Diamine ligands in copper-catalyzed reactions
David S.
Surry
and
Stephen L.
Buchwald
*
Department of Chemistry, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139, USA. E-mail: sbuchwal@mit.edu; Fax: +1-617-253-3297; Tel: +1-617-253-1885
First published on 12th May 2010
Abstract
The utility of copper-mediated cross-coupling reactions has been significantly increased by the development of mild reaction conditions and the ability to employ catalytic amounts of copper. The use of diamine-based ligands has been important in these advances and in this perspective we discuss these systems, including the choice of reaction conditions and applications in the synthesis of pharmaceuticals, natural products and designed materials.
 David S. Surry | David S. Surry was born in London and carried out his undergraduate studies at the University of Cambridge, UK. During this time he performed research in the laboratories of Prof. I. Fleming. He then stayed on at Cambridge to complete a PhD with Dr D. R. Spring and spent a period at GlaxoSmithKline, UK. He is currently conducting postdoctoral research at the Massachusetts Institute of Technology with Prof. S. L. Buchwald. |
 Stephen L. Buchwald | Stephen L. Buchwald is the Camille Dreyfus Professor of Chemistry at MIT where he has been on the faculty since 1984. He has received numerous honors, the latest being the Gustavus J. Esselen Award for Chemistry in the Public Interest. During the period January 1999–June 2009, he was the most cited chemist in the world (per paper). He lives in Newton, Massachusetts with his wife, Susan Haber, and their two children, Sara (age 10) and Nathan (age 13), along with their three cats, Trixie, Rocket and the famous orange tabby, Rufus. |
Introduction
Copper-mediated cross-coupling reactions that form C–C, C–N and C–O bonds have a long history in organic chemistry and have been used in industrial processes.1 A key limitation of these protocols, however, is the requirement for harsh reaction conditions (high temperatures and polar solvents) and the need to use stoichiometric or greater quantities of copper. Following important earlier contributions,2–6 a major improvement came in 2001 when Buchwald showed that diamine ligands enable the coupling of aryl halides and amides (the Goldberg reaction) to be performed under much milder conditions using a weak base, non-polar solvent and temperatures as low as room temperature with a catalytic amount of copper.7 These new conditions significantly increased the utility of these reactions and there has since been an avalanche of research into Cu-catalyzed cross-coupling.8–15 These Cu-catalyzed reactions are robust, can be performed with simple ligands and can be used in the presence of a plethora of functional groups, making them highly attractive to those engaged in the synthesis of complex molecular architectures. A range of different useful ligands have emerged including phenanthrolines,3,16,17 α-amino acids,4,12,18,19 1,3-diketones,20,21 imines,22,23 salicylamides24 and others6,25–39 as well as “ligand-free” systems.40–45 A key advantage of diamine-based ligand systems (Fig. 1) for copper over some others that have been brought forward is the large scale access to the ethylenediamine and cyclohexanediamine backbones (it should be noted there have also been studies with BINAM46–48) and the commercial availability of the ligands.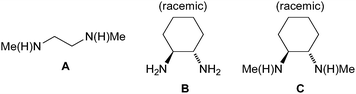 | ||
| Fig. 1 Commercially available diamine ligands. | ||
The mechanistic details of these reactions have remained somewhat obscure, however, several recent experimental49–57 and computational studies58 have begun to shed light on this area. Recently, excellent reviews on Cu-catalyzed cross-coupling have focused on applications in synthesis,13 developments in methodology11 and the use of amino acid-based ligands.12 It is the goal of this perspective to discuss the utilization of diamine ligands in Cu catalysis and to illustrate the power of these methods with examples drawn from the synthesis of natural products, pharmaceuticals and novel materials.
A complementary approach, pioneered by Lam, Chan59,60 and Evans,61 is provided by the coupling of heteroatom-based nucleophiles with boronic acids in the presence of a copper salt, typically under an atmosphere of air. These reactions have the advantage of taking place under mild conditions (often at room temperature under neutral conditions)9,62,63 and have been utilized in some elegant natural product syntheses.64–66 A number of aryl donors have been employed in these reactions including aryl stannanes,67 aryl bismuth reagents,68 aryl siloxanes69 and diaryl iodonium salts.70 These reactions can offer unrivalled functional group tolerance, however, the expense and difficulty of access to the arylating agents can represent a serious limitation.
Copper-catalyzed methods
In 2001 Buchwald7 showed that the use of diamine ligands allows the Goldberg reaction of aryl halides to be performed under mild conditions, employing a weak base and non-polar solvent (Scheme 1).71–75 This discovery has had a significant impact on the synthesis of natural products and pharmaceuticals (vide infra) and other catalyst systems have since been described by others.19,22,26,76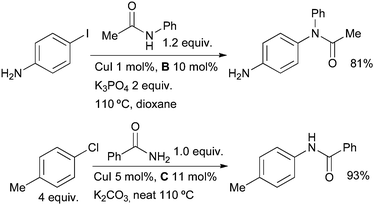 | ||
| Scheme 1 Development of Cu-catalyzed amidation of aryl halides by Buchwald. | ||
These early studies laid out the parameters which have proved useful in later applications of these catalyst systems. The best solvents are toluene and dioxane, although DMF is better for very polar amides. A range of different copper sources are active, CuI gives the highest yields and has the advantage of being cheap and air stable. The choice of base, however, is much more important. For aryl iodides K3PO4 is best, K2CO3 giving a much slower reaction. On the other hand, for aryl bromides, which generally react more slowly that aryl iodides, K3PO4 is often unsuccessful, but K2CO3 is effective. This disparity is thought to result from the need to match the rate of deprotonation of the amide and the rate of C–N bond formation. If the rate of deprotonation is relatively too high, then deprotonated amide accumulates and forms an unreactive cuprate complex.49 Evidence to support this hypothesis comes from the observation that strong bases such as KHMDS are only efficacious if added slowly to the reaction mixture. Indeed both K3PO4 and K2CO3 behave as strong bases in aprotic solvents, but their low solubility ensures slow formation of the deprotonated amide. Studies were also performed to determine the optimal ligand structure. A wide range of diamines were examined in these reactions and it was found that ligands based on either ethylenediamine or cyclohexanediamine provide favorable reactivity.71,77 Ligands derived from the former are generally preferred due to their lower cost. In some cases the unsubstituted diamines can be used, however, N,N′-dimethyl-substituted ligands generally provide higher rates and avoid problems associated with undesired N-arylation of the ligand under the reaction conditions.77 Further substitution at N or the use of larger groups in place of methyl results in substantially less efficient catalysts (Fig. 2).71 Kinetic studies have indicated that the role of the ligand is to help prevent the formation of less reactive, multiply ligated cuprate structures. Indeed diamine coordination to the isolated copper(I) amidate has been shown to generate a species that is both chemically and kinetically competent for N-arylation.49,52
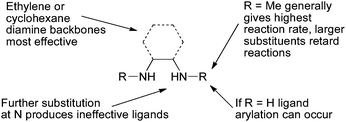 | ||
| Fig. 2 Critical features of diamine-based ligands. | ||
The reaction is applicable to a range of electron-rich and electron-deficient aryl and heteroaryl bromides and iodides and even some aryl chlorides. Carbamates are also useful coupling partners, but the reaction remains most suitable for lactams and primary amides. Acyclic secondary amides are much more challenging, an efficient reaction can only be realized for N-aryl or N-methyl amides, except in the case of formamides (Scheme 2).
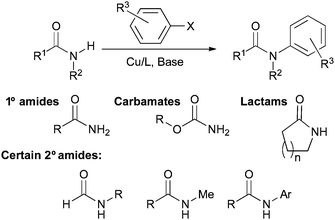 | ||
| Scheme 2 Substrate scope of intermolecular Cu-catalyzed amidation. | ||
In a notable application, the Cu-catalyzed arylation of trifluoroacetamide followed by hydrolysis has been used to allow the use of trifluoroacetamide as an ammonia equivalent in the synthesis of anilines from aryl halides.78 The same ligand system can be used for the synthesis of enamides by the coupling of amides with vinyl halides (Scheme 3).79 This method has seen considerable application in the synthesis of various natural products (vide infra). Very similar conditions are also applicable to the coupling of azoles with vinyl halides.80
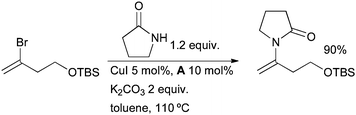 | ||
| Scheme 3 Synthesis of enamides from vinyl halides. | ||
Porco had previously detailed the use of copper(I) thiophenecarboxylate (CuTC) as a catalyst for the amidation of vinyl halides, however this method is limited to terminal vinyl iodides.81 Porco subsequently recognized that the combination of CuTC and A as stoichiometric reagents constitutes a useful system.82 Ma has shown that an alternative catalyst system exploiting N,N-dimethylglycine as a ligand is also quite effective.83
Hsung found that diamine ligands allow the successful amidation of alkynyl bromides to give ynamides (Scheme 4).84 This method permits a complementary access to ynamides, which are proving to be useful synthons.85
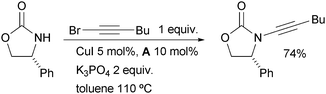 | ||
| Scheme 4 Synthesis of ynamides according to Hsung. | ||
The method was later extended to include sulfonamides by Urabe and Sato.86 Subsequently, Danheiser87 and Hsung88,89 have discovered alternative conditions which have served to increase the substrate scope. Recently Evano has documented a new method for the synthesis of ynamides from 1,1-dibromo-1-alkenes and sulfonamides or carbamates using a copper-diamine catalyst (Scheme 5).90
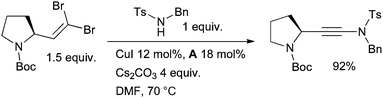 | ||
| Scheme 5 Synthesis of ynamides according to Evano. | ||
The reaction is thought to proceed via initial amidation of the trans C–Br bond of the dibromoalkene followed by elimination of HBr to yield the ynamide. Evano has also shown that the formation of either a ketene N,N-acetal or an ynamide from the reaction of a 1,1-dibromo-1-alkene with amides can be controlled by the choice of solvent—when dioxane is used the ynamide results, with toluene the N,N-acetal.91 Allenamides are another important class of synthetic intermediate and can be accessed by the Cu-catalyzed coupling of nucleophiles with allenyl halides. The first reaction of this class was described by Trost (Scheme 6).92
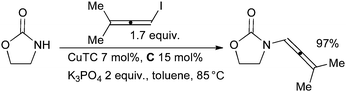 | ||
| Scheme 6 Synthesis of allenamides according to Trost. | ||
Hsung studied a very similar catalyst system and showed that enantiomerically enriched allenyl halides undergo coupling without isomerization.93 Recently, Bäckvall showed that the use of a closely related catalyst system employing A as ligand allows the synthesis of allylic allenamides by the coupling of N-tosyl allylic amines with allenyl halides.94
A number of ligands have been used for the Cu-catalyzed arylation of anilines,18,25,95,96 however, the arylation of heteroarylamines remains challenging as reduction of the aryl halide electrophile competes with the desired amination.97 Heteroaryl diarylamines have emerged as an important class of molecules in a range pharmaceuticals, notably protein kinase inhibitors such as imatinib.98 One of the best systems for the arylation of heteroarylamines was disclosed by Liu using diamine ligand A; phenanthroline and proline proved much less efficient (Scheme 7).99 In order for the reaction of aryl bromides to proceed efficiently, it is necessary to add potassium iodide to the reaction mixture, presumably to effect an in situ halide exchange reaction.100
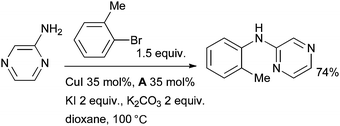 | ||
| Scheme 7 Arylation of heteroarylamines according to Liu. | ||
Buchwald has shown that these catalytic systems can also be used for the N-arylation of indoles77 and for a range of other NH heterocycles (Scheme 8).7,101 For simple substrates the unsubstituted ligand B is serviceable. However, for more difficult cases (for example, ortho-substituted aryl halides), N,N′-substituted ligands A and C give better results (in the case of ligand C, the trans isomer is more effective than the cis). It was demonstrated that for challenging substrates the N-arylation of the unsubstituted ligand B can occur and the formation of such N-aryl ligands decreases the rate of the desired arylation as well as consuming one of the substrates. Aryl iodides and bromides undergo the desired reaction with the indole nitrogen and selective arylation is possible in the presence of free alcohols, anilines, amides and primary amines. A considerable variety of alternative ligand systems has since become available.16,18,22,23,26,39,44,45,102,103
 | ||
| Scheme 8 Cu-catalyzed arylation of NH heterocycles. | ||
The use of diamine ligands can also give a favorable outcome for the Cu-catalyzed vinylation of NH heterocycles.104–108 This approach was applied to the enantioselective synthesis of β-azaheterocyclic acid derivatives109 by coupling of iodoenoates followed by enantioselective Cu-catalyzed conjugate reduction (Scheme 9).
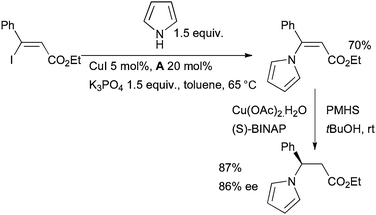 | ||
| Scheme 9 Synthesis of β-azaheterocyclic acid derivatives. | ||
Later Xi studied the use of diamine ligands in this reaction in more detail and found that ethylenediamine is the most efficient, allowing the coupling of a variety of azoles with vinyl bromides.80 Movassaghi has developed a complementary Pd-catalyzed coupling of pyrroles and indoles with vinyl triflates.110
Saito disclosed the Cu-catalyzed coupling of aryl iodides and some aryl bromides with tetra-n-butylammonium nitrite to yield aromatic nitro compounds (Scheme 10).111 This was the first example of a cross-coupling route to these important compounds. The scope of this approach has recently been significantly extended by the use of a Pd-based catalyst.112
 | ||
| Scheme 10 Synthesis of aryl nitro compounds according to Saito. | ||
Diamine ligands can also be used in the Cu-catalyzed coupling of sodium azide with aryl halides, the reaction proving suitable for a range of electron-rich and electron-deficient aryl bromides.113 Ma has shown that proline is also an effective ligand for copper for this reaction.114 In a recent study by Molander and Ham, diamine ligands and proline proved equally efficient in bringing about the coupling of azide with haloaryltrifluoroborates (Scheme 11).115
 | ||
| Scheme 11 Synthesis of aryl azides by coupling of aryl halides and inorganic azide. | ||
The synthesis of aryl azides by Cu-catalyzed coupling is significant because of the utility of aryl azides in click chemistry116 and the reported difficulty in using Pd-based catalysts to effect this transformation.117
Diamine ligands are also useful in Cu-catalyzed C–P bond construction (Scheme 12).118 It was found that with these ligands the efficiency of the coupling of secondary phosphites with aryl halides is significantly increased, although the coupling of secondary phosphines can be conducted without the addition of ligand. Subsequently it has been shown that both proline and picolinic acid can also be employed as ligands in bringing about this transformation.119
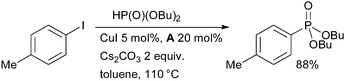 | ||
| Scheme 12 Cu-catalyzed coupling of secondary phosphites and aryl halides. | ||
Li has studied the Cu-catalyzed C- or O-arylation of enolates using diamine ligands (Scheme 13).120 In initial screening these authors showed that diamine ligand A provides a far more active catalyst system than proline or phenanthroline.
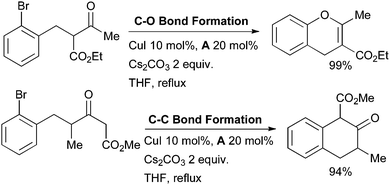 | ||
| Scheme 13 Selective, intramolecular arylation of enolates studied by Li. | ||
Researchers at Merck Frosst have demonstrated that diamine ligands can be useful for the synthesis of sulfones from sulfinate salts and aryl halides (Scheme 14).121 This represents a considerable improvement over previous conditions which required the use of stoichiometric CuI.122
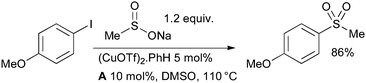 | ||
| Scheme 14 Merck Frosst synthesis of aryl sulfones. | ||
It has subsequently been shown by Ma123 that proline is also a competent ligand in this transformation and allows the use of some aryl bromides.
The classic Rosenmund–von Braun reaction (the conversion of aryl halides to benzonitriles with CuCN)124 can be performed in a catalytic manner by using diamine ligand A and NaCN as the source of cyanide (Scheme 15).125 This method is applicable to aryl iodides and by addition of 20 mol% KI can be extended to aryl bromides by exploiting an in situ Finkelstein reaction.100
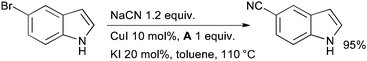 | ||
| Scheme 15 Synthesis of benzonitriles according to Buchwald. | ||
These conditions have been used in the synthesis of biologically active molecules126,127 and a number of variants have since been documented using alternative ligand systems and cyanide sources.128–131
Applications in the synthesis of heterocycles
Transition metal-catalyzed reactions have seen extensive use in the synthesis of heterocycles.132,133 Interest in these processes stems from their ability to make heterocycles with unusual substitution patterns that are inaccessible by traditional approaches. The facility with which Cu/diamine systems can bring about carbon–heteroatom bond formation has led to the use of these catalysts in a number of novel methods, allowing access to a range of functionalized heterocycles, typically under mild conditions.One of the earliest examples of heterocycle synthesis with a copper-diamine catalyst system was the synthesis of indolines by intramolecular aryl amidation (Scheme 16).71
 | ||
| Scheme 16 Synthesis of indolines according to Buchwald. | ||
It was found that the presence of the ligand was essential for reaction to occur. Intramolecular amidation proceeds much more easily than the intermolecular process, a phenomenon which has also been observed in analogous Pd-catalyzed reactions.134 Subsequently other systems have been described for such intramolecular amidations.18,135 Evano extended this method to the synthesis of pyrroloindoles (a structural motif found in a range of biologically active alkaloids) from iodotryptophans (Scheme 17).136
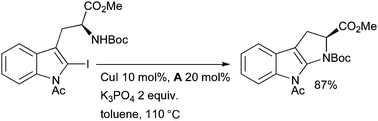 | ||
| Scheme 17 Synthesis of pyrroloindoles according to Evano. | ||
Li has investigated intramolecular amidation reactions of vinyl halides. Initially the synthesis of β-lactams and alkylideneazetidines was studied (Scheme 18).137 The outcome is highly dependent on the nature of the substituent on nitrogen, the reaction is successful for N-tosyl amines but not for unsubstituted or Boc-protected amines. The choice of ligand for copper is also significant: diamine ligand A is very effective, while the use of amino acid derivatives and phenanthroline gives much lower yields. This method was later extended to the preparation of larger ring systems.138
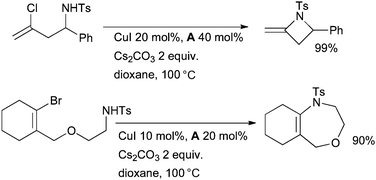 | ||
| Scheme 18 Li's investigation of intramolecular amidation of vinyl halides. | ||
It is also possible to synthesize medium-ring nitrogen heterocycles by Cu-catalyzed arylation of a β-lactam, followed by ring-opening by a pendant amino group (Scheme 19).139 The realization of the reaction is dependent on the competing rates of the desired arylation of the amide against the undesired intramolecular reaction of the amine to form an indoline. It was shown that the ratio of these products is strongly influenced by the presence of the diamine ligand.
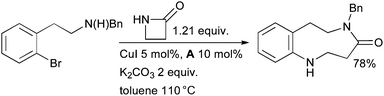 | ||
| Scheme 19 Synthesis of medium-ring nitrogen heterocycles by tandem cross-coupling/ring expansion. | ||
Pabba at Albany Molecular Research has shown that N-aryl indazoles may be accessed in a two-step, one pot protocol from 2-halobenzaldehydes (Scheme 20).140 An aryl hydrazine is first treated with a 2-halobenzaldehyde under microwave irradiation to furnish an intermediate aryl hydrazone which is not isolated, CuI and diamine ligand are then added to the reaction mixture and irradiation continued to afford the indazole. Importantly, it was shown that in the absence of the diamine ligand only a trace of product is formed.
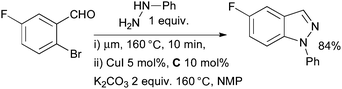 | ||
| Scheme 20 Albany Molecular Research synthesis of indazoles. | ||
A two step reaction sequence has also been employed to make indolines via a domino Cu-catalyzed amidation/nucleophilic substitution process (Scheme 21). The clean inversion observed at the mesylate center strongly indicates the occurrence of an SN2 substitution process.141 The reaction can also be used to synthesize 6- and 7-membered rings and a range of nitrogen protecting groups including Boc, Cbz and acetyl are compatible.
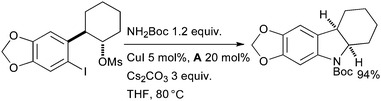 | ||
| Scheme 21 Synthesis of indolines according to Buchwald. | ||
Buchwald used a Cu/diamine catalyst system in the synthesis of N-(2-ketoaryl)amides by amidation of o-halophenones (Scheme 22).142 The reaction still proceeds in the absence of ligand, but at a much lower rate. These intermediates can subsequently be engaged in a Camps cyclization to generate 2-aryl-4-quinolones or in a McMurry coupling to access indoles.
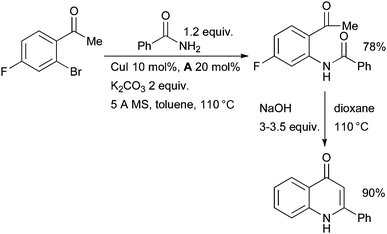 | ||
| Scheme 22 Synthesis of quinolones according to Buchwald. | ||
Li143,144 and Buchwald145 have both delineated a route to substituted pyrroles based on the Cu-catalyzed double alkenylation of amides with 1,4-diiodo-1,3-dienes which are available from the zirconium-146 or titanium-mediated147 coupling of two alkynes (Scheme 23).
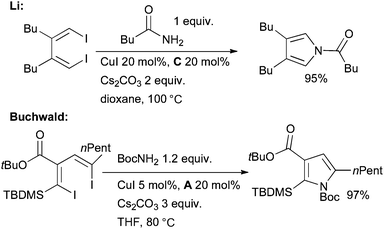 | ||
| Scheme 23 Synthesis of pyrroles according to Li and Buchwald. | ||
Willis has used this method in the synthesis of indoles by the tandem coupling of 2-(2-haloalkenyl)aryl halides with carbamates, amides or anilines (Scheme 24).148 This author had previously published a complementary procedure using Pd-based catalysts.149,150
 | ||
| Scheme 24 Synthesis of indoles according to Willis. | ||
A Cu-catalyzed amidation of vinyl halides followed by cyclization with iodine provides a route to oxazoles (Scheme 25).151 This method grants entry to trisubstituted oxazoles, but is much less efficient for the preparation of mono- and disubstituted oxazoles. The latter could, however, be accessed via a domino C–N/C–O bond-forming reaction of 1,2-dihaloalkenes. A related method had been used previously by Glorius in the synthesis of benzoxazoles.152
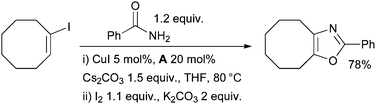 | ||
| Scheme 25 Synthesis of oxazoles according to Buchwald. | ||
Scientists at Johnson & Johnson have developed a conceptually similar approach for the synthesis of benzimidazoles from 1,2-dihaloarenes and amidines (Scheme 26).153
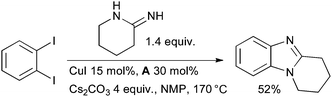 | ||
| Scheme 26 Johnson & Johnson synthesis of benzimidazoles. | ||
In 2006 Buchwald disclosed a method for the synthesis of pyrroles by a domino Cu-catalyzed amidation/hydroamidation sequence of haloenynes.154 Pyrazoles could also be accessed by this method by utilizing bis-Boc hydrazine as nucleophile (Scheme 27). Preliminary mechanistic studies indicated that the hydroamidation step is both Cu-catalyzed and base-assisted. Ackermann has applied an allied approach to the synthesis of indoles.155
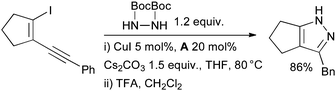 | ||
| Scheme 27 Synthesis of pyrazoles according to Buchwald. | ||
Lautens has used a diamine ligand in a tandem intramolecular amidation of gem-dibromoalkenes to generate imidazoindolones (Scheme 28). Such 2-aminoindoles are hard to generate by more traditional approaches such as the Fischer indole synthesis, but are structural components of a range of alkaloids.156
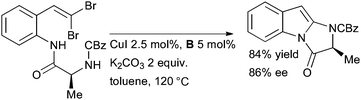 | ||
| Scheme 28 Synthesis of imidazoindolones according to Lautens. | ||
In 2007 Buchwald showed that N-alkyl benzimidazoles can be accessed from o-haloanilides by Cu-catalyzed amidation, followed by acid- or base-mediated cyclization; diamine ligands proved to be the most effective of those examined (Scheme 29).157
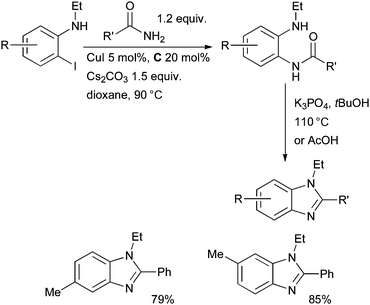 | ||
| Scheme 29 Synthesis of benzimidazoles according to Buchwald. | ||
This is an important method as it allows access to regioisomerically pure, substituted, N-alkyl benzimidazoles. At approximately the same time Ma published a similar process for the synthesis of benzimidazoles using proline as a ligand.158 A complementary Pd-catalyzed method allows access to N-aryl benzimidazoles by exploiting anilines as the nucleophile.159
Copper-catalyzed halide exchange
In 2002 Buchwald showed that diamine ligands exhibit an accelerating effect on the copper-catalyzed halide exchange between aryl bromides and inorganic iodides, sometimes referred to as the aromatic Finkelstein reaction (Scheme 30).100 | ||
| Scheme 30 Cu-catalyzed halide exchange. | ||
The reaction provides access to a range of electron-rich and electron-deficient aryl iodides and has excellent functional group tolerance because of the lack of a strong base in the reaction medium. The process is efficient for heteroaryl bromides and preliminary studies also indicated that vinyl bromides are viable substrates. In analogy to the Finkelstein reaction, this is an equilibrium reaction, the driving force for conversion to the aryl iodide being provided by the difference in solubility of the sodium halides. The equilibrium conversion to the aryl iodide is higher in solvents in which NaBr has relatively low solubility, such as dioxane or pentanol, than in polar solvents in which it is more freely soluble such as DMF. These conditions represent a significant improvement over previously published procedures which required high temperatures (>150 °C), polar solvents and an excess of copper160,161 or other metal162,163 (although these traditional methods are still preferred in some contexts164). Later this reaction was incorporated into a domino halide exchange, cyanation method, allowing the direct conversion of aryl bromides to nitriles125 and in an analogous procedure to produce aryl azides.113 The conditions which make use of diamine ligands have seen considerable application, particularly in the field of materials science where it is often advantageous to perform multiple consecutive cross-coupling reactions on functionalized substrates, thus providing an iterative approach to oligomeric structures. Aryl bromides are often less reactive than aryl iodides under classical cross-coupling conditions, this therefore allows an aryl iodide to be “protected” as an aryl bromide during a synthesis and unmasked as necessary. Vicente showcased this tactic in the synthesis of thioether phenylene aryl iodide monomers that could participate in further cross-coupling reactions (Scheme 31).165
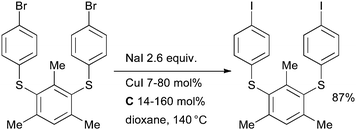 | ||
| Scheme 31 Vicente's approach to thioether phenylene aryl iodide monomers | ||
In a particularly impressive example, Rapenne executed this approach in the synthesis of ruthenium complexes which have potential utility as molecular motors (Scheme 32).166 This is the first example of the halogen–metal exchange procedure of an organometallic compound. If the Ru center is coordinated to hydrotris(indazolyl)borate ligand, however, the reaction is much less successful. The aryl iodide was subsequently used in a Sonogashira reaction.
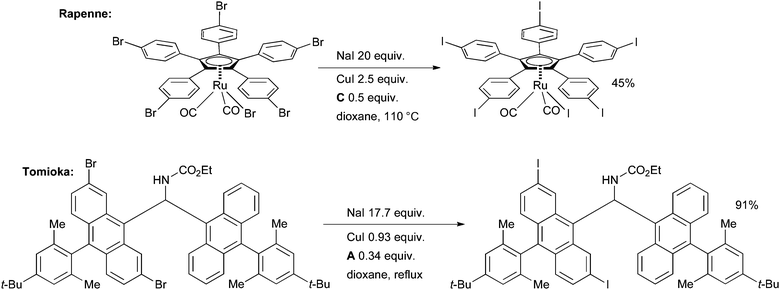 | ||
| Scheme 32 Applications of the aromatic Finkelstein reaction. | ||
Tomioka examined this method in the synthesis of copper complexes of sterically congested diaryldiazomethanes as part of a study on molecular magnets (Scheme 32).167 The aryl iodides were later employed in a Suzuki reaction on a stable diazonium intermediate. In an example which illustrates the functional group tolerance of the reaction, Whitehead made a complex vinyl iodide intermediate en route to the antibacterial agent (6S)-6-fluoroshikimic acid (Scheme 33).168
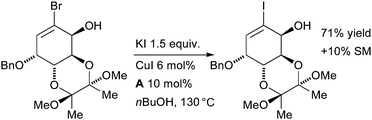 | ||
| Scheme 33 Whitehead's synthesis of fluoroshikimic acid. | ||
The choice of ligand was crucial to the success of the halide exchange, the authors found in a simplified model system that ligand A was far more effective than B. Once again, the product was subsequently engaged in a Pd-catalyzed cross-coupling reaction.
Spencer also employed this approach in the synthesis of photoactivatable cholesterol surrogates for studies of HDL formation and cholesterol metabolism (Scheme 34).169
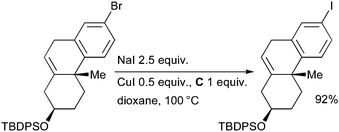 | ||
| Scheme 34 Synthesis of photoactivatable cholesterol surrogates according to Spencer. | ||
Aryl iodides can be key agents in medically important imaging techniques such as positron emission tomography (PET) and single photon emission computed tomography (SPECT). With a view to investigating such agents, Sutherland utilized an aromatic Finkelstein reaction in the synthesis of an iodinated analogue of the noradrenaline reuptake inhibitor reboxetine which has potential as an imaging agent for the noradrenaline transporter (Scheme 35).170 The ability to visualize this receptor within the brain has implications in the treatment of neurodegenerative and psychiatric disorders.
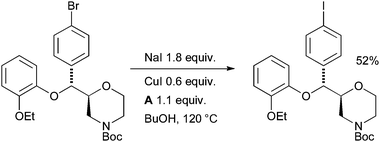 | ||
| Scheme 35 Synthesis of an iodinated reboxetine analogue according to Sutherland. | ||
Sutherland has also used this method in the synthesis of an iodo analogue of the peripheral benzodiazepine receptor ligand PK11195, which could function as a radiolabelled ligand for the study of the physiological role of this receptor (Scheme 36).171 It must be noted, however, that the application of the Cu-catalyzed halide exchange in the synthesis of radioiodine-labeled compounds may require the reaction to proceed at a higher rate, typical reaction times for this process are 20–24 h, which could present difficulties when synthesizing radioactive compounds with nuclei with short half-lives.
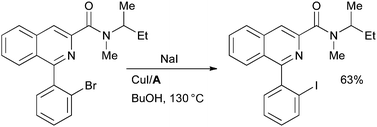 | ||
| Scheme 36 Sutherland's synthesis of a peripheral benzodiazepine receptor ligand. | ||
Applications in pharmaceutical research
Copper-mediated coupling reactions with diamine ligands have been extensively employed in the synthesis of novel pharmaceutical agents. Cu-catalyzed arylation is especially efficient for lactams and these substrates are salient in medicinal chemistry programs. Koert used such a reaction in the synthesis of new factor Xa inhibitors based on dianhydrohexitoles for the treatment of thrombosis (Scheme 37).172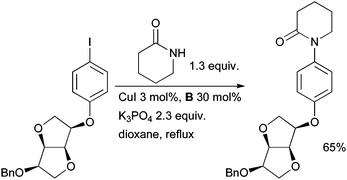 | ||
| Scheme 37 Koert's synthesis of factor Xa inhibitors. | ||
At Eli Lilly a Cu-catalyzed aryl amidation was valuable in SAR studies of selective norepinephrine reuptake inhibitors that have potential in the treatment of depression and attention-deficit/hyperactivity disorder (Scheme 38).173 A number of derivatives were prepared with both electron-deficient and electron-neutral aryl halides and the reaction was amenable to gram scale preparation of intermediates.
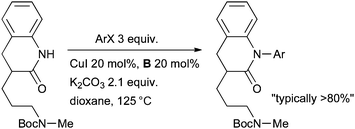 | ||
| Scheme 38 Eli Lilly synthesis of norepinephrine reuptake inhibitors. | ||
Researchers at Abbott have also examined an aryl amidation in the synthesis of dipeptidyl peptidase IV inhibitors (Scheme 39).174 The somewhat low yield perhaps stems from undesired deprotonation α to the nitro group leading to side reactions.
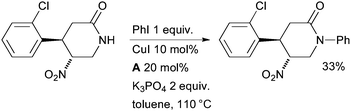 | ||
| Scheme 39 Abbott synthesis of dipeptidyl peptidase IV inhibitors. | ||
Beghyn explored Cu-catalyzed amidation of aryl iodides in the synthesis of arylated derivatives of the PDE5 inhibitor tadalafil (Scheme 40).175 The low temperature of the process, long reaction time and high Cu loading are noteworthy. The authors found that more conventional conditions (for example, performing the reaction at 110 °C) resulted in complete isomerization from the cis to the thermodynamically more stable trans isomer of the tetrahydrocarboline (which shows much lower biological activity). Only by running the reaction at 14 °C was a 95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 ratio of cis:trans isomers obtained. It was also noted that under none of the conditions examined was any N-arylation of the indole nitrogen observed.
:5 ratio of cis:trans isomers obtained. It was also noted that under none of the conditions examined was any N-arylation of the indole nitrogen observed.
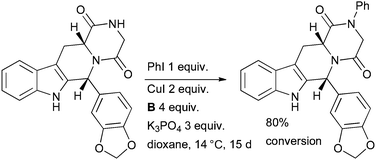 | ||
| Scheme 40 Beghyn's study of tadalafil analogues. | ||
At the Genomics Institute of the Novartis Research Foundation a Cu-catalyzed amidation reaction was employed in the synthesis of novel non-nucleoside reverse transcriptase inhibitors (NNRTIs) for the treatment of HIV (Scheme 41).176 The generality of the amidation was valuable in the synthesis of a wide range of derivatives with differing substitution patterns on the N-aryl ring.
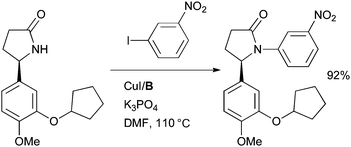 | ||
| Scheme 41 Synthesis of non-nucleoside reverse transcriptase inhibitors performed at the Genomics Institute of the Novartis Research Foundation. | ||
At GlaxoSmithKline researchers investigated the amidation of an aryl iodide in the synthesis of non-brain-penetrant histamine H3 receptor antagonists for the treatment of allergy conditions (Scheme 42).177
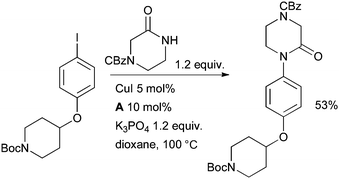 | ||
| Scheme 42 GlaxoSmithKline synthesis of histamine H3 receptor antagonists. | ||
The Cu-catalyzed vinylation of amides is also apposite in medicinal chemistry. Scientists at Bristols-Myers Squibb exploited diamine ligands in the Cu-catalyzed synthesis of enamides with aminopeptidase activity from α-amino amides (Scheme 43).178 Both N-Boc protected and unprotected α-amino amides were suitable substrates in the reaction. N-Cbz protected substrates, however, gave extensive hydantoin formation. A range of vinyl iodides could be used, although internal vinyl iodides gave lower conversion.
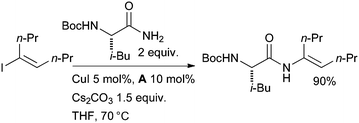 | ||
| Scheme 43 Bristols-Myers Squibb synthesis of enamides with aminopeptidase activity. | ||
At Pfizer, a Cu-catalyzed arylation of an oxazolidinone was achieved in one route to the compound CJ-15,161 which showed promise as a κ-opioid receptor agonist as a non-addictive analgesic (Scheme 44).179,180 The authors went on to explore the generality of the method and found the reaction to be effective with a range of electron-deficient and electron-neutral aryl bromides and iodides and a variety of oxazolidinones, providing a complementary method to other reported metal-catalyzed arylation reactions of these substrates with aryl halides.181–184
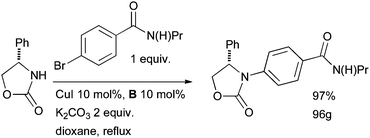 | ||
| Scheme 44 Pfizer synthesis of CJ-15,161, a κ-opioid receptor agonist. | ||
At Wyeth the amidation of a bromopyridine with a vinylogous amide was used in the synthesis of squarate inhibitors of mitogen activated protein kinase-activated protein kinase 2 (MK-2), which are of interest as therapeutic agents for various inflammatory conditions (Scheme 45).185
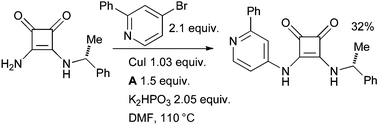 | ||
| Scheme 45 Wyeth synthesis of histamine H3 receptor antagonists. | ||
At Pfizer the Cu-catalyzed O-arylation of pyridinones with A was expedient in the synthesis of CRF1 receptor antagonists which have potential for the treatment of stress-related disorders (Scheme 46).186
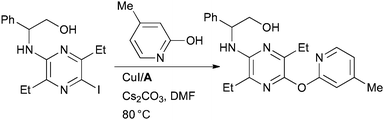 | ||
| Scheme 46 Pfizer synthesis of CRF1 receptor antagonists. | ||
This is an interesting result as previous studies of the arylation of 2-pyridinones with Cu/diamine catalyst systems have indicated that N-arylation dominates.36,187 Such an N-arylation was utilized at Amgen in the synthesis of inhibitors of receptor tyrosine kinase c-Kit for the treatment of fibrotic diseases (Scheme 47).188
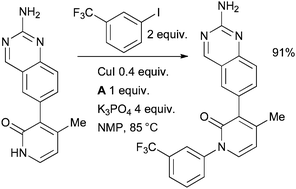 | ||
| Scheme 47 Amgen synthesis of inhibitors of receptor tyrosine kinase c-Kit. | ||
A number of analogues were synthesized with this method; notably the presence of the unprotected aminoquinazoline did not interfere with the outcome of the reaction. In a related example, Steinmetzer performed an intermolecular N-arylation of a pyridazinone in the synthesis of a matriptase inhibitor which might find application in cancer therapy (Scheme 48).189
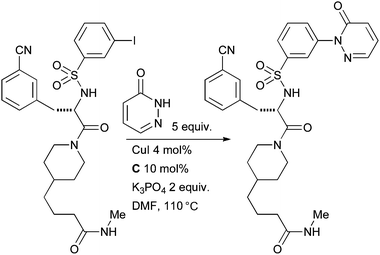 | ||
| Scheme 48 Steinmetzer's synthesis of a matriptase inhibitor. | ||
Another example of the utilization of a Cu/diamine catalyst system in the arylation of a pyridazinone has been provided by research at Neurocrine on the synthesis of thienopyridazinone-based melanin-concentrating hormone receptor 1 antagonists (Scheme 49).190 These molecules have potential as anorectic agents for the treatment of obesity. Multi-gram batches of material were prepared with this protocol.
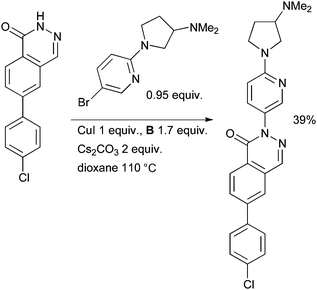 | ||
| Scheme 49 Neurocrine synthesis of melanin-concentrating hormone receptor 1 antagonists. | ||
Cu-catalyzed arylation of sulfonamides191–193 is also a valuable tool in medicinal chemistry. At Merck, chemists employed a Cu-mediated arylation of benzene sulfonamide with a complex aryl bromide substrate in the synthesis of dipeptidyl peptidase IV inhibitors for the treatment of diabetes (Scheme 50).194 A full equivalent of copper was necessary in order to ensure reproducible yields, although extensive optimization was not attempted.
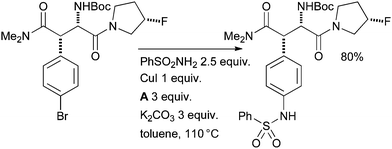 | ||
| Scheme 50 Merck synthesis of dipeptidyl peptidase IV inhibitors. | ||
The coupling of tert-butyl carbamate with aryl halides provides a convenient method of directly synthesizing Boc-protected anilines. At Schering-Plough a combination of CuI and A was used to catalyze this reaction in the synthesis of melanin-concentrating hormone receptor 1 antagonists for the treatment of obesity (Scheme 51).195
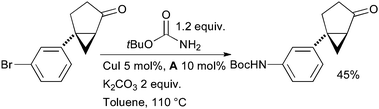 | ||
| Scheme 51 Schering-Plough synthesis of melanin-concentrating hormone receptor 1 antagonists. | ||
The arylation of NH heterocycles with Cu/diamine systems77,101 is also pertinent in the synthesis of pharmaceuticals. At Athersys, Cu-catalyzed arylation of indoles has been practised in the synthesis of ramatroban analogues that can act as selective CRTH-2 (chemoattractant receptor-homologous) antagonists; such compounds have potential in the treatment of asthma (Scheme 52).196
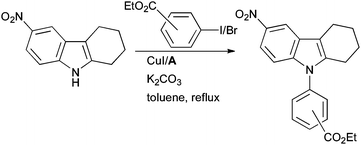 | ||
| Scheme 52 Athersys synthesis of ramatroban analogues. | ||
This method is also illustrated by the work of scientists at IRBM-Merck Research Laboratories in the synthesis of inhibitors of the hedgehog signaling pathway for the treatment of various cancers (Scheme 53).197 Note that the difficulty in coupling acyclic secondary amides allows selective arylation of the NH heterocycle in the presence of the free amide.
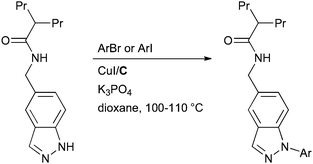 | ||
| Scheme 53 IRBM-Merck Research Laboratories synthesis of inhibitors of the hedgehog signaling pathway. | ||
At Merck, an intramolecular Cu-catalyzed arylation of a pyrazole found purpose in the synthesis of novel tricyclic full agonists for the G-protein-coupled niacin receptor 109A for the treatment of hyperlipidaemia (Scheme 54).198
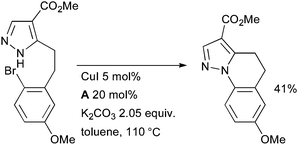 | ||
| Scheme 54 Merck synthesis of agonists of G-protein-coupled niacin receptor 109A. | ||
Applications in materials science
Metal-catalyzed cross-coupling has assumed a central role in the synthesis of novel materials. Cu-catalyzed processes are especially attractive in this area due to their robustness, allowing them to be applied to complex, designed molecular architectures without resorting to specialized equipment (such as dry-box or Schlenk line techniques).Wang made use of a Cu-catalyzed arylation of an azaindole in the synthesis of molecular stars with six-fold symmetry (Scheme 55). These molecules exhibit luminescence and form self-assembled structures in the solid state and as such could be useful in OLEDs.199 The overall isolated yield of 40% corresponds to an average yield of 86% per C–N coupling reaction. A Pd-based catalyst, however, was more effective in the amination of this substrate with a diarylamine. The arylation of carbazoles was also investigated by Yamamoto in the formation of dendrimers possessing a potential gradient; such materials are important in organic electronics (Scheme 55).200 Ligands B and C were valuable in synthesizing a range of dendrimers of varying size. The authors note that the milder reaction conditions provided by the diamine ligands in the coupling reaction permitted access to large dendrimers which were inaccessible under the harsher, more oxidizing conditions of the classical Ullmann reaction.201
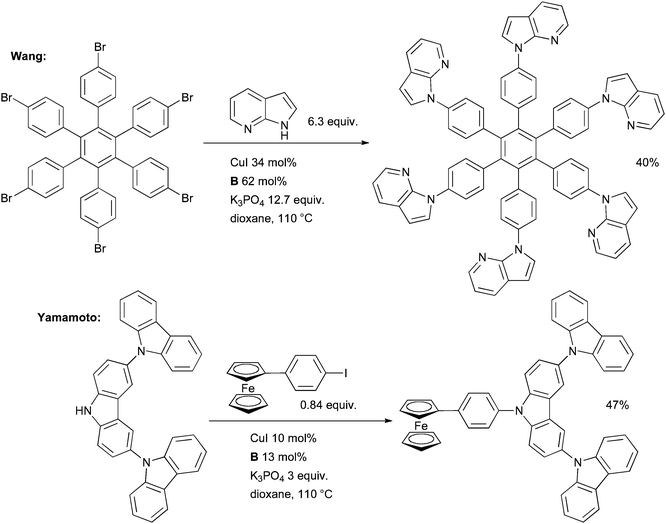 | ||
| Scheme 55 Applications of Cu/diamine-catalyzed cross-coupling in materials science. | ||
Burgess used a Cu-catalyzed arylation of carbazole in the synthesis of energy transfer systems based on squaraines (Scheme 56).202 The goal of this work was to investigate donor acceptor cassettes connected by a conjugated linker. Pd-catalyzed coupling reactions were not of merit in effecting this transformation, even though Pd-based methods had proven efficacious for simpler substrates.
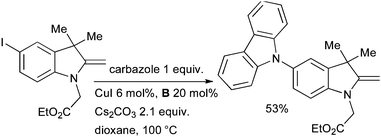 | ||
| Scheme 56 Synthesis of energy transfer systems by Burgess. | ||
Applications in natural product synthesis
The mild conditions engendered by the application of diamine ligands in Cu-catalyzed coupling reactions has increasingly encouraged the practitioners of total synthesis to employ these reactions. It is noticeable that these cross-coupling reactions are often performed at the later stages of a synthesis on labile intermediates. The synthesis of macrocyclic enamides by intramolecular Cu-catalyzed vinylation of amides is an especially valuable tool in this context.14 Cu-mediated methods provide access to simple enamides from amides and aryl halides in the presence of a weak base,203 facilitating the use of enamides as protecting groups for ketones. It should be noted that other catalyst systems have also proven effective in complex molecule synthesis;13–15 notable syntheses include ziziphine (Ma),204 yatakemycin (Fukuyama)205 and vancomycin (Nicolaou).206Nishikawa and Isobe took advantage of a Cu/diamine-mediated vinylation of a β-lactam in the synthesis of model compounds for chartellines (Scheme 57).207 Surprisingly, a mixture of products resulting from either reaction with the vinyl iodide or vinyl chloride of the electrophile was obtained, however, under the optimized conditions the desired terminal product dominated.
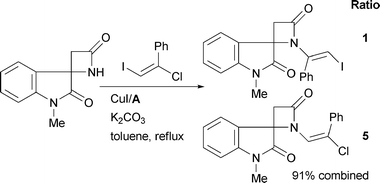 | ||
| Scheme 57 Nishikawa and Isobe's synthesis of chartelline alkaloids. | ||
Evano investigated a Cu-catalyzed coupling in the synthesis of the sedative cyclopeptide alkaloid paliurine F (Scheme 58).208
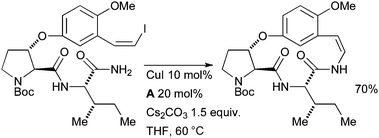 | ||
| Scheme 58 Evano's synthesis of paliurine F. | ||
These authors examined a range of reaction conditions, however, the combination of CuI/A was the most efficacious; the only other system to provide a reasonable quantity of product was CuTC/NMP. The same author has used very similar procedures in the syntheses of abyssenine209 and mucronine E.210
Huang at Schering-Plough applied a Cu-mediated copper coupling of an amide and a vinyl iodide in the synthesis of the potent anticancer natural product psymberin (Scheme 59).211 The same method was later exemplified in the synthesis of a series of analogues.212 This example is noteworthy because of the complexity of the amide coupling partner. A similarly complex nucleophile was utilized by Maier in the synthesis of cruentaren A analogues in order to perform cytotoxicity studies (Scheme 59).213 This represents an especially challenging substrate as a result of the risk of the amide undergoing an undesired E1cb reaction under the basic reaction conditions. Nicolaou and Chen capitalized on the high functional group tolerance of these methods in the amidation of another sensitive vinyl iodide in the synthetic campaign which led to the structural revision of the cytotoxic marine natural product palmerolide A (Scheme 59).214 This approach was later deployed extensively in the synthesis of analogues.215
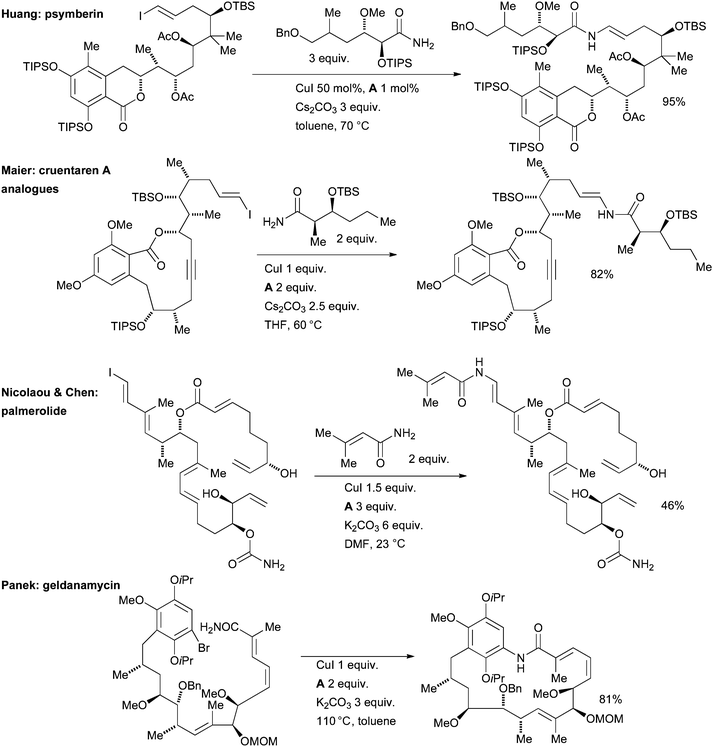 | ||
| Scheme 59 Applications of Cu/diamine-catalyzed cross-coupling in natural product synthesis. | ||
A copper-mediated coupling of a vinyl bromide and an oxazolidinone was part of Movassaghi's approach to (−)-galbulimima alkaloid 13 (Scheme 60).216 The same author used an analogous strategy in the synthesis of the related alkaloid himandrine.217 In both cases the enamide was carried forward as a masked ketone that was revealed later in the synthesis by treatment with acid.
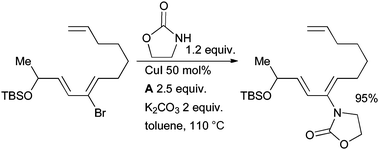 | ||
| Scheme 60 Movassaghi's synthesis of galbulimima alkaloid 13. | ||
Charette explored a Cu-catalyzed coupling between tert-butyl carbamate and a vinyl iodide in the synthesis of barrenazine alkaloids (Scheme 61).218 In this case the resulting enamide was subsequently engaged in a diastereoselective Cu-catalyzed reduction to generate the desired 2-amino ketone.
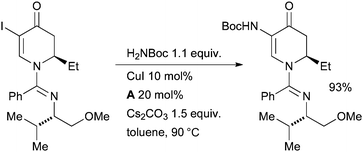 | ||
| Scheme 61 Charette's synthesis of barrenazine alkaloids. | ||
A Cu-catalyzed coupling of a vinyl iodide with a protected prolinamide was employed by Stanovnik in the synthesis of analogues of the prenylated alkaloid tryprostatin B, a natural product which has been shown to exhibit cell cycle control (Scheme 62).219
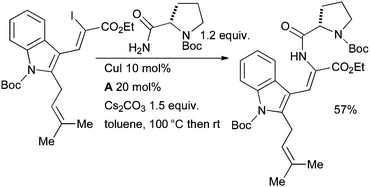 | ||
| Scheme 62 Stanovnik's synthesis of tryprostatin B. | ||
Andersen used an intramolecular Cu-mediated amidation to form a 7-membered ring as a key step in the synthesis of microtubule-stabilizing ceratamine alkaloids (Scheme 63).220
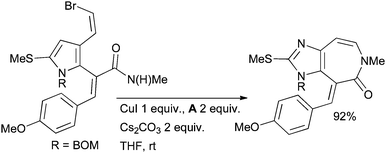 | ||
| Scheme 63 Andersen's synthesis of ceratamine alkaloids. | ||
Cu/diamine catalyst systems have also been exploited for aryl amidation in natural product syntheses. Panek successfully tested an intramolecular amidation of an aryl bromide as a key step in the synthesis of geldanamycin, a natural product that shows activity against Hsp90 and is under development for the treatment of cancer (Scheme 59).221 Kobayashi engaged an intermolecular Cu-catalyzed arylation of an amide in the synthesis of hasubanan alkaloids, a group closely related to the morphine alkaloids (Scheme 64).222
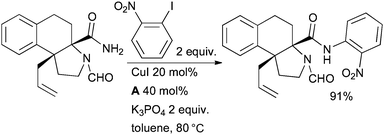 | ||
| Scheme 64 Kobayashi's synthesis of hasubanan alkaloids. | ||
Evano also applied this method in an intramolecular fashion in the synthesis of the modified tripeptide natural product chaetominine, which shows promising cytotoxicity against a variety of human cancer cell lines (Scheme 65).223 The authors note that the formation of this 6-membered ring by amidation proved much more difficult than the 5-membered ring in an analogous system136 and that the exact combination of ligand, base and solvent was crucial in ensuring the desired outcome. Attempts to affect the cyclization with catalysts based on other metals (Pd and Fe) were also unsuccessful.
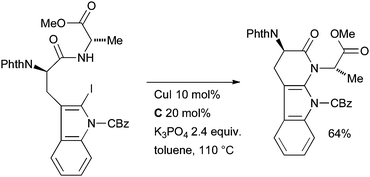 | ||
| Scheme 65 Evano's synthesis of chaetominine. | ||
An intramolecular amidation of an aryl chloride was an important synthetic step in Nakada's formal synthesis of physostigmine, an alkaloid of potential medical importance (Scheme 66).224 This example illustrates the greater facility of performing intramolecular reactions, compared to intermolecular reactions, of aryl chlorides.
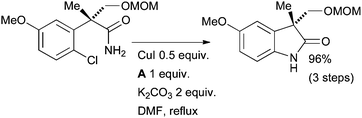 | ||
| Scheme 66 Nakada's formal synthesis of physostigmine. | ||
Bergman exploited a Cu-catalyzed amidation of an aryl chloride en route to the alkaloid vasicoline (Scheme 67).225 Trifluoroacetamide was used as a masked ammonia equivalent. All attempts at a Pd-catalyzed cross-coupling of this substrate in conjunction with a range of different ammonia equivalents were fruitless. Note that an intermolecular Cu-catalyzed coupling of an aryl chloride is made feasible here because of the presence of a heteroatom-directing group.
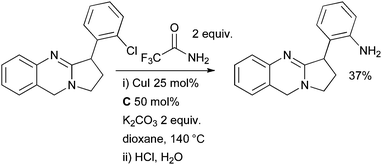 | ||
| Scheme 67 Bergman's synthesis of vasicoline. | ||
Conclusions
There is continuing interest in Cu-catalyzed cross-coupling reactions. Recent advances have included the demonstration of these reactions at low catalyst loading,226 improved methods for performing intermolecular reactions at room temperature20,96 and promising studies of enantioselective reactions.227 Future challenges concern both increasing the scope of existing reactions and the discovery of catalytic conditions for new coupling reactions. Another important goal in enabling more efficient syntheses of complex molecular architectures will be in improving the selectivity of these reactions228,229 in order to reduce the dependence on protecting groups. The application and development of new diamine ligands is sure to be important in these advances.Acknowledgements
We thank the National Institutes of Health (NIH) for support of work in this area (Grant GM-58160).Notes and references
- J. Lindley, Tetrahedron, 1984, 40, 1433–1456 CrossRef CAS.
- H. Weingarten, J. Org. Chem., 1964, 29, 3624–3626 CAS.
- H. B. Goodbrand and N. X. Hu, J. Org. Chem., 1999, 64, 670–674 CrossRef CAS.
- D. W. Ma, Y. D. Zhang, J. C. Yao, S. H. Wu and F. G. Tao, J. Am. Chem. Soc., 1998, 120, 12459–12467 CrossRef CAS.
- R. Oi, C. Shimakawa and S. Takenaka, Chem. Lett., 1988, 899–900 CrossRef CAS.
- P. J. Fagan, E. Hauptman, R. Shapiro and A. Casalnuovo, J. Am. Chem. Soc., 2000, 122, 5043–5051 CrossRef CAS.
- A. Klapars, J. C. Antilla, X. H. Huang and S. L. Buchwald, J. Am. Chem. Soc., 2001, 123, 7727–7729 CrossRef CAS.
- I. P. Beletskaya and A. V. Cheprakov, Coord. Chem. Rev., 2004, 248, 2337–2364 CrossRef CAS.
- S. V. Ley and A. W. Thomas, Angew. Chem., Int. Ed., 2003, 42, 5400–5449 CrossRef CAS.
- K. Kunz, U. Scholz and D. Ganzer, Synlett, 2003, 2428–2439 CrossRef CAS.
- F. Monnier and M. Taillefer, Angew. Chem., Int. Ed., 2009, 48, 6954–6971 CrossRef CAS.
- D. W. Ma and Q. A. Cai, Acc. Chem. Res., 2008, 41, 1450–1460 CrossRef CAS.
- G. Evano, N. Blanchard and M. Toumi, Chem. Rev., 2008, 108, 3054–3131 CrossRef CAS.
- G. Evano, M. Toumi and A. Coste, Chem. Commun., 2009, 4166–4175 RSC.
- Q. Cai, H. Zhang, B. L. Zou, X. Xie, W. Zhu, G. He, J. Wang, X. H. Pan, Y. Chen, Q. Yuan, F. Liu, B. A. Lu and D. W. Ma, Pure Appl. Chem., 2009, 81, 227–234 CrossRef CAS.
- R. A. Altman, E. D. Koval and S. L. Buchwald, J. Org. Chem., 2007, 72, 6190–6199 CrossRef CAS.
- A. Kiyomori, J. F. Marcoux and S. L. Buchwald, Tetrahedron Lett., 1999, 40, 2657–2660 CrossRef CAS.
- H. Zhang, Q. Cai and D. W. Ma, J. Org. Chem., 2005, 70, 5164–5173 CrossRef CAS.
- W. Deng, Y. F. Wang, W. Zou, L. Liu and Q. X. Guo, Tetrahedron Lett., 2004, 45, 2311–2315 CrossRef CAS.
- A. Shafir and S. L. Buchwald, J. Am. Chem. Soc., 2006, 128, 8742–8743 CrossRef CAS.
- B. de Lange, M. H. Lambers-Verstappen, L. S. van de Vondervoort, N. Sereinig, R. de Rijk, A. H. M. de Vries and J. G. de Vries, Synlett, 2006, 3105–3109 CrossRef CAS.
- H. J. Cristau, P. P. Cellier, J. F. Spindler and M. Taillefer, Chem.–Eur. J., 2004, 10, 5607–5622 CrossRef CAS.
- Y. Wang, Z. Q. Wu, L. X. Wang, Z. K. Li and X. G. Zhou, Chem.–Eur. J., 2009, 15, 8971–8974 CrossRef CAS.
- F. Y. Kwong and S. L. Buchwald, Org. Lett., 2003, 5, 793–796 CrossRef CAS.
- H. H. Rao, Y. Jin, H. Fu, Y. Y. Jiang and Y. F. Zhao, Chem.–Eur. J., 2006, 12, 3636–3646 CrossRef CAS.
- X. Lv and W. L. Bao, J. Org. Chem., 2007, 72, 3863–3867 CrossRef CAS.
- Q. Zhang, D. P. Wang, X. Y. Wang and K. Ding, J. Org. Chem., 2009, 74, 7187–7190 CrossRef CAS.
- H. C. Ma and X. Z. Jiang, J. Org. Chem., 2007, 72, 8943–8946 CrossRef CAS.
- F. Y. Kwong, A. Klapars and S. L. Buchwald, Org. Lett., 2002, 4, 581–584 CrossRef CAS.
- Z. J. Zhang, J. C. Mao, D. Zhu, F. Wu, H. L. Chen and B. S. Wan, Tetrahedron, 2006, 62, 4435–4443 CrossRef CAS.
- D. S. Jiang, H. Fu, Y. Y. Jiang and Y. F. Zhao, J. Org. Chem., 2007, 72, 672–674 CrossRef CAS.
- M. Yang and F. Liu, J. Org. Chem., 2007, 72, 8969–8971 CrossRef CAS.
- Y. Z. Huang, J. Gao, H. Ma, H. Miao and J. Xu, Tetrahedron Lett., 2008, 49, 948–951 CrossRef CAS.
- P. Suresh and K. Pitchumani, J. Org. Chem., 2008, 73, 9121–9124 CrossRef CAS.
- A. S. Gajare, K. Toyota, M. Yoshifuji and F. Ozawa, Chem. Commun., 2004, 1994–1995 RSC.
- L. Xu, D. Zhu, F. Wu, R. L. Wang and B. S. Wan, Tetrahedron, 2005, 61, 6553–6560 CrossRef CAS.
- A. K. Verma, J. Singh and R. C. Larock, Tetrahedron, 2009, 65, 8434–8439 CrossRef CAS.
- L. Liang, Z. K. Li and X. G. Zhou, Org. Lett., 2009, 11, 3294–3297 CrossRef CAS.
- L. B. Zhu, L. Cheng, Y. X. Zhang, R. G. Xie and J. S. You, J. Org. Chem., 2007, 72, 2737–2743 CrossRef CAS.
- B. Sreedhar, R. Arundhathi, P. L. Reddy and M. L. Kantam, J. Org. Chem., 2009, 74, 7951–7954 CrossRef CAS.
- T. Kubo, C. Katoh, K. Yamada, K. Okano, H. Tokuyama and T. Fukuyama, Tetrahedron, 2008, 64, 11230–11236 CrossRef CAS.
- H. H. Xu and C. Wolf, Chem. Commun., 2009, 1715–1717 RSC.
- E. Sperotto, J. G. de Vries, G. P. M. van Klink and G. van Koten, Tetrahedron Lett., 2007, 48, 7366–7370 CrossRef CAS.
- L. B. Zhu, P. Guo, G. C. Li, J. B. Lan, R. G. Xie and J. S. You, J. Org. Chem., 2007, 72, 8535–8538 CrossRef CAS.
- L. B. Zhu, G. C. Li, L. Luo, P. Guo, J. B. Lan and J. S. You, J. Org. Chem., 2009, 74, 2200–2202 CrossRef CAS.
- A. B. Naidu, O. R. Raghunath, D. J. C. Prasad and G. Sekar, Tetrahedron Lett., 2008, 49, 1057–1061 CrossRef CAS.
- A. B. Naidu and G. Sekar, Tetrahedron Lett., 2008, 49, 3147–3151 CrossRef CAS.
- D. J. C. Prasad, A. B. Naidu and G. Sekar, Tetrahedron Lett., 2009, 50, 1411–1415 CrossRef CAS.
- E. R. Strieter, D. G. Blackmond and S. L. Buchwald, J. Am. Chem. Soc., 2005, 127, 4120–4121 CrossRef CAS.
- L. M. Huffman and S. S. Stahl, J. Am. Chem. Soc., 2008, 130, 9196–9197 CrossRef CAS.
- J. W. Tye, Z. Weng, A. M. Johns, C. D. Incarvito and J. F. Hartwig, J. Am. Chem. Soc., 2008, 130, 9971–9983 CrossRef CAS.
- E. R. Strieter, B. Bhayana and S. L. Buchwald, J. Am. Chem. Soc., 2009, 131, 78–88 CrossRef CAS.
- A. Ouali, M. Taillefer, J. F. Spindler and A. Jutand, Organometallics, 2007, 26, 65–74 CrossRef CAS.
- A. Ouali, J. F. Spindler, A. Jutand and M. Taillefer, Adv. Synth. Catal., 2007, 349, 1906–1916 CrossRef CAS.
- M. Mansour, R. Giacovazzi, A. Ouali, M. Taillefer and A. Jutand, Chem. Commun., 2008, 6051–6053 RSC.
- H. Kaddouri, V. Vicente, A. Ouali, F. Ouazzani and M. Taillefer, Angew. Chem., Int. Ed., 2009, 48, 333–336 CrossRef CAS.
- B. Yao, D. X. Wang, Z. T. Huang and M. X. Wang, Chem. Commun., 2009, 2899–2901 RSC.
- S. L. Zhang, L. Liu, Y. Fu and Q. X. Guo, Organometallics, 2007, 26, 4546–4554 CrossRef CAS.
- D. M. T. Chan, K. L. Monaco, R. P. Wang and M. P. Winters, Tetrahedron Lett., 1998, 39, 2933–2936 CrossRef CAS.
- P. Y. S. Lam, C. G. Clark, S. Saubern, J. Adams, M. P. Winters, D. M. T. Chan and A. Combs, Tetrahedron Lett., 1998, 39, 2941–2944 CrossRef CAS.
- D. A. Evans, J. L. Katz and T. R. West, Tetrahedron Lett., 1998, 39, 2937–2940 CrossRef CAS.
- D. M. T. Chan and P. Y. S. Lam, in Boronic Acids, ed. D. G. Hall, Wiley-VCH, Weinheim, 2005, pp. 205–240 Search PubMed.
- J. P. Collman, M. Zhong, L. Zeng and S. Costanzo, J. Org. Chem., 2001, 66, 1528–1531 CrossRef CAS.
- D. A. Evans, J. L. Katz, G. S. Peterson and T. Hintermann, J. Am. Chem. Soc., 2001, 123, 12411–12413 CrossRef CAS.
- C. P. Decicco, Y. Song and D. A. Evans, Org. Lett., 2001, 3, 1029–1032 CrossRef CAS.
- O. Skaff, K. A. Jolliffe and C. A. Hutton, J. Org. Chem., 2005, 70, 7353–7363 CrossRef CAS.
- P. Y. S. Lam, G. Vincent, D. Bonne and C. G. Clark, Tetrahedron Lett., 2002, 43, 3091–3094 CrossRef CAS.
- D. M. T. Chan, Tetrahedron Lett., 1996, 37, 9013–9016 CrossRef CAS.
- P. Y. S. Lam, S. Deudon, K. M. Averill, R. H. Li, M. Y. He, P. DeShong and C. G. Clark, J. Am. Chem. Soc., 2000, 122, 7600–7601 CrossRef CAS.
- S.-K. Kang, S.-H. Lee and D. Lee, Synlett, 2000, 1022–1024 CAS.
- A. Klapars, X. H. Huang and S. L. Buchwald, J. Am. Chem. Soc., 2002, 124, 7421–7428 CrossRef CAS.
- K. R. Crawford and A. Padwa, Tetrahedron Lett., 2002, 43, 7365–7368 CrossRef CAS.
- S. K. Kang, D. W. Kim and J. N. Park, Synlett, 2002, 427–430 CrossRef CAS.
- D. P. Phillips, A. R. Hudson, B. Nguyen, T. L. Lau, M. H. McNeill, J. E. Dalgard, J. H. Chen, R. J. Penuliar, T. A. Miller and L. Zhi, Tetrahedron Lett., 2006, 47, 7137–7138 CrossRef CAS.
- D. P. Phillips, X. F. Zhu, T. L. Lau, X. H. He, K. Y. Yang and H. Liu, Tetrahedron Lett., 2009, 50, 7293–7296 CrossRef CAS.
- Y. J. Chen and H. H. Chen, Org. Lett., 2006, 8, 5609–5612 CrossRef CAS.
- J. C. Antilla, A. Klapars and S. L. Buchwald, J. Am. Chem. Soc., 2002, 124, 11684–11688 CrossRef CAS.
- C. Z. Tao, J. Li, Y. Fu, L. Liu and Q. X. Guo, Tetrahedron Lett., 2008, 49, 70–75 CrossRef CAS.
- L. Jiang, G. E. Job, A. Klapars and S. L. Buchwald, Org. Lett., 2003, 5, 3667–3669 CrossRef CAS.
- Q. Liao, Y. X. Wang, L. Y. Zhang and C. J. Xi, J. Org. Chem., 2009, 74, 6371–6373 CrossRef CAS.
- R. C. Shen and J. A. Porco, Org. Lett., 2000, 2, 1333–1336 CrossRef CAS.
- X. Wang and J. A. Porco, J. Am. Chem. Soc., 2003, 125, 6040–6041 CrossRef CAS.
- X. H. Pan, Q. Cai and D. W. Ma, Org. Lett., 2004, 6, 1809–1812 CrossRef CAS.
- M. O. Frederick, J. A. Mulder, M. R. Tracey, R. P. Hsung, J. Huang, K. C. M. Kurtz, L. C. Shen and C. J. Douglas, J. Am. Chem. Soc., 2003, 125, 2368–2369 CrossRef CAS.
- C. A. Zificsak, J. A. Mulder, R. P. Hsung, C. Rameshkumar and L. L. Wei, Tetrahedron, 2001, 57, 7575–7606 CrossRef CAS.
- S. Hirano, R. Tanaka, H. Urabe and F. Sato, Org. Lett., 2004, 6, 727–729 CrossRef CAS.
- J. R. Dunetz and R. L. Danheiser, Org. Lett., 2003, 5, 4011–4014 CrossRef CAS.
- Y. S. Zhang, R. P. Hsung, M. R. Tracey, K. C. M. Kurtz and E. L. Vera, Org. Lett., 2004, 6, 1151–1154 CrossRef CAS.
- X. J. Zhang, Y. S. Zhang, J. Huang, R. P. Hsung, K. C. M. Kurtz, J. Oppenheimer, M. E. Petersen, I. K. Sagamanova, L. C. Shen and M. R. Tracey, J. Org. Chem., 2006, 71, 4170–4177 CrossRef CAS.
- A. Coste, G. Karthikeyan, F. Couty and G. Evano, Angew. Chem., Int. Ed., 2009, 48, 4381–4385 CrossRef CAS.
- A. Coste, F. Couty and G. Evano, Org. Lett., 2009, 11, 4454–4457 CrossRef CAS.
- B. M. Trost and D. T. Stiles, Org. Lett., 2005, 7, 2117–2120 CrossRef CAS.
- L. C. Shen, R. P. Hsung, Y. S. Zhang, J. E. Antoline and X. J. Zhang, Org. Lett., 2005, 7, 3081–3084 CrossRef CAS.
- A. K. A. Persson, E. V. Johnston and J. E. Bäckvall, Org. Lett., 2009, 11, 3814–3817 CrossRef CAS.
- H. H. Rao, H. Fu, Y. Y. Jiang and Y. F. Zhao, J. Org. Chem., 2005, 70, 8107–8109 CrossRef CAS.
- C. T. Yang, Y. Fu, Y. B. Huang, J. Yi, Q. X. Guo and L. Liu, Angew. Chem., Int. Ed., 2009, 48, 7398–7401 CrossRef CAS.
- R. A. Altman, K. W. Anderson and S. L. Buchwald, J. Org. Chem., 2008, 73, 5167–5169 CrossRef CAS.
- J. A. Bikker, N. Brooijmans, A. Wissner and T. S. Mansour, J. Med. Chem., 2009, 52, 1493–1509 CrossRef CAS.
- Y. Liu, Y. Bai, J. Zhang, Y. Li, J. Jiao and X. Qi, Eur. J. Org. Chem., 2007, 6084–6088 CrossRef CAS.
- A. Klapars and S. L. Buchwald, J. Am. Chem. Soc., 2002, 124, 14844–14845 CrossRef CAS.
- J. C. Antilla, J. M. Baskin, T. E. Barder and S. L. Buchwald, J. Org. Chem., 2004, 69, 5578–5587 CrossRef CAS.
- H. J. Cristau, P. P. Cellier, J. F. Spindler and M. Taillefer, Eur. J. Org. Chem., 2004, 695–709 CrossRef CAS.
- Z. X. Xi, F. H. Liu, Y. B. Zhou and W. Z. Chen, Tetrahedron, 2008, 64, 4254–4259 CrossRef CAS.
- M. Taillefer, A. Ouali, B. Renard and J. F. Spindler, Chem.–Eur. J., 2006, 12, 5301–5313 CrossRef CAS.
- A. Ouali, R. Laurent, A. M. Caminade, J. P. Majoral and M. Taillefer, J. Am. Chem. Soc., 2006, 128, 15990–15991 CrossRef CAS.
- W. L. Bao, Y. Y. Liu and X. Lv, Synthesis, 2008, 1911–1917 CrossRef CAS.
- Z. M. Wang, W. L. Bao and Y. Jiang, Chem. Commun., 2005, 2849–2851 RSC.
- J. C. Mao, Q. Q. Hua, J. Guo, D. Q. Shi and S. J. Ji, Synlett, 2008, 2011–2016 CAS.
- M. P. Rainka, Y. Aye and S. L. Buchwald, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5821–5823 CrossRef CAS.
- M. Movassaghi and A. E. Ondrus, J. Org. Chem., 2005, 70, 8638–8641 CrossRef CAS.
- S. Saito and Y. Koizumi, Tetrahedron Lett., 2005, 46, 4715–4717 CrossRef CAS.
- B. P. Fors and S. L. Buchwald, J. Am. Chem. Soc., 2009, 131, 12898–12899 CrossRef CAS.
- J. Andersen, U. Madsen, F. Bjorkling and X. F. Liang, Synlett, 2005, 2209–2213 CAS.
- W. Zhu and D. W. Ma, Chem. Commun., 2004, 888–889 RSC.
- Y. A. Cho, D. S. Kim, H. R. Ahn, B. Canturk, G. A. Molander and J. Ham, Org. Lett., 2009, 11, 4330–4333 CrossRef CAS.
- M. Meldal and C. W. Tornoe, Chem. Rev., 2008, 108, 2952–3015 CrossRef CAS.
- J. Barluenga, C. Valdes, G. Beltran, M. Escribano and F. Aznar, Angew. Chem., Int. Ed., 2006, 45, 6893–6896 CrossRef CAS.
- D. Gelman, L. Jiang and S. L. Buchwald, Org. Lett., 2003, 5, 2315–2318 CrossRef CAS.
- C. Huang, X. Tang, H. A. Fu, Y. Y. Jiang and Y. F. Zhao, J. Org. Chem., 2006, 71, 5020–5022 CrossRef CAS.
- Y. W. Fang and C. Z. Li, J. Org. Chem., 2006, 71, 6427–6431 CrossRef CAS.
- J. M. Baskin and Z. Y. Wang, Org. Lett., 2002, 4, 4423–4425 CrossRef CAS.
- H. Suzuki and H. Abe, Tetrahedron Lett., 1995, 36, 6239–6242 CrossRef CAS.
- W. Zhu and D. W. Ma, J. Org. Chem., 2005, 70, 2696–2700 CrossRef CAS.
- G. P. Ellis and T. M. Romneyalexander, Chem. Rev., 1987, 87, 779–794 CrossRef CAS.
- J. Zanon, A. Klapars and S. L. Buchwald, J. Am. Chem. Soc., 2003, 125, 2890–2891 CrossRef CAS.
- H. D. King, Z. X. Meng, D. Denhart, R. Mattson, R. Kimura, D. D. Wu, Q. Gao and J. E. Macor, Org. Lett., 2005, 7, 3437–3440 CrossRef CAS.
- C. Enguehard-Gueiffier, H. Hubner, A. El Hakmaoui, H. Allouchi, P. Gmeiner, A. Argiolas, M. R. Melis and A. Gueiffier, J. Med. Chem., 2006, 49, 3938–3947 CrossRef CAS.
- H. J. Cristau, A. Ouali, J. F. Spindler and M. Taillefer, Chem.–Eur. J., 2005, 11, 2483–2492 CrossRef CAS.
- T. Schareina, A. Zapf, W. Magerlein, N. Muller and M. Beller, Chem.–Eur. J., 2007, 13, 6249–6254 CrossRef CAS.
- D. Wang, L. Kuang, Z. Li and K. Ding, Synlett, 2008, 69–72.
- Y. L. Ren, W. Wang, S. Zhao, X. Z. Tian, J. J. Wang, W. P. Yin and L. Cheng, Tetrahedron Lett., 2009, 50, 4595–4597 CrossRef CAS.
- I. Nakamura and Y. Yamamoto, Chem. Rev., 2004, 104, 2127–2198 CrossRef CAS.
- N. T. Patil and Y. Yamamoto, Chem. Rev., 2008, 108, 3395–3442 CrossRef CAS.
- J. P. Wolfe, R. A. Rennels and S. L. Buchwald, Tetrahedron, 1996, 52, 7525–7546 CrossRef CAS.
- K. Yamada, T. Kubo, H. Tokuyama and T. Fukuyama, Synlett, 2002, 231–234 CrossRef CAS.
- A. Coste, M. Toumi, K. Wright, V. Razafimahaleo, F. Couty, J. Marrot and G. Evano, Org. Lett., 2008, 10, 3841–3844 CrossRef CAS.
- H. J. Lu and C. Z. Li, Org. Lett., 2006, 8, 5365–5367 CrossRef CAS.
- H. J. Lu, X. T. Yuan, S. Zhu, C. H. Sun and C. Z. Li, J. Org. Chem., 2008, 73, 8665–8668 CrossRef CAS.
- A. Klapars, S. Parris, K. W. Anderson and S. L. Buchwald, J. Am. Chem. Soc., 2004, 126, 3529–3533 CrossRef CAS.
- C. Pabba, H. J. Wang, S. R. Mulligan, Z. J. Chen, T. M. Stark and B. T. Gregg, Tetrahedron Lett., 2005, 46, 7553–7557 CrossRef CAS.
- A. Minatti and S. L. Buchwald, Org. Lett., 2008, 10, 2721–2724 CrossRef CAS.
- C. P. Jones, K. W. Anderson and S. L. Buchwald, J. Org. Chem., 2007, 72, 7968–7973 CrossRef CAS.
- X. Y. Yuan, X. B. Xu, X. B. Zhou, J. W. Yuan, L. G. Mai and Y. Z. Li, J. Org. Chem., 2007, 72, 1510–1513 CrossRef CAS.
- E. D. Li, X. B. Xu, H. F. Li, H. M. Zhang, X. L. Xu, X. Y. Yuan and Y. Z. Li, Tetrahedron, 2009, 65, 8961–8968 CrossRef CAS.
- R. Martin, C. H. Larsen, A. Cuenca and S. L. Buchwald, Org. Lett., 2007, 9, 3379–3382 CrossRef CAS.
- S. L. Buchwald and R. B. Nielsen, J. Am. Chem. Soc., 1989, 111, 2870–2874 CrossRef CAS.
- T. Hamada, D. Suzuki, H. Urabe and F. Sato, J. Am. Chem. Soc., 1999, 121, 7342–7344 CrossRef CAS.
- R. C. Hodgkinson, J. Schulz and M. C. Willis, Org. Biomol. Chem., 2009, 7, 432–434 RSC.
- M. C. Willis, G. N. Brace and I. P. Holmes, Angew. Chem., Int. Ed., 2005, 44, 403–406 CrossRef CAS.
- A. J. Fletcher, M. N. Bax and M. C. Willis, Chem. Commun., 2007, 4764–4766 RSC.
- R. Martin, A. Cuenca and S. L. Buchwald, Org. Lett., 2007, 9, 5521–5524 CrossRef CAS.
- G. Altenhoff and F. Glorius, Adv. Synth. Catal., 2004, 346, 1661–1664 CrossRef CAS.
- X. H. Deng, H. McAllister and N. S. Mani, J. Org. Chem., 2009, 74, 5742–5745 CrossRef CAS.
- R. Martin, M. R. Rivero and S. L. Buchwald, Angew. Chem., Int. Ed., 2006, 45, 7079–7082 CrossRef CAS.
- L. Ackermann, S. Barfusser and H. K. Potukuchi, Adv. Synth. Catal., 2009, 351, 1064–1072 CrossRef CAS.
- J. Yuen, Y. Q. Fang and M. Lautens, Org. Lett., 2006, 8, 653–656 CrossRef CAS.
- N. Zheng and S. L. Buchwald, Org. Lett., 2007, 9, 4749–4751 CrossRef CAS.
- B. L. Zou, Q. L. Yuan and D. W. Ma, Angew. Chem., Int. Ed., 2007, 46, 2598–2601 CrossRef CAS.
- N. Zheng, K. W. Anderson, X. H. Huang, H. N. Nguyen and S. L. Buchwald, Angew. Chem., Int. Ed., 2007, 46, 7509–7512 CrossRef CAS.
- H. Suzuki, A. Kondo and T. Ogawa, Chem. Lett., 1985, 411–412 CrossRef CAS.
- H. Suzuki, A. Kondo, M. Inouye and T. Ogawa, Synthesis, 1986, 121–122 CrossRef CAS.
- T. D. Sheppard, Org. Biomol. Chem., 2009, 7, 1043–1052 RSC.
- K. Takagi, N. Hayama and S. Inokawa, Bull. Chem. Soc. Jpn., 1980, 53, 3691–3695 CAS.
- N. Masuda, S. Tanba, A. Sugie, D. Monguchi, N. Koumura, K. Hara and A. Mori, Org. Lett., 2009, 11, 2297–2300 CrossRef CAS.
- J. Vicente, J. A. Abad and R. M. Lopez-Nicolas, Tetrahedron, 2008, 64, 6281–6288 CrossRef CAS.
- A. Carella, J. P. Launay, R. Poteau and G. Rapenne, Chem.–Eur. J., 2008, 14, 8147–8156 CrossRef CAS.
- T. Itoh, M. Matsuno, E. Kamiya, K. Hirai and H. Tomioka, J. Am. Chem. Soc., 2005, 127, 7078–7093 CrossRef CAS.
- J. L. Humphreys, D. J. Lowes, K. A. Wesson and R. C. Whitehead, Tetrahedron, 2006, 62, 5099–5108 CrossRef CAS.
- Y. H. Gan and T. A. Spencer, J. Org. Chem., 2006, 71, 5870–5875 CrossRef CAS.
- N. K. Jobson, R. Spike, A. R. Crawford, D. Dewar, S. L. Pimlott and A. Sutherland, Org. Biomol. Chem., 2008, 6, 2369–2376 RSC.
- L. Stevenson, S. L. Pimlott and A. Sutherland, Tetrahedron Lett., 2007, 48, 7137–7139 CrossRef CAS.
- M. Vogler, U. Koert, K. Harms, D. Dorsch, J. Gleitz and P. Raddatz, Synthesis, 2004, 1211–1228 CAS.
- C. D. Beadle, J. Boot, N. P. Camp, N. Dezutter, J. Findlay, L. Hayhurst, J. J. Masters, R. Penariol and M. W. Walter, Bioorg. Med. Chem. Lett., 2005, 15, 4432–4437 CrossRef CAS.
- Z. H. Pei, X. F. Li, T. W. von Geldern, K. Longenecker, D. Pireh, K. D. Stewart, B. J. Backes, C. Q. Lai, T. H. Lubben, S. J. Ballaron, D. W. A. Beno, A. J. Kempf-Grote, H. L. Sham and J. M. Trevillyan, J. Med. Chem., 2007, 50, 1983–1987 CrossRef CAS.
- T. Beghyn, C. Hounsou and B. P. Deprez, Bioorg. Med. Chem. Lett., 2007, 17, 789–792 CrossRef CAS.
- B. G. Wu, K. Kuhen, T. N. Nguyen, D. Ellis, B. Anaclerio, X. H. He, K. Y. Yang, D. Karanewsky, H. Yin, K. Wolff, K. Bieza, J. Caldwell and Y. He, Bioorg. Med. Chem. Lett., 2006, 16, 3430–3433 CrossRef CAS.
- P. A. Procopiou, R. A. Ancliff, M. J. Bamford, C. Browning, H. Connor, S. Davies, Y. C. Fogden, S. T. Hodgson, D. S. Holmes, B. E. Looker, K. M. L. Morriss, C. A. Parr, E. A. Pickup, S. S. Sehmi, G. V. White, C. J. Watts, D. M. Wilson and M. D. Woodrow, J. Med. Chem., 2007, 50, 6706–6717 CrossRef CAS.
- R. R. Cesati, G. Dwyer, R. C. Jones, M. P. Hayes, P. Yalamanchili and D. S. Casebier, Org. Lett., 2007, 9, 5617–5620 CrossRef CAS.
- A. Ghosh, J. E. Sieser, S. Caron, M. Couturier, K. Dupont-Gaudet and M. Girardin, J. Org. Chem., 2006, 71, 1258–1261 CrossRef CAS.
- B. M. Andresen, S. Caron, M. Couturier, K. M. DeVries, N. M. Do, K. Dupont-Gaudet, A. Ghosh, M. Girardin, J. M. Hawkins, T. M. Makowski, M. Riou, J. E. Sieser, J. L. Tucker, B. C. Vanderplas and T. J. N. Watson, Chimia, 2006, 60, 554–560 CrossRef CAS.
- D. J. Madar, H. Kopecka, D. Pireh, J. Pease, M. Pliushchev, R. J. Sciotti, P. E. Wiedeman and S. W. Djuric, Tetrahedron Lett., 2001, 42, 3681–3684 CrossRef.
- S. Cacchi, G. Fabrizi, A. Goggiamani and G. Zappia, Org. Lett., 2001, 3, 2539–2541 CrossRef CAS.
- B. Mallesham, B. M. Rajesh, P. R. Reddy, D. Srinivas and S. Trehan, Org. Lett., 2003, 5, 963–965 CrossRef CAS.
- A. Ghosh, J. E. Sieser, M. Riou, W. L. Cai and L. Rivera-Ruiz, Org. Lett., 2003, 5, 2207–2210 CrossRef CAS.
- F. Lovering, S. Kirincich, W. H. Wang, K. Combs, L. Resnick, J. E. Sabalski, J. Butera, J. L. Liu, K. Parris and J. B. Telliez, Bioorg. Med. Chem., 2009, 17, 3342–3351 CrossRef CAS.
- J. W. Corbett, M. R. Rauckhorst, F. Qian, R. L. Hoffman, C. S. Knauer and L. W. Fitzgerald, Bioorg. Med. Chem. Lett., 2007, 17, 6250–6256 CrossRef CAS.
- C. S. Li and D. D. Dixon, Tetrahedron Lett., 2004, 45, 4257–4260 CrossRef.
- E. Hu, A. Tasker, R. D. White, R. K. Kunz, J. Human, N. Chen, R. Buerli, R. Hungate, P. Novak, A. Itano, X. X. Zhang, V. Yu, Y. Nguyen, Y. Tudor, M. Plant, S. Flynn, Y. Xu, K. L. Meagher, D. A. Whittington and G. Y. Ng, J. Med. Chem., 2008, 51, 3065–3068 CrossRef CAS.
- A. Schweinitz, D. Donnecke, A. Ludwig, P. Steinmetzer, A. Schulze, J. Kotthaus, S. Wein, B. Clement and T. Steinmetzer, Bioorg. Med. Chem. Lett., 2009, 19, 1960–1965 CrossRef CAS.
- B. Dyck, S. Markison, L. R. Zhao, J. Tamiya, J. Grey, M. W. Rowbottom, M. Z. Zhang, T. Vickers, K. Sorensen, C. Norton, J. Wen, C. E. Heise, J. Saunders, P. Conlon, A. Madan, D. Schwarz and V. S. Goodfellow, J. Med. Chem., 2006, 49, 3753–3756 CrossRef CAS.
- D. Steinhuebel, M. Palucki, D. Askin and U. Dolling, Tetrahedron Lett., 2004, 45, 3305–3307 CrossRef CAS.
- H. He and Y. J. Wu, Tetrahedron Lett., 2003, 44, 3385–3386 CrossRef CAS.
- W. Deng, L. Liu, C. Zhang, M. Liu and Q. X. Guo, Tetrahedron Lett., 2005, 46, 7295–7298 CrossRef CAS.
- J. L. Duffy, B. A. Kirk, L. P. Wang, G. J. Eiermann, H. B. He, B. Leiting, K. A. Lyons, R. A. Patel, S. B. Patel, A. Petrov, G. Scapin, J. K. Wu, N. A. Thornberry and A. E. Weber, Bioorg. Med. Chem. Lett., 2007, 17, 2879–2885 CrossRef CAS.
- M. D. McBriar, H. Guzik, S. Shapiro, J. Paruchova, R. Xu, A. Palani, J. W. Clader, K. Cox, W. J. Greenlee, B. E. Hawes, T. J. Kowalski, K. O'Neill, B. D. Spar, B. Weig, D. J. Weston, C. Farley and J. Cook, J. Med. Chem., 2006, 49, 2294–2310 CrossRef CAS.
- M. J. Robarge, D. C. Bom, L. N. Tumey, N. Varga, E. Gleason, D. Silver, J. P. Song, S. M. Murphy, G. Ekema, C. Doucette, D. Hanniford, M. Palmer, G. Pawlowski, J. Danzig, M. Loftus, K. Hunady, B. A. Sherf, R. W. Mays, A. Stricker-Krongrad, K. R. Brunden, J. J. Harrington and Y. L. Bennani, Bioorg. Med. Chem. Lett., 2005, 15, 1749–1753 CrossRef CAS.
- G. Dessole, D. Branca, F. Ferrigno, O. Kinzel, E. Muraglia, M. C. Palumbi, M. Rowley, S. Serafini, C. Steinkuhler and P. Jones, Bioorg. Med. Chem. Lett., 2009, 19, 4191–4195 CrossRef CAS.
- H. C. Shen, F. X. Ding, Q. L. Deng, L. C. Wilsie, M. L. Krsmanovic, A. K. Taggart, E. Carballo-Jane, N. Ren, T. Q. Cai, T. J. Wu, K. K. Wu, K. Cheng, Q. Chen, M. S. Wolff, X. C. Tong, T. G. Holt, M. G. Waters, M. L. Hammond, J. R. Tata and S. L. Colletti, J. Med. Chem., 2009, 52, 2587–2602 CrossRef CAS.
- W. L. Jia, R. Y. Wang, D. T. Song, S. J. Ball, A. B. McLean and S. N. Wang, Chem.–Eur. J., 2005, 11, 832–842 CrossRef.
- K. Albrecht and K. Yamamoto, J. Am. Chem. Soc., 2009, 131, 2244–2251 CrossRef CAS.
- A. Hameurlaine and W. Dehaen, Tetrahedron Lett., 2003, 44, 957–959 CrossRef CAS.
- G. S. Jiao, A. Loudet, H. B. Lee, S. Kalinin, L. B. A. Johansson and K. Burgess, Tetrahedron, 2003, 59, 3109–3116 CrossRef CAS.
- J. R. Dehli, J. Legros and C. Bolm, Chem. Commun., 2005, 973–986 RSC.
- G. He, J. Wang and D. W. Ma, Org. Lett., 2007, 9, 1367–1369 CrossRef CAS.
- K. Okano, H. Tokuyama and T. Fukuyama, J. Am. Chem. Soc., 2006, 128, 7136–7137 CrossRef CAS.
- K. C. Nicolaou, A. E. Koumbis, M. Takayanagi, S. Natarajan, N. F. Jain, T. Bando, H. Li and R. Hughes, Chem.–Eur. J., 1999, 5, 2622–2647 CrossRef CAS.
- T. Nishikawa, S. Kajii and M. Isobe, Chem. Lett., 2004, 33, 440–441 CrossRef CAS.
- M. Toumi, F. Couty and G. Evano, Angew. Chem., Int. Ed., 2007, 46, 572–575 CrossRef CAS.
- M. Toumi, F. Couty and G. Evano, J. Org. Chem., 2007, 72, 9003–9009 CrossRef CAS.
- M. Toumi, F. Couty and G. Evano, Synlett, 2008, 29–32 CAS.
- X. H. Huang, N. Shao, A. Palani, R. Aslanian and A. Buevich, Org. Lett., 2007, 9, 2597–2600 CrossRef CAS.
- X. H. Huang, N. Shao, R. Huryk, A. Palani, R. Aslanian and C. Seidel-Dugan, Org. Lett., 2009, 11, 867–870 CrossRef CAS.
- V. V. Vintonyak, M. Cala, F. Lay, B. Kunze, F. Sasse and M. E. Maier, Chem.–Eur. J., 2008, 14, 3709–3720 CrossRef CAS.
- K. C. Nicolaou, Y. P. Sun, R. Guduru, B. Banerji and D. Y. K. Chen, J. Am. Chem. Soc., 2008, 130, 3633–3644 CrossRef CAS.
- K. C. Nicolaou, G. Y. C. Leung, D. H. Dethe, R. Guduru, Y. P. Sun, C. S. Lim and D. Y. K. Chen, J. Am. Chem. Soc., 2008, 130, 10019–10023 CrossRef CAS.
- M. Movassaghi, D. K. Hunt and M. Tjandra, J. Am. Chem. Soc., 2006, 128, 8126–8127 CrossRef CAS.
- M. Movassaghi, M. Tjandra and J. Qi, J. Am. Chem. Soc., 2009, 131, 9648–9650 CrossRef CAS.
- T. Focken and A. B. Charette, Org. Lett., 2006, 8, 2985–2988 CrossRef CAS.
- J. Wagger, J. Svete and B. Stanovnik, Synthesis, 2008, 1436–1442 CAS.
- M. Nodwell, A. Pereira, J. L. Riffell, C. Zimmerman, B. O. Patrick, M. Roberge and R. J. Andersen, J. Org. Chem., 2009, 74, 995–1006 CrossRef CAS.
- H.-L. Qin and J. S. Panek, Org. Lett., 2008, 10, 2477–2479 CrossRef CAS.
- T. X. Nguyen and Y. Kobayashi, J. Org. Chem., 2008, 73, 5536–5541 CrossRef CAS.
- M. Toumi, F. Couty, J. Marrot and G. Evano, Org. Lett., 2008, 10, 5027–5030 CrossRef CAS.
- K. Asakawa, N. Noguchi, S. Takashima and M. Nakada, Tetrahedron: Asymmetry, 2008, 19, 2304–2309 CrossRef CAS.
- S. H. Wiedemann, J. A. Ellman and R. G. Bergman, J. Org. Chem., 2006, 71, 1969–1976 CrossRef CAS.
- P. F. Larsson, A. Correa, M. Carril, P. O. Norrby and C. Bolm, Angew. Chem., Int. Ed., 2009, 48, 5691–5693 CrossRef CAS.
- X. A. Xie, Y. Chen and D. W. Ma, J. Am. Chem. Soc., 2006, 128, 16050–16051 CrossRef.
- A. Shafir, P. A. Lichtor and S. L. Buchwald, J. Am. Chem. Soc., 2007, 129, 3490–3491 CrossRef CAS.
- D. Maiti and S. L. Buchwald, J. Am. Chem. Soc., 2009, 131, 17423–17429 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |