, and the activities in toluene increased in the order: iBu2AlCl (52000 kg-PE/mol-V h) > EtAlCl2 (37400) > Me2AlCl (27500) > Et2AlCl (11700) > MAO (2930) ≫ Et2Al(OEt), Me3Al, Et3Al (trace or less). The activity did not decrease after 30 min. The activity was highly affected by the solvent employed; the activity of 584000 kg-PE/mol-V h (TOF 20800000 h−1, 5780 s−1) was attained in CH2Cl2 in the presence of EtAlCl2. The resultant polymers prepared in toluene possessed ultrahigh molecular weights with unimodal molecular weight distributions (the Mη values in the resultant polymers prepared in the presence of iBu2AlCl and Me2AlCl were 9.87–12.5 × 106 and 8.98 × 106, respectively). The activity decreased upon addition of CCl3CO2Et, which can be commonly used as an effective additive (for restarting the catalytic cycle from the deactivated catalyst by re-oxidation to a higher oxidation state) in the ethylene polymerisation using vanadium(III) and/or vanadium(IV) complexes such as V(acac)3 (acac = acetylacetonato), V(β-diketonate)3.5,25 The results clearly suggest that the catalytically active species were thus different from those prepared from vanadium(III),(IV) complexes. We assumed that a plausible reason for the observed difference in the catalytic activities in the presence of MAO and Et2AlCl cocatalysts might be due to the different catalytically-active species and catalyst/cocatalyst nuclearity effect26 generated in the two catalyst systems.
2. Synthesis of (imido)vanadium(V)-alkyl complexes, and some reactions with alcohols, thiols, borates
2-1. Synthesis of (imido)vanadium(V)-dialkyl, trialkyl complexes
The reaction of 2a with LiCH2SiMe3 in n-hexane afforded V(NAr)(CH2SiMe3)2(NCtBu2) (7) in high yield (95%, Scheme 3).21 The coordination of PMe3 to 7 was not observed even by addition of an excess amount (7.0 equiv.) at 25 °C, and this observation might be due to the steric hindrance of the rather bulky CH2SiMe3 group around the V metal center. The dialkyl complexes containing (2-anilidomethyl)pyridine ligand 8 were also isolated by adopting this approach (yields 60, 73% for R = Me, iPr, respectively).21 However, attempts at isolating the dialkyl complexes from the other dichloro analogues (1, 3, 5, 6) failed due to difficulties in the isolation of the pure, desired complexes by recrystallisation in most cases. For example, reaction of V(NAr)Cl2(O-2,6-iPrC6H3) with 2.0 equiv. of LiCH2SiMe3 in n-hexane afforded the corresponding dialkyl complex (>90%) containing a tiny amount of unidentified by-product (in the 1H NMR spectrum) which caused difficulties for separation.27 As described below, these dialkyl complexes are key intermediates for the synthesis of the alkylidene complexes by α-hydrogen elimination, therefore, we had to find another possibility for the isolation of the dialkyl complexes.
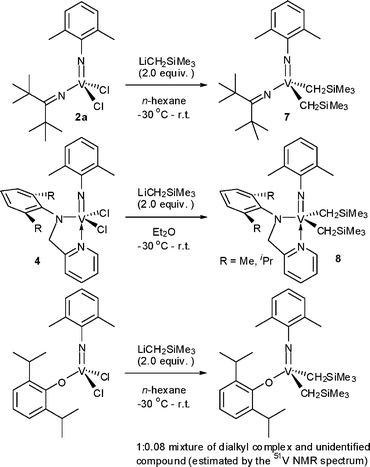 | ||
| Scheme 3 Synthesis of dialkyl complexes.19,21,27 | ||
In contrast, the analytically pure trialkyl analogues, V(NAr)(CH2SiMe3)3 (9) and V(NAd)(CH2SiMe3)3 (10), could be isolated in high yields (Scheme 4).27,28 Although these complexes (9, 10) are low coordinate unsaturated vanadium(V) complexes, the coordination of PMe3 to V was not observed (by 51V NMR spectroscopy), even by addition of an excess amount (7.0 equiv.) in C6D6 at 25 °C, and this observation would be due to the steric hindrance of three CH2SiMe3 ligands around the vanadium(V) metal center. Resonance in the 51V NMR spectrum (δ 1070 ppm) is relatively close to that in V(N-2,6-iPr2C6H3)(CH2Ph)3 (1008 ppm),12b and the resonances in a series of (imido)vanadium(V)-trialkyl complexes are found to be influenced by the imido substituent employed [ca. V(N-4-MeC6H4)(CH2SiMe3)3 (δ 1028),29 V(NAd)(CH2SiMe3)3 (δ 895),28 V(NtBu)(CH2SiMe3)3 (δ 877)30]. The crystallographic result (Fig. 2)28 revealed that 10 folds in a rather distorted tetrahedral geometry around V. The V–N–C bond angle is 180.00(17)° and the V–N distance is 1.6317(14) Å, and the three V–C bond distances and angles are the same [112.55(7)°, 2.0267(18) Å]; these are in unique contrast to those for the arylimido-tribenzyl complex, V(N-2,6-iPr2C6H3)(CH2Ph)3, [ex. V–N–C 169.0(5)° and 1.641(6) Å] reported by Turner and Murphy.12b As described below, these complexes are important intermediates for the synthesis of various dialkyl analogues containing anionic ancillary donor ligands.
 | ||
| Scheme 4 Synthesis of (imido)trialkyl complexes (9, 10).27,28 | ||
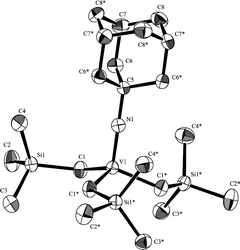 | ||
| Fig. 2 ORTEP drawing of 10.28 Thermal ellipsoids are drawn at the 50% probability level and H atoms are omitted for clarity. | ||
2-2. Reaction of (imido)vanadium-alkyl complexes with various alcohols, thiols, and borates
In general, metal-alkyls, especially metal-alkyls with early transition metals, possess a more nucleophilic nature than those with late transition metals, and are thus highly reactive toward Brønsted/Lewis acids.1,5,31,32 For instance, cationic alkyl complexes, which have been proposed to be the catalytically-active species in olefin coordination polymerisation, are generated from their dialkyl analogues by reacting them with cocatalysts via facile protonolysis or alkyl abstraction;31 some organometallic complexes can be thus grafted onto a silica surface by reaction of the alkyl compounds with silanol groups on the surface.33,34In contrast, some exceptions (that showed remarkable stability toward alcohols), exemplified by reactions of Zr(CH2Ph)4 or Zr(CH2tBu)4 with tBu3COH requiring longer hours even under refluxing conditions, are known.32 Although the reactions of early transition metal-alkyls with alcohols (phenols) should be important basic reactions in organometallic chemistry, a detailed mechanistic study, especially including direct isolation and/or observation of the intermediates, had not so far been explored.
Reactions of the (arylimido)vanadium(V) trialkyl analogue, V(N-2,6-iPr2C6H3)(CH2Ph)3, with 2,6-iPr2C6H3OH or (CF3)3COH in CH2Cl2 or n-hexane afforded the corresponding aryloxide/alkoxide in high yield.12b The similar reaction of V(NAr)(CH2SiMe3)3 (9), with 2,6-Me2C6H3OH, 2,6-iPr2C6H3OH, C6F5OH (in n-hexane at 25 °C) cleanly afforded corresponding vanadium(V)-aryloxide complexes (11a–c) in high yields (90–93%, Scheme 5).27 The reactions of 9 with 1.0 equiv. of tBuOH, (CF3)2MeCOH in n-hexane also gave the corresponding alkoxides (11d,e) in high yields (92–93%).27 Substitution of the alkyl group in 9 with alcohol or phenol leads to upfield chemical shifts (δ 406–659 ppm) from the trialkyl complex (9, 1070 pm) in the 51V NMR spectra, and the degree of the upfield shift in the dialkyl complexes (11a–e) increased in the order: δ 406 (11d, OtBu) > 506–507 (11a,b, O-2,6-Me2C6H3, O-2,6-iPr2C6H3) > 595 [11e, OCMe(CF3)2] > 659 (11c, OC6F5). These resonances seem to be influenced mostly by an electronic factor in the anionic donor ligand employed. Reaction of V(NAd)(CH2SiMe3)3 (10) with 1.0 equiv. of C6F5OH in n-hexane afforded a mixture of V(NAd)(CH2SiMe3)2(OC6F5) (62% yield) and [V(NAd)(CH2SiMe3)(OC6F5)(μ2-OC6F5)]2 (4% yield).28 The present route from the trialkyl analogues should thus be more suited for the synthesis of a series of vanadium(V) dialkyl derivatives containing aryloxo/alkoxo ligands, because, as described above, isolations of these complexes were difficult from the dichloro analogues (1, 5) by alkylation.
 | ||
| Scheme 5 Reaction of V(NAr)(CH2SiMe3)3 (9) with phenols, alcohols.27 | ||
Note that the reaction of 9 with 2,6-tBu2-4-MeC6H2OH did not take place under the same conditions; the reaction did not occur even if the C2D2Cl4 solution containing 9 was heated at 50 °C in the presence of 2,6-tBu2-4-MeC6H2OH with an excess amount (2.6 equiv.).27 The above fact is interesting, because V(NAr)Cl2(O-2,6-tBu2-4-MeC6H2) was prepared from V(NAr)Cl3 by treatment with 1.0 equiv. of 2,6-tBu2-4-MeC6H2OH (yield 94%).18 We believe that the observed fact would be due to the steric bulk of both the phenol and three CH2SiMe3 ligands in the coordination of oxygen to the vanadium(V) metal center, because, as described below, we propose that the reaction of V(NAr)Me(NCtBu2)2 with phenols proceeds via a pentacoordinated trigonal bipyramidal species by coordination of the phenol to the vanadium.35,36
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) CtBu2)2 with various phenols, alcohols.
Reactions of V(NAr)Me(NCtBu2)2 (12), which was prepared by treating V(NAr)Cl(NCtBu2)2 (1a) with 1.0 equiv. of MeMgBr,35 with 1.0 equiv. of phenols exclusively gave another vanadium(V)-methyl complex of the type, V(NAr)Me(NCtBu2)(OAr′) [13, Ar′ = 2,6-Me2C6H3 (a), 4-tBu-2,6-iPr2C6H2 (b), Ph (c), C6F5 (d)], by replacement with the ketimide ligand (Scheme 6).35,36 Note that the reaction with the methyl group did not proceed under these conditions, and the selectivity in the reaction was not dependent upon the kind of phenol employed. Treatment of 12 with iPrOH, 3-buten-1-ol or 5-hexen-1-ol in n-hexane cleanly afforded V(NAr)Me(NCtBu2)(OiPr) (13e), VMe(NAr)(NCtBu2)[OCH2(CH2)nCHCH2] [n = 1 (13f), 3 (13g)], respectively.35,36 The reaction of 12 with 2-Me-6-{(2,6-iPr2C6H3)NCH}C6H3OH (1.1 equiv. to V) in C6D6 at 25 °C reached completion after 3 days, although the above reaction with 2,6-iPr2C6H3OH, 2,6-Me2C6H3OH completed within 3 h. The product was V(NAr)Me(NCtBu2)[O-2-Me-6-{(2,6-iPr2C6H3)NCH}C6H3] (13h) (yield 92%),20 indicating that a unique reactivity toward alcohol in the vanadium-methyl complex was preserved even in the reaction with the imino-phenol.
CtBu2)2 with various phenols, alcohols.
Reactions of V(NAr)Me(NCtBu2)2 (12), which was prepared by treating V(NAr)Cl(NCtBu2)2 (1a) with 1.0 equiv. of MeMgBr,35 with 1.0 equiv. of phenols exclusively gave another vanadium(V)-methyl complex of the type, V(NAr)Me(NCtBu2)(OAr′) [13, Ar′ = 2,6-Me2C6H3 (a), 4-tBu-2,6-iPr2C6H2 (b), Ph (c), C6F5 (d)], by replacement with the ketimide ligand (Scheme 6).35,36 Note that the reaction with the methyl group did not proceed under these conditions, and the selectivity in the reaction was not dependent upon the kind of phenol employed. Treatment of 12 with iPrOH, 3-buten-1-ol or 5-hexen-1-ol in n-hexane cleanly afforded V(NAr)Me(NCtBu2)(OiPr) (13e), VMe(NAr)(NCtBu2)[OCH2(CH2)nCHCH2] [n = 1 (13f), 3 (13g)], respectively.35,36 The reaction of 12 with 2-Me-6-{(2,6-iPr2C6H3)NCH}C6H3OH (1.1 equiv. to V) in C6D6 at 25 °C reached completion after 3 days, although the above reaction with 2,6-iPr2C6H3OH, 2,6-Me2C6H3OH completed within 3 h. The product was V(NAr)Me(NCtBu2)[O-2-Me-6-{(2,6-iPr2C6H3)NCH}C6H3] (13h) (yield 92%),20 indicating that a unique reactivity toward alcohol in the vanadium-methyl complex was preserved even in the reaction with the imino-phenol.
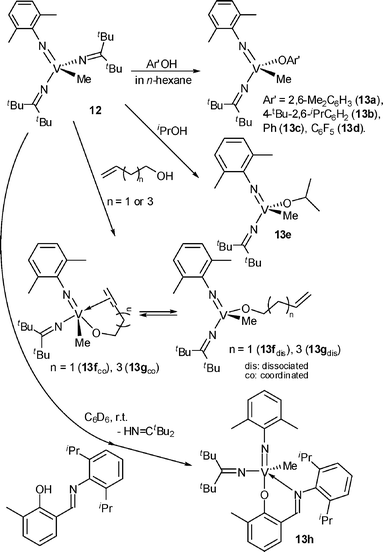 | ||
| Scheme 6 Reaction of V(NAr)Me(NCtBu2)2 (12) with various phenols, alcohols.20,35,36 | ||
Mechanistic studies for reaction of V(NAr)Me(N
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/i_char_e001.gif) CtBu2)2 (12) with alcohols. Exploration for unique reactivity of the vanadium-methyl bonds toward alcohols.
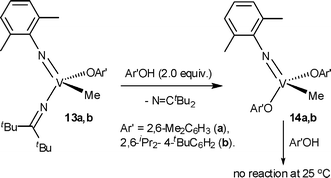
The reaction of 12 with 1.0 equiv. of 2,6-Me2C6H3OH or 2,6-iPr2-4-tBuC6H2OH at 25 °C afforded a mixture of 13a,b and the corresponding bis(phenoxide) (14a,b), and the quantitative conversion of 14a,b could be achieved by reacting with 2.0 equiv. of Ar′OH at 25 °C after 24 h (Scheme 7).36 Note that the formation of methane was not observed even under these conditions.
CtBu2)2 (12) with alcohols. Exploration for unique reactivity of the vanadium-methyl bonds toward alcohols.
The reaction of 12 with 1.0 equiv. of 2,6-Me2C6H3OH or 2,6-iPr2-4-tBuC6H2OH at 25 °C afforded a mixture of 13a,b and the corresponding bis(phenoxide) (14a,b), and the quantitative conversion of 14a,b could be achieved by reacting with 2.0 equiv. of Ar′OH at 25 °C after 24 h (Scheme 7).36 Note that the formation of methane was not observed even under these conditions.
A rapid scrambling of 13a and 13b was observed when 13a was treated with 1.0 equiv. of 4-tBu-2,6-iPr2C6H2OH in CDCl3 at 25 °C or when 1.0 equiv. of 2,6-Me2C6H3OH was added into a CDCl3 solution containing 13b (Scheme 8); the resultant solution eventually gave a ca. 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of 13a and 13b within 30 min at 25 °C.36 The solution then gave 3 species upon heating at 60 °C for 12 h which were assigned as 14a, 14b, and the mixed bis(aryloxo) complex, (ArN)VMe(O-4-tBu-2,6-iPr2C6H2)(O-2,6-Me2C6H3) (14c). These results clearly explain the unique reactivity of both 12 and 13a,b toward alcohols according to Scheme 8. Both phenol-scrambling and the phenol/ketimide exchange reaction should be preferred if the phenol approaches the electrophilic vanadium metal center trans to the Me group (NNN face in 12 or NNO face in 13a,b, and not the NNC faces in 12 or 13a,b), to give pentacoordinated trigonal bipyramidal intermediates (shown in brackets in Scheme 8).36 The reaction with the Me group would not occur if the reaction takes place via this proposed intermediate. A similar assumption was also proposed by Schrock et al.37 in the alkoxide exchange reaction of Mo(NAr)(CHtBu)(CH2tBu)(OAr) with ROH (OR = OCMe3, OAd, OC6F5) and the presence of a similar intermediate was assumed.
:1 mixture of 13a and 13b within 30 min at 25 °C.36 The solution then gave 3 species upon heating at 60 °C for 12 h which were assigned as 14a, 14b, and the mixed bis(aryloxo) complex, (ArN)VMe(O-4-tBu-2,6-iPr2C6H2)(O-2,6-Me2C6H3) (14c). These results clearly explain the unique reactivity of both 12 and 13a,b toward alcohols according to Scheme 8. Both phenol-scrambling and the phenol/ketimide exchange reaction should be preferred if the phenol approaches the electrophilic vanadium metal center trans to the Me group (NNN face in 12 or NNO face in 13a,b, and not the NNC faces in 12 or 13a,b), to give pentacoordinated trigonal bipyramidal intermediates (shown in brackets in Scheme 8).36 The reaction with the Me group would not occur if the reaction takes place via this proposed intermediate. A similar assumption was also proposed by Schrock et al.37 in the alkoxide exchange reaction of Mo(NAr)(CHtBu)(CH2tBu)(OAr) with ROH (OR = OCMe3, OAd, OC6F5) and the presence of a similar intermediate was assumed.
 | ||
| Scheme 8 Proposed reaction pathways.36 | ||
Based on our experiments, we propose that the reaction with alcohols proceeds in the following steps: 1) the alcohols initially approaches the electron-deficient metal center trans to the methyl group to give a pentacoordinated trigonal bipyramidal species, and 2) proton transfer to the aryloxide/ketimide occurs to give ketimine/phenol dissociation.36
CtBu2)2 (12) with various thiols, borates.
In contrast, the reaction of 12 with 1.0 equiv. of 2,6-Me2C6H3SH afforded V(NAr)(NCtBu2)2(S-2,6-Me2C6H3) (15a) via cleavage of the V–Me bond (Scheme 9).36 The reaction with n-C6H13SH in n-hexane afforded V(NAr)(NCtBu2)2(S-n-C6H13) (15b) as the sole product.36 These facts clearly indicate that the reaction mechanism should differ between alcohol and thiol.
 | ||
| Scheme 9 Reactions of V(NAr)Me(NCtBu)2 (12) with thiols, borates.36 | ||
The reaction of 12 with 1.0 equiv. of [PhN(H)Me2][B(C6F5)4] in THF afforded cationic [V(NAr)(NCtBu)2(THF)][B(C6F5)4] (16a) (Scheme 9). The crystallographic analysis of 16a (Fig. 3) indicates that 16a folds in a pseudo-tetrahedral geometry around the vanadium center with the coordination of one THF molecule, and the V–N(CtBu2) distances (1.802–1.808 Å) are comparable to those found in V(NAr)Cl(NCtBu2)2 and somewhat shorter than those in 12. The position of another THF molecule could not be defined/determined. The same compound (16a) could also be cleanly isolated (90%) by the reaction of 12 with 1.0 equiv. of [Ph3C][B(C6F5)4], and the reaction of 12 with B(C6F5)3 also gave [V(NAr)(NCtBu)2(THF)][MeB(C6F5)3] (16b) exclusively.36 Moreover, the reaction of 14a with 1.0 equiv. of [PhN(H)Me2][B(C6F5)4] also gave the corresponding cationic species [V(NAr)(NCtBu2)(O-2,6-Me2C6H3)(THF)2][B(C6F5)4] (16c). These results clearly indicate that cleavage of the V–Me bond was favored in all cases.36
 | ||
| Fig. 3 ORTEP drawing (50% probability ellipsoids) for 16a.36 All hydrogen atoms are omitted for clarity. | ||
CMe}C5H3N] was prepared from V(NAd)(CH2SiMe3)3 (10) with the corresponding imino-phenol (1.0 equiv. in C6D6 at 50 °C for 3 days).38 The complex (17) folds in a trans (not cis) geometry, and is thermally stable in C6D6 at 80 °C (even after 72 h). Dissociation of neutral donor(s) in the chelate ligand (L′) did not take place even in the presence of NHC [2.1 equiv. in C6D6 at 80 °C, 72 h, NHC = 1,3-bis(2,4,6-trimethylphenyl)imidazol-2-ylidene].38
The reaction of 17 with 1.0 equiv. of (CF3)2CHOH (in n-hexane at 25 °C) did not take place even after >12 h, and the reaction with 2.0 equiv. of (CF3)2CHOH did not take place even after several hours. The fact should be an interesting contrast to the observed fact that the reaction of the other (imido)vanadium(V)-alkyl complexes [such as V(NAr)Me(NCtBu2)2 (12), V(NAr)(CH2SiMe3)3 (9), V(NAd)(CH2SiMe)3 (10)] with alcohols (phenols) took place cleanly even at room temperature.
Monitoring the reaction mixture by NMR spectroscopy revealed that the reaction of 17 with (CF3)2CHOH first generated an intermediate, and was converted to the original complex (17) when the mixed solution was placed in vacuo (to remove volatiles). On the basis of 1H, 19F, and 51V NMR spectra, the intermediate should be V(NAd)(CH2SiMe3)2(L′)[(CF3)2CHOH] (T1) formed by coordination of oxygen in (CF3)2CHOH into the vanadium in 17, accompanied as well by dissociation of the two imino groups (Scheme 10). The reaction completed upon heating or by stirring for a longer time (48 h) to afford the monoalkyl-alkoxo species, V(NAd)(CH2SiMe3)(L′)[OCH(CF3)2] (18a). The structure determined by X-ray crystallography indicates that 18a folds in a distorted tetrahedral geometry around vanadium, and that the two neutral nitrogen donors in L′ are dissociated.38
 | ||
| Scheme 10 Proposed scheme for reaction of V(NAd)(CH2SiMe3)2(L′) (17) with alcohol.38 | ||
Moreover, a reaction mixture of the dialkyl complex (17) and 2 equiv. of (CF3)2CHOH (25 °C after 48 h) afforded two resonances in the 51V NMR spectrum including a resonance ascribed to the monoalkyl complex (18a). Only one resonance due to 18a was observed when the reaction mixture was placed in vacuo (to remove volatiles). The fact also suggests the formation of another intermediate, assumed to be V(NAd)(CH2SiMe3)(L)[OCH(CF3)2][(CF3)2CHOH] (T2), generated by coordination of (CF3)2CHOH into 18a.38 The reaction of 17 with 2.0 equiv. of (CF3)2(CH3)COH in place of (CF3)2CHOH did not take place at 25 °C, and a similar observation (such as formation of an intermediate that could be returned to 17 after removal of volatiles) was made. The reaction completed to afford V(NAd)(CH2SiMe3)[OC(CH3)(CF3)2] (18b), when the mixture was heated at 80 °C for 22 h.38
On the basis of the above results, we propose that reactions of the dialkyl complex (17) with ROH [R = CH(CF3)2, C(CH3)(CF3)2] proceeded via five coordinate intermediates formed by coordination of the oxygen in ROH (not via addition of H+) accompanied by dissociation of two neutral nitrogen donors (imino groups) in the chelate tridentate ligand (Scheme 10). Confirmation of the intermediates, V(NAd)(CH2SiMe3)2(L′)[ROH] (T1), is potentially important not only for better understanding of the reactions of vanadium(V)-alkyls with alcohols, but also to explain the observed unique reactivities of the vanadium(V)-alkyls in V(NAr)(Me)(NCtBu2)2 (12),35,36 V(NAr)(CH2SiMe3)3 (9),27 V(NAd)(CH2SiMe)3 (10)28 with various alcohols. We believe that this hypothesis should be applied to the reactions of early transition metal alkyls (especially in high oxidation states) with alcohols.
3. Synthesis of vanadium(V)-alkylidene complexes and their use as olefin metathesis catalysts
As described in the introduction, transition metal-alkylidene complexes play important key roles in olefin metathesis, which introduces promising possibilities for the synthesis of functional polymers as well as valuable organic compounds,14 as demonstrated especially by molybdenum15 and ruthenium.16 High-oxidation-state early transition metal alkylidene complexes attract considerable attention,15,17 because they play essential roles as catalysts in olefin metathesis and Wittig-type or group transfer reactions,14,15,17 as demonstrated especially by molybdenum (so-called Schrock-type carbene complexes).14,15,17Although classical Ziegler type vanadium catalysts display unique characteristics such as use in the synthesis of high molecular weight polymers with rapid propagation in olefin coordination insertion polymerisation3a,b,5 and the syntheses of some vanadium(III),(IV)-alkylidene39,40 as well as vanadium(V)-alkylidene complexes41 are known, examples for olefin metathesis with vanadium-alkylidene complexes have been limited. Since no examples were known until recently concerning the synthesis of ‘olefin metathesis active’ vanadium(V)-alkylidenes,39b their synthesis and reaction chemistry are thus promising subjects in terms of the basic understanding of organometallic chemistry as well in their application in catalysis.
3-1. Approaches for the synthesis of vanadium-alkylidene complexes: previous reports and selected recent examples
The most common method to prepare high-oxidation state metal alkylidenes is by promoting α-H abstraction or α-H deprotonation reactions from metal alkyl complexes lacking β-hydogens.17 Some additional assistance (that can stimulate photochemically and/or sterically, or with a base) is commonly used to promote/induce the abstraction/deprotonation. Scheme 11 shows selected reported examples for the synthesis of vanadium-alkylidene complexes till 2000. Vanadium(III)-alkylidene complex, CpV(CHtBu)(dmpe), was prepared by α-hydrogen abstraction upon addition of dmpe (Scheme 11, top equation).39b Reaction of CpV(CHtBu)(dmpe) with norbornene was attempted but showed extremely low (catalytic) activity (TON 0.92 after 96 h at 20 °C).39b
Transfer of a benzylidene from phosphorus to vanadium(III) afforded the first vanadium(V)-alkylidene complex (Scheme 11, middle).41b However, no reaction between CpV(CHPh)(N-2,6-iPr2C6H3)(PMe3) and norbornene or acetone was observed. Reaction of a V(III) borohydride complex with diphenylacetylene yielded a vanadium(V) complex containing a V=C double bond, although the mechanism was not clear (Scheme 11, bottom).41c No data concerning the reactivity of this carbene species were given.
Another approach recently employed for the synthesis of a vanadium(IV)-alkylidene was the so-called oxidatively induced α-hydrogen abstraction reported by Mindiola et al.39c,e As shown in Scheme 12, a vanadium(III)-dialkyl complex, (nacnac)V(CH2tBu)2 [nacnac− = {(Ar′)NC(Me)CHC(Me)CN(Ar′)}−], was oxidised to afford neutral or cationic vanadium(IV)-alkylidene complexes, [(nacnac)V(CHtBu)(I)] or [(nacnac)V(CHtBu)(THF)]+[BPh4]−.39c Reaction of [(nacnac)V(CHtBu)(I)] with LiPR (R = 2,4,6-iPr3C6H2, 2,4,6-tBu3C6H2) afforded the corresponding vanadium(IV)-alkyl, phosphinidene complex, [(nacnac)V(CH2tBu)(PR)],39d and the vanadium(V)-alkylidyne complex, [(nacnac)V(CtBu)(L)], was formed via the corresponding vanadium(IV)-alkyl,alkylidene complex upon oxidation (Scheme 12).39e
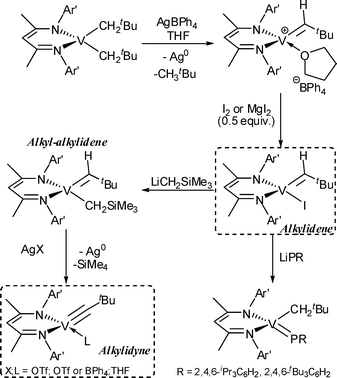 | ||
| Scheme 12 Synthesis of a vanadium-alkylidene by oxidatively induced α-hydrogen abstraction.39c–e | ||
More recently, the synthesis of a series of vanadium(V)-alkylidene complexes from a vanadium(III) dialkyl complex containing a monoanionic tridentate chelate ligand (PNP), [V(CH2tBu)2(PNP)] PNP = N[4-Me-2-(PiPr2)C6H3]2−, by treating with π-acids or two electron oxidants was reported (Scheme 13).42 The reaction of [V(CH2tBu)2(PNP)] with chalcogen sources resulted in the formation of the vanadium(V)-alkylidene, chalcogenide species, [V(X)(CHtBu)(PNP)] (X = O, S, Se, Te). The vanadium(III)-alkylidene complex was also formed upon addition of 2,2′-bipyridine by α-hydrogen abstraction, and a similar reaction with N2CPh2 in Et2O afforded the alkylidene-hydrazido complex, V(CHtBu)(NNCPh2)(PNP). It has been proposed that these reactions take place via a transient vanadium(III)-alkylidene complex.42
 | ||
| Scheme 13 Synthesis of vanadium(V)-alkylidenes containing a PNP ligand.42 | ||
3-2. Generation of catalytically-active species for ring-opening metathesis polymerisation (ROMP) from (imido)vanadium(V)-dialkyl complexes containing anionic ancillary donor ligands
Although the initial attempt at reaction of CpV(CHCMe3)(dmpe) with norbornene (NBE) afforded polymers with extremely low (catalytic) activity (TON 0.92 after 96 h at 20 °C), it was revealed that ring-opening metathesis polymerisation (ROMP) of NBE took place in the presence of V(NAr)Cl2(OAr′) (1) and AlMe3.18a Since 1 showed remarkable catalytic activities for ethylene polymerisation in the presence of halogenated Al alkyls, is was thus assumed that the aluminium promoters directly control the olefin coordination insertion or metathesis pathways (Scheme 14). Moreover, the dibenzyl complex (20) initiated ROMP of NBE catalytically at 25 °C, affording the ring-opened poly(NBE) with high molecular weight with uniform molecular weight distribution.18a As far as we know, this is apparently the first catalytic example of vanadium-initiated ROMP of NBE, and formation of the alkylidene species was thus assumed in this catalysis.18a The dibenzyl complex is, however, not stable and decomposed gradually; an attempted isolation of the assumed alkylidene was not successful. | ||
| Scheme 14 Ring-opening metathesis polymerisation (ROMP) of norbornene (NBE) by (arylimido)vanadium(V) complex catalysts containing an aryloxo ligand.18a | ||
Later, we reported that V(NAr)(CH2SiMe3)2(NCtBu2) (7), which showed better thermal stability than V(NAr)(CH2Ph)2(O-2,6-iPr2C6H3) (20), initiated ROMP of NBE in toluene.19 The activity increased especially by addition of PMe3 (3 equiv.) at higher temperature (80 °C) but decreased on further addition (5 equiv.); the activity was extremely low in the absence of PMe3 even at 80 °C.19 The resultant polymer possessed a ring-opened structure containing a mixture of cis- and trans-olefinic double bonds. These results clearly suggest the formation of vanadium(V)-alkylidene by α-hydrogen elimination from the dialkyl species in the presence of PMe3.
3-3. Synthesis of (imido)vanadium(V)-alkylidene complexes and their use as catalysts for ring-opening metathesis polymerisation (ROMP)
It was then revealed that the reaction of 7 with PMe3 (7.0 equiv.) in benzene-d6 at 80 °C afforded the alkylidene complex, V(CHSiMe3)(NAr)(NCtBu2)(PMe3) (21, a mixture of syn/anti forms in solution), by α-hydrogen elimination (Scheme 15).19,43 This is the common method to prepare high-oxidation state metal alkylidenes by promoting α-hydrogen abstraction reactions from metal alkyl complexes lacking β-hydrogens, and addition of PMe3 was required to promote the abstraction by steric crowding.17 The crystal structure (Fig. 4) indicates that 21 folds in a distorted tetrahedral geometry around the vanadium metal center, and the V–CHSiMe3 bond distance (1.860 Å) is close to that in the bicyclic carbene-amide complex (1.876 Å)41c and is shorter than that in the benzylidene complex (1.922 Å).41b The 1H and 13C NMR spectra showed resonances corresponding to VCHSiMe3 at 14.52, 302.0 ppm, respectively. These results clearly indicate that 21 is a nucleophilic 16-electron vanadium(V)-alkylidene complex.
 | ||
| Scheme 15 Synthesis of vanadium(V)-alkylidene complex (21).19,43 | ||
 | ||
| Fig. 4 ORTEP drawing of 21 Thermal ellipsoids are drawn at the 50% probability level and H atoms are omitted for clarity.19 | ||
The complex (21) exhibited remarkable catalytic activity for the ROMP of NBE without a cocatalyst (Table 2).19,43 The activity (turnover number, TON) was low when the ROMP was conducted at 25 °C, whereas a standard Schrock type initiator, Mo(CHCMe2Ph)(N-2,6-iPr2C6H3)(OtBu)2 (Mo), exhibited especially remarkable catalytic activity under similar conditions.43,44 The activity of 21 increased at 50 °C, whereas the activity of Mo was negligible due to the decomposition of the catalytically active species.44 The activity markedly increased at higher temperature (80 °C), and the observed activity was higher than that of Ru(CHPh)(Cl)2(PCy)2 under the same conditions due to the improved thermal stability.19,43
CtBu2)(PMe3) (21), Ru(CHPh)Cl2(PCy3)2 (Ru, Cy = cyclohexyl), and Mo(CHCMe2Ph)(N-2,6-iPr2C6H3)(OtBu)2 (Mo).a
| Complex (μmol) | Solvent | Temperature/°C | Time/min | TONb | M w × 10−4 | M w/Mnc |
|---|---|---|---|---|---|---|
| a Selected data cited from ref. 43, conditions: catalyst 0.2 or 1.0 μmol, NBE 2.12 mmol, benzene or toluene 9.6 mL, initial NBE conc. 0.22 mmol mL−1. b TON = NBE consumed (mmol)/V (mmol). c GPC data vs. polystyrene standards. | ||||||
| 21 (1.0) | Benzene | 25 | 360 | 267 | 46 | 2.3 |
| Ru (1.0) | Toluene | 25 | 60 | 1306 | 54 | 1.7 |
| Mo (0.20) | Toluene | 25 | 60 | 8550 | 160 | 1.2 |
| 21 (1.0) | Benzene | 50 | 180 | 1275 | 49 | 1.6 |
| 21 (1.0) | Benzene | 50 | 180 | 166 | ||
| Mo (0.20) | Toluene | 50 | 40 | Trace | ||
| 21 (1.0) | Benzene | 80 | 30 | 967 | 140 | 1.3 |
| 21 (1.0) | Benzene | 80 | 60 | 1583 | 133 | 1.4 |
| 21 (1.0) | Toluene | 80 | 60 | 1244 | 32 | 2.8 |
| Ru (1.0) | Toluene | 80 | 60 | 350 |
The aryloxo-alkylidene analogue (22) was also isolated from the corresponding dialkyl complex (11b), which also showed remarkable catalytic activities for ROMP of NBE in the presence of PMe3, upon heating in C6D6 in the presence of PMe3 (Scheme 16).27 PMe3 was partially dissociated, as expressed in V(CHSiMe3)(NAr)(O-2,6-iPr2C6H3)(PMe3)x (22, x = 0.89), from the metal center during the purification procedure (by placing the grown microcrystals in vacuo to remove solvent), probably due to the steric bulk of the two iPr groups on the aryloxo ligand and the presence of both the PMe3 coordinated and the PMe3 free species in solution. This can also be confirmed by the observation that two resonances in both 51V and 1H spectra (corresponding to VCHSiMe3) became one resonance on addition of PMe3. Complex 22 could be prepared from the dichloro analogue without isolation of the dialkyl analogue (11b).
 | ||
| Scheme 16 Synthesis of (arylimido)vanadium(V)-alkylidene containing aryloxo ligand (22).27 | ||
Other vanadium(V)-dialkyl complexes containing an aryloxo ligand like V(NAr)(CH2SiMe3)2(O-2,6-Me2C6H3) (11a) and other complexes containing a (2-anilidomethyl)pyridine ligand (8 in Scheme 3) showed notable catalytic activities for the ROMP of NBE in the presence of PMe3, but attempts at isolating the corresponding vanadium(V)-alkylidene were not successful, although the presence of resonances that may be ascribed to the assumed alkylidene species was observed in the 1H NMR spectra.21,27 One probable reason for this difficulty could be assumed as due to the formed vanadium(V)-alkylidene species being highly reactive even in C6D6 and affording another species [such as V[CH(D)SiMe3](C6D5) etc.] by C–H activation.45 Some reactions with the isolated vanadium(V)-alkylidene are thus in progress, and will be introduced soon.
The aryloxo-alkylidene (22) showed higher catalytic activities at 25 °C than the ketimide-alkylidene (21), and the resultant polymers obtained at 25 °C possessed narrow molecular weight distributions and the Mn value increased over the time course (Fig. 5). The Mn values increased upon increasing the TON values (polymer yields) with consistently narrow molecular weight distributions (Mw/Mn = 1.1–1.2) during the polymerisation, clearly indicating that the ROMP of NBE by 22 proceeded in a living manner at 25 °C.
 | ||
| Fig. 5 Plots of TON vs Mn, Mw/Mn in the ring-opening metathesis polymerisation (ROMP) of norbornene by V(NAr)(CHSiMe3)(O-2,6-iPr2C6H3)(PMe3)x (22) in benzene (conditions: 22 5.0 μmol, NBE 21.2 mmol, benzene 24 mL at 25 °C).27 | ||
Coordination of NHC was not observed if 10 was added with 1.0 equiv. of NHC in C6D6 at 25 °C, but a new resonance ascribed to the alkylidene proton (13.4 ppm) appeared when the solution was heated at 80 °C for 18 h.28 The targeted alkyl-alkylidene complex, V(CHSiMe3)(NAd)(CH2SiMe3)(NHC) (23), could be isolated (22% based on NHC, 15% based on V) when 10 was treated with 2/3 equiv. of NHC at 80 °C in C6D6 (Scheme 17).28 Similarly, the reaction of the arylimido analogue (9) with NHC afforded V(CHSiMe3)(NAr)(CH2SiMe3)(NHC) (24) in better yield (75% based on NHC, 60% based on V, Scheme 17). Thermolysis of the (adamantylimido)vanadium(V) trialkyl analogue, V(NAd)(CH2SiMe3)3 (10), in C6D6 at 80 °C afforded a dimeric species, [V(μ2-NAd)(CH2SiMe3)2]2 (25) determined by X-ray crystallography (Fig. 6). However, the reaction of 10 with (more bulky and stable) 1,3-di-tert-butylimidazole-2-ylidene did not show any changes in the 51V NMR spectra even at 80 °C for 3 days. The fact would thus suggest that this NHC stabilises 10 probably by weak coordination but did not promote α-hydrogen elimination under these conditions.
 | ||
| Scheme 17 Reactions of (imido)vanadium(V)-trialkyl complexes.28 | ||
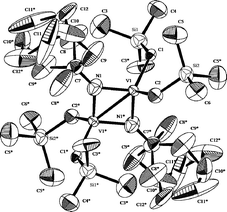 | ||
| Fig. 6 ORTEP drawing of 25. Thermal ellipsoids are drawn at the 50% probability level and H atoms are omitted for clarity.28 | ||
The (adamantylimido)vanadium(V)-alkyl, alkylidene complex containing NHC (23) folds in a rather distorted tetrahedral geometry around vanadium (Fig. 7), and the V–C bond distances for the alkyl, the alkylidene and the NHC are 2.069(3), 1.829(3) and 2.172(2) Å, respectively; these values (alkyl, alkylidene) are similar to those in previous reports.19,47 The V–C(NHC) distance is close to that in VOCl3(IMes) (2.137 Å); the coordination seems likely as neutral carbene and a similar explanation would thus be possible. Ring-opening metathesis polymerisation of norbornene by 23 (and 24) took place, but the activities (at 80 °C in benzene) were lower than that of V(CHSiMe3)(NAr)(NCtBu2)(PMe3),19,43 and the activities at 25 °C were negligible.
 | ||
| Fig. 7 ORTEP drawing of 23. Thermal ellipsoids are drawn at the 50% probability level and H atoms are omitted for clarity.28 | ||
The present approaches should be widely applied for the preparation of various early transition metal-alkylidene complexes. Moreover, the present alkyl-alkylidenes would also be promising intermediates for syntheses of not only the alkylidynes but also of the cationic alkylidenes or the alkylidene-containing series of anionic donor ligands. Further studies concerning their application as catalysts, including the effect of the adamantyl imido ligand are now underway.
Conclusions
In this account, recent examples of the syntheses and reactions of vanadium(V)-alkyl, and alkylidene complexes containing imido ligands have been summarised. In particular, unique promising characteristics, such as the synthesis of ultra high molecular weight polymers in coordination polymerisation, unique reactivity with alcohols (ROH) that proceed via intermediates formed by coordination of ROH, and olefin metathesis active vanadium(V)-alkylidene complexes that exhibit better thermal stability, have been introduced. On the basis of these efforts, we believe this subject will expand the possibilities for the establishment of more efficient/precise catalytic processes as well as the better understanding of organometallic chemistry with vanadium.Acknowledgements
K.N. expresses his heartfelt thanks to former/present group members who contributed to this project as coauthors, and Mr Shohei Katao (Nara Institute of Science and Technology, NAIST) for his assistance in the crystallographic analysis. The research by K.N. was partly supported by a Grant-in-Aid for Scientific Research on Priority Areas (No. 19028047, “Chemistry of Concerto Catalysis”) from the Ministry of Education, Culture, Sports, Science and Technology, Japan. W.Z. expresses her sincere thanks to JSPS for a postdoctoral fellowship (P06349). The authors also express their thanks to Dr Shu Zhang (NAIST) for help in the preparation of this manuscript.Notes and references
- For example (general text of metal-alkyl chemistry): (a) R. H. Crabtree, The Organometallic Chemistry of the Transition Metals, 4th edn, John Wiley & Sons, Inc., Hoboken, New Jersey, USA, 2005, p. 53 Search PubMed; (b) Comprehensive Organometallic Chemistry III, ed. R. H. Crabtree and D. M. P. Mingos, Elsevier Science/Pergamon US, USA, 2006 Search PubMed; (c) Synthesis of Organometallic Compounds: A Practical Guide, ed. S. Komiya, John Wiley & Sons Ltd., West Sussex, England, 1997 Search PubMed; (d) Organometallics in Synthesis: A Manual, 2nd edn, ed. M. Schlosser, John Wiley & Sons Ltd, West Sussex, England, 2002 Search PubMed; (e) D. Astruc, Organometallic Chemistry and Catalysis, Springer-Verlag, Berlin Heidelberg, Germany, 2007 Search PubMed.
- A. F. Mason, G. W. Coates, in Macromolecular Engineering, ed. K. Matyjaszewski, Y. Gnanou and L. Leibler, Wiley-VCH, Weinheim, Germany, 2007, vol. 1, p. 217 Search PubMed.
- For selected recent reviewing articles, accounts, see: (a) V. C. Gibson and S. K. Spitzmesser, Chem. Rev., 2003, 103, 283 CrossRef CAS; (b) P. D. Bolton and P. Mountford, Adv. Synth. Catal., 2005, 347, 355 CrossRef; (c) D. W. Stephan, Organometallics, 2005, 24, 2548 CrossRef CAS; (d) K. Nomura, J. Liu, S. Padmanabhan and B. Kitiyanan, J. Mol. Catal. A: Chem., 2007, 267, 1 CrossRef CAS; (e) J. Cano and K. Kunz, J. Organomet. Chem., 2007, 692, 4411 CrossRef CAS; (f) G. J. Domski, J. M. Rose, G. W. Coates, A. D. Bolig and M. Brookhart, Prog. Polym. Sci., 2007, 32, 30 CrossRef CAS; (g) K. Nomura, Dalton Trans., 2009, 8811 RSC.
- For recent special issues, see: (a) Metallocene Complexes as Catalysts for Olefin Polymerisation, Coord. Chem. Rev., ed. H. G. Alt, 2006, 250(1–2), 1 Search PubMed; (b) Metal-catalysed Polymerisation, ed. B. Milani and C. Claver, Dalton Trans., 2009, (41), 8769 Search PubMed.
- For recent reviews (vanadium catalysts), see: (a) H. Hagen, J. Boersma and G. van Koten, Chem. Soc. Rev., 2002, 31, 357 RSC; (b) S. Gambarotta, Coord. Chem. Rev., 2003, 237, 229 CrossRef CAS; (c) K. Nomura, in New Developments in Catalysis Research, ed. L. P. Bevy, NOVA Science Publishers, New York, USA, 2005, p. 199 Search PubMed.
- (a) W. L. Carrick, J. Am. Chem. Soc., 1958, 80, 6455 CrossRef CAS; (b) W. L. Carrick, R. W. Kluiber, E. F. Bonner, L. H. Wartman, F. M. Rugg and J. J. Smyth, J. Am. Chem. Soc., 1960, 82, 3883 CrossRef CAS; (c) G. W. Phillips and W. L. Carrick, J. Polym. Sci., 1962, 59, 401 CrossRef CAS.
- (a) E. Junghanns, O. Gumboldt and G. Bier, Makromol. Chem., 1962, 58, 18 CrossRef; (b) G. Natta, G. Mazzanti, A. Valvassori, G. Sartori and D. Fiumani, J. Polym. Sci., 1961, 51, 411 CrossRef CAS.
- For example: D. L. Christman and G. I. Keim, Macromolecules, 1968, 1, 358 Search PubMed.
- Y. Doi, S. Ueki and T. Keii, Macromolecules, 1979, 12, 814 CrossRef CAS.
- Y. Doi, T. Koyama and K. Soga, Makromol. Chem., 1985, 186, 11 CrossRef CAS.
- Some structural characterizations, reaction chemistry of V(III),(IV) methyl complex: (a) B. Hessen, J. H. Teuben, T. H. Lemmen, J. C. Huffman and K. G. Caulton, Organometallics, 1985, 4, 946 CrossRef CAS; (b) B. Hessen, T. H. Lemmen, H. J. G. Luttikhedde, J. H. Teuben, J. L. Petersen, S. Jagner, J. C. Huffman and K. G. Caulton, Organometallics, 1987, 6, 2354 CrossRef CAS; (c) B. Hessen, A. Meetama and J. H. Teuben, J. Am. Chem. Soc., 1989, 111, 5977 CrossRef CAS; (d) C. P. Gerlach and J. Arnold, Organometallics, 1996, 15, 5260 CrossRef CAS; (e) G. Aharonian, K. Feghali, S. Gambarotta and G. P. A. Yap, Organometallics, 2001, 20, 2616 CrossRef CAS; (f) K. Feghali, D. J. Harding, D. Reardon, S. Gambarotta, G. Yap and Q. Wang, Organometallics, 2002, 21, 968 CrossRef CAS; (g) R. Choukroun, C. Lorber and B. Donnadieu, Organometallics, 2002, 21, 1124 CrossRef CAS; (h) G. Liu, D. J. Beetstra, A. Meetsma and B. Hessen, Organometallics, 2004, 23, 3914 CrossRef CAS.
- Selected pioneering examples for structurally characterized V(V) alkyls: (a) J. de With, A. D. Horton and A. G. Orpen, Organometallics, 1990, 9, 2207 CrossRef CAS; (b) V. J. Murphy and H. Turner, Organometallics, 1997, 16, 2495 CrossRef CAS.
- Examples: (a) F. Preuss and L. Ogger, Z. Naturforsch., B: Anorg. Chem., Org. Chem., 1982, 37B, 957 CAS; (b) D. D. Devore, J. D. Lichtenhan, F. Takusagawa and E. Maatta, J. Am. Chem. Soc., 1987, 109, 7408 CrossRef CAS; (c) F. Preuss, H. Becker, J. Kraub and W. J. Sheldrick, Z. Naturforsch., B: Chem. Sci., 1988, 43B, 1195; (d) F. Preuss, H. Becker and T. Wieland, Z. Naturforsch., B: Chem. Sci., 1990, 45B, 191; (e) G. A. Solan, P. G. Cozzi, C. Floriani, A. Chiesi-Villa and C. Rizzoli, Organometallics, 1994, 13, 2572 CrossRef CAS; (f) M. C. W. Chan, J. M. Cole, V. C. Gibson and J. A. K. Howard, Chem. Commun., 1997, 2345 RSC.
- For examples, see: (a) M. R. Buchmeiser, Chem. Rev., 2000, 100, 1565 CrossRef CAS; (b) Ring-Opening Metathesis Polymerisation and Related Chemistry, ed. E. Khosravi and T. Szymanska-Buzar, Kluwer, Dordrecht, The Netherlands, 2002 Search PubMed; (c) Handbook of Metathesis, ed. R. H. Grubbs, Wiley-VCH, Weinheim, Germany, 2003, vol. 1–3 Search PubMed; (d) Novel Metathesis Chemistry: Well-Defined Initiator Systems for Specialty Chemical Synthesis, Tailored Polymer and Advanced Material Applications, ed. Y. Imamoglu and L. Bencze, Kluwer, Dordrecht, 2003 Search PubMed; (e) Metathesis Chemistry, ed. Y. Imamoglu and V. Dragutan, Springer, Dordrecht, The Netherlands, 2007 Search PubMed.
- For examples, see: (a) R. R. Schrock, Acc. Chem. Res., 1990, 23, 158 CrossRef CAS; (b) R. R. Schrock, in Alkene Metathesis in Organic Synthesis, ed. A. Fürstner, Springer, Berlin, Germany, 1998, p. 1 Search PubMed.
- For examples, see: (a) T. M. Trnka and R. H. Grubbs, Acc. Chem. Res., 2001, 34, 18 CrossRef CAS; (b) S. T. Nguyen, T. M. Trnka, in Handbook of Metathesis, ed. R. H. Grubbs, Wiley-VCH, Weinheim, 2003, vol. 3, p. 61 Search PubMed.
- For examples, see: (a) R. R. Schrock, Acc. Chem. Res., 1979, 12, 98 CrossRef CAS; (b) R. R. Schrock, Chem. Rev., 2002, 102, 145 CrossRef CAS; (c) R. R. Schrock, in Handbook of Metathesis, ed. R. H. Grubbs, Wiley-VCH, Weinheim, 2003, vol. 1, p. 8 Search PubMed; (d) D. Mindiola, Acc. Chem. Res., 2006, 39, 813 CrossRef; (e) D. Mindiola, B. Bailey and F. Basuli, Eur. J. Inorg. Chem., 2006, 3135 CrossRef CAS; (f) R. R. Schrock, Chem. Rev., 2009, 109, 3211 CrossRef CAS.
- (a) K. Nomura, A. Sagara and Y. Imanishi, Macromolecules, 2002, 35, 1583 CrossRef CAS; (b) W. Wang, J. Yamada, M. Fujiki and K. Nomura, Catal. Commun., 2003, 4, 159 CrossRef CAS.
- J. Yamada, M. Fujiki and K. Nomura, Organometallics, 2005, 24, 2248 CrossRef CAS.
- Y. Onishi, S. Katao, M. Fujiki and K. Nomura, Organometallics, 2008, 27, 2590 CrossRef CAS.
- S. Zhang, S. Katao, H. W,-Sun and K. Nomura, Organometallics, 2009, 28, 5925 CrossRef CAS.
- W. Zhang and K. Nomura, Inorg. Chem., 2008, 47, 6482 CrossRef CAS.
- (a) W. Wang and K. Nomura, Macromolecules, 2005, 38, 5905 CrossRef CAS; (b) W. Wang and K. Nomura, Adv. Synth. Catal., 2006, 348, 743 CrossRef CAS.
- K. Nomura, W. Wang and J. Yamada, Stud. Surf. Sci. Catal., 2006, 161, 123 CAS.
- For example D. L. Christman, J. Polym. Sci., Part A-1, 1972, 10, 471 Search PubMed.
- For example (review): A. Macchioni, Chem. Rev., 2005, 105, 2039 Search PubMed.
- K. Nomura, Y. Onishi, M. Fujiki and J. Yamada, Organometallics, 2008, 27, 3818 CrossRef CAS.
- W. Zhang and K. Nomura, Organometallics, 2008, 27, 6400 CrossRef CAS.
- The data including syntheses of V(N-4-MeC6H4)Cl(CH2SiMe3) and V(N-4-MeC6H4)Cl2(CH2SiMe3) are shown in ref. 13b. Calculation chemistry concerning the effect of the imido substituent in V(NR)Me3 is as follows: M. Bühl, Organometallics, 1999, 18, 4894 Search PubMed.
- F. Preuss, G. Overhoff, H. Becker, H. J. Häusler, W. Frank and G. Reiss, Z. Anorg. Allg. Chem., 1993, 619, 1827 CrossRef CAS.
- For example (reviews): (a) R. F. Jordan, Adv. Organomet. Chem., 1991, 32, 325 CAS; (b) H. H. Brintzinger, D. Fischer, R. Mülhaupt, B. Rieger and R. M. Waymouth, Angew. Chem., Int. Ed. Engl., 1995, 34, 1143 CrossRef CAS; (c) E. Y-. X. Chen and T. J. Marks, Chem. Rev., 2000, 100, 1391 CrossRef CAS.
- (a) T. V. Lubben, P. T. Wolczanski and G. D. van Duyne, Organometallics, 1984, 3, 977 CrossRef CAS . In this report, the reaction with Zr(CH2Ph)4 with 1.14 equiv. of tBu3COH in benzene under reflux conditions for 7 h afforded Zr(CH2Ph)3(OCtBu3), whereas synthesis of Zr(CH2Ph)3(OCtBu3) by the reaction of Zr(CH2tBu)4 with tBu3COH in benzene required 30 h at 93–95 °C; (b) it is also known that certain chromium(IV)-tetra(alkyl)s are stable even in alcohols under reflux conditions.
- For related reviewing articles, see: (a) C. Copéret, M. Chavanas, R. P. Saint-Arroman and J. M. Basset, Angew. Chem., Int. Ed., 2003, 42, 156 CrossRef CAS; (b) J. M. Thomas, R. Raja and D. W. Lewis, Angew. Chem., Int. Ed., 2005, 44, 6456 CrossRef CAS; (c) for an example of a proposed mechanism for the reaction of Cr(CH2tBu)4 with a silica surface, see: J. A. N. Ajou and S. L. Scott, Organometallics, 1997, 16, 86 Search PubMed.
- For recent examples, see: (a) P. Nicholas, H. S. Ahn and T. J. Marks, J. Am. Chem. Soc., 2003, 125, 4325 CrossRef CAS; (b) M. W. McKittrick and C. W. Jones, J. Am. Chem. Soc., 2004, 126, 3052 CrossRef CAS; (c) B. Rhers, A. Salameh, A. Baudouin, E. A. Quadrelli, M. Taoufik, C. Copéret, F. Lefebvre, J. -M. Basset, X. Solans-Monfort, O. Eisenstein, W. W. Lukens, L. P. H. Lopez, A. Sinha and R. R. Schrock, Organometallics, 2006, 25, 3554 CrossRef CAS; (d) F. Blanc, J.-M. Basset, C. Copéret, A. Sinha, Z. J. Tonzetich, R. R. Schrock, X. Solans-Monfort, E. Clot, O. Eisenstein, A. Lesage and L. Emsley, J. Am. Chem. Soc., 2008, 130, 5886 CrossRef CAS.
- J. Yamada and K. Nomura, Organometallics, 2005, 24, 3621 CrossRef CAS.
- J. Yamada, M. Fujiki and K. Nomura, Organometallics, 2007, 26, 2579 CrossRef CAS.
- A. Sinha, L. P. H. Lopez, R. R. Schrock, A. S. Hock and P. Müller, Organometallics, 2006, 25, 1412 CrossRef CAS.
- W. Zhang, S. Katao, W.-H. Sun and K. Nomura, Organometallics, 2009, 28, 1558 CrossRef CAS.
- Examples for isolation of terminal vanadium(III),(IV) alkylidenes: (a) ref. 11c ; (b) B. Hessen, J.-K. F. Buijink, A. Meetsma, J. H. Teuben, G. Helgesson, M. Hakansson, S. Jagner and A. L. Spek, Organometallics, 1993, 12, 2268 CrossRef CAS . Reaction of CpV(CHCMe3)(dmpe) with norbornene was attempted but showed extremely low (catalytic) activity (TON 0.92 after 96 h at 20 °C). No GPC data were described; (c) F. Basuli, U. J. Kilgore, X. Hu, K. Meyer, M. Pink, J. C. Huffman and D. J. Mindiola, Angew. Chem., Int. Ed., 2004, 43, 3156 CrossRef CAS; (d) F. Basuli, B. C. Bailey, J. C. Huffman, M.-H. Baik and D. J. Mindiola, J. Am. Chem. Soc., 2004, 126, 1924 CrossRef CAS. Synthesis of vanadium(IV)-phosphinidene complex from the vanadium(IV)-alkylidene; (e) F. Basuli, B. C. Bailey, D. Brown, J. Tomaszewski, J. C. Huffman, M.-H. Baik and D. J. Mindiola, J. Am. Chem. Soc., 2004, 126, 10506 CrossRef CAS. Synthesis of vanadium(V)-alkylidyne via oxidatively induced α-hydrogen abstraction from vanadium(IV)-alkyl, alkylidene complex.
- Related isolated examples for vanadium-carbene complexes: (a) G. Erker, R. Lecht, R. Schlund, K. Angermund and C. Krüger, Angew. Chem., Int. Ed. Engl., 1987, 26, 666 CrossRef; (b) R. Milczarek, W. Rüsseler, P. Binger, K. Jonas, K. Angermund, C. Kruger and M. Regitz, Angew. Chem., Int. Ed. Engl., 1987, 26, 908 CrossRef.
- (a) B. Hessen, A. Meetsma, F. van Bolhuis and J. H. Teuben, Organometallics, 1990, 9, 1925 CrossRef CAS; (b) J.-K. F. Buijink, J. H. Teuben, H. Kooijman and A. L. Spek, Organometallics, 1994, 13, 2922 CrossRef CAS. No reaction between CpV(CHPh)(NAr)(PMe3) and norbornene or acetone was observed (c) M. Moore, S. Gambarotta, G. Yap, L. M. Liable-Sands and A. L. Rheingold, Chem. Commun., 1997, 643 RSC.
- (a) U. J. Kilgore, C. A. Sengelaub, M. Pink, A. R. Fout and D. J. Mindiola, Angew. Chem., Int. Ed., 2008, 47, 3769 CrossRef CAS; (b) U. J. Kilgore, C. A. Sengelaub, H. Fan, J. Tomaszewski, J. A. Karty, M.-H. Baik and D. J. Mindiola, Organometallics, 2009, 28, 843 CrossRef CAS.
- W. Zhang, J. Yamada and K. Nomura, Organometallics, 2008, 27, 5353 CrossRef CAS.
- K. Nomura, T. Atsumi, M. Fujiki and J. Yamada, J. Mol. Catal. A: Chem., 2007, 275, 1 CrossRef CAS.
- S. Zhang, K. Nomura, unpublished results. Formation of the phenyl complex was confirmed by the crystallographic analysis. These results will be submitted soon.
- For example: (a) N-Heterocyclic Carbenes in Synthesis, ed. S. P. Nolan, Wiley-VCH, Weinheim, Germany, 2006 Search PubMed; (b) N-Heterocyclic Carbenes in Transition Metal Catalysis, ed. F. Glorius, Springer, Berlin, Germany, 2007 Search PubMed; (c) D. Bourissou, O. Guerret, F. P. Gabbai and G. Bertrand, Chem. Rev., 2000, 100, 39 CrossRef; (d) D. Enders, O. Niemeier and A. Henseler, Chem. Rev., 2007, 107, 5605.
- C. D. Abernethy, G. M. Codd, M. D. Spicer and M. K. Taylor, J. Am. Chem. Soc., 2003, 125, 1128 CrossRef.
| This journal is © The Royal Society of Chemistry 2010 |