Clickable initiators, monomers and polymers in controlled radical polymerizations – a prospective combination in polymer science
Ulrich
Mansfeld
abc,
Christian
Pietsch
abc,
Richard
Hoogenboom
b,
C. Remzi
Becer
*ab and
Ulrich S.
Schubert
*abc
aLaboratory of Organic and Macromolecular Chemistry, Friedrich-Schiller-University Jena, Humboldtstr. 10, 07743, Jena, Germany. E-mail: ulrich.schubert@uni-jena.de; c.r.becer@warwick.ac.uk
bLaboratory of Macromolecular Chemistry and Nanoscience, Eindhoven University of Technology, P. O. Box 513, 5600 MB, Eindhoven, The Netherlands
cDutch Polymer Institute (DPI), P. O. Box 902, 5600 AX, Eindhoven, The Netherlands
First published on 20th September 2010
Abstract
Preparation of multifunctional and well-defined macromolecules requires a smart selection of the most suitable controlled polymerization technique in combination with appropriate click reactions. In this review, we provide an overview on the use of various “clickable” initiators and monomers as well as on the postpolymerization modifications that have been widely used to construct clickable macromolecules. As such, this contribution will aid polymer chemists to select a suitable combination of CRP and click methodologies to design the target structures.
 Ulrich Mansfeld | Ulrich Mansfeld studied chemistry at the Friedrich-Schiller-University Jena (Germany; 2003–2008) and accomplished the master thesis at the Eindhoven University of Technology (Netherlands) under the supervision of Prof. Ulrich S. Schubert. In 2009, he began his PhD studies working in the fields of controlled radical polymerizations and supramolecular chemistry. |
 Christian Pietsch | Christian Pietsch studied chemistry at the Friedrich-Schiller-University Jena (Germany). He completed his MSc in 2008 at the University of Technology in Eindhoven (Netherlands), where he worked on the synthesis of stimuli-responsive copolymers under the supervision of Prof. Ulrich S. Schubert. He continued as a PhD student in Jena working on responsive copolymers as well as living radical polymerization and dye-labeled polymers. Recently, he spent two months at CSIRO (Melbourne, Australia), where he carried out research on RAFT polymerizations. |
 Richard Hoogenboom | Richard Hoogenboom was born in 1978 in Rotterdam (Netherlands) and studied chemical engineering at the Eindhoven University of Technology (TU/e; Netherlands). In 2005, he obtained his PhD under the supervision of Ulrich S. Schubert (TU/e) and continued working as project leader for the Dutch Polymer Institute. After postdoctoral training with Martin Möller at the RWTH Aachen (Humboldt fellowship) and Roeland J. M. Nolte at the Radboud University Nijmegen (NWO veni-grant), he was appointed as associate professor at Ghent University from July 2010. His research interests include stimuli-responsive polymers, supramolecular polymers, and poly(2-oxazoline)s. |
 C. Remzi Becer | C. Remzi Becer was born in 1980 in Izmir, Turkey. He received his BSc degree in 2003 at the Chemistry Department of the Istanbul Technical University (ITU). In 2005, he received his MSc degree in Polymer Science and Technology at the ITU. He completed his PhD study titled as “Controlling Polymer Architectures” in 2009 under the supervision of Ulrich S. Schubert at the Eindhoven University of Technology (Netherlands) and the Friedrich-Schiller-University Jena (Germany). Since late 2009, he has been a Marie Curie Research Fellow in the University of Warwick (United Kingdom). His research interests include controlled living polymerization techniques, click reactions and glycopolymers. |
 Ulrich S. Schubert | Ulrich S. Schubert was born in Tübingen in 1969. He studied chemistry at the Universities of Frankfurt and Bayreuth (both Germany) and the Virginia Commonwealth University, Richmond (USA). His PhD thesis was executed at the University of Bayreuth and the University of South Florida/Tampa. After postdoctoral training with Professor Lehn at the Université Strasbourg (France) he moved to the Technische Universität München (Germany) to obtain his Habilitation in 1999. From 1999 to 2000 he held a temporal position as professor at the Center for NanoScience, Universität München (Germany). From 2000 to 2007 he was Full-Professor at the Eindhoven University of Technology. Currently he holds a chair at the Friedrich-Schiller-University Jena. |
1. Introduction
The synthesis of well-defined polymers has been the ultimate challenge of polymer chemists in the last decades. The development of anionic polymerization by Szwarc et al. opened new avenues and a new field of materials research.1,2 Besides, polymeric materials have improved the quality of our lives in all areas from engineering to electronics and medical applications.3–5 Following the invention of anionic polymerization, other possible types of living and/or controlled polymerizations, cationic and radical, have been widely studied.6–9 The most significant controlled radical polymerization (CRP) techniques to date could be listed as atom transfer radical polymerization (ATRP),6–8 nitroxide-mediated radical polymerization (NMP)9,10 and reversible addition-fragmentation chain transfer polymerization (RAFT).11,12 Each of these techniques requires the use of a dedicated metal/ligand complex, chain transfer agent (CTA) or nitroxide mediator to gain control over the polymerization of various monomeric structures. All these parameters have been extensively investigated in detail and reported by numerous research groups. Consequently, nowadays well-defined polymers can be successfully synthesized and characterized by performing these techniques under specific conditions.After significant advances in the controlled/“living” radical polymerization techniques over the last years, functionalization of the macromolecules has been the following challenge for polymer chemists. Synthesis of end- or side-functional macromolecules has been achieved by using functional initiators, monomers or end capping techniques. However, these specific functional groups might have enormous effects on the polymerization rate, control over the polydispersity index and the composition of the polymers. Fortunately, a decade ago, the click chemistry concept was introduced by Sharpless et al. that enables nowadays the preparation of not only telechelic polymers but also side-group functionalized polymers using clickable initiators, monomers or polymers.13–21 Sharpless and coworkers drew attention to several highly efficient organic reactions, such as the copper-catalyzed [3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) +2] Huisgen cycloaddition reaction (CuAAC), which has developed into the most widely employed click reaction.22,23 This reaction requires a copper salt and a ligand as catalysts but proceeds very rapidly and selectively at room temperature.24 Several other efficient organic reactions have been claimed to be “click” reactions since they fulfilled all or some of the click chemistry criteria, which can be listed as modular and wide in scope, high efficiency and high yields, no or inoffensive byproducts, stereospecific, readily available starting materials and reagents, no solvent or a benign solvent, and simple purification techniques.17
+2] Huisgen cycloaddition reaction (CuAAC), which has developed into the most widely employed click reaction.22,23 This reaction requires a copper salt and a ligand as catalysts but proceeds very rapidly and selectively at room temperature.24 Several other efficient organic reactions have been claimed to be “click” reactions since they fulfilled all or some of the click chemistry criteria, which can be listed as modular and wide in scope, high efficiency and high yields, no or inoffensive byproducts, stereospecific, readily available starting materials and reagents, no solvent or a benign solvent, and simple purification techniques.17
Metal-free click reactions have attracted the greatest attention in recent years since they eliminate the main disadvantage of CuAAC click reactions: the use of a copper catalyst.17,25 This opens new avenues to rapid and efficient reactions that can be employed in, e.g., living organisms.26 Several types of metal-free click-like reactions have been developed and the most prominent ones are thiol-ene,19,27,28 thiol-yne,29,30 thiol-para-fluoro,31,32 nitrile oxide-alkyne cycloaddition,33 pyridyl disulfide exchange,34–36 and Diels–Alder reactions.37–39 These reactions have pros and cons in comparison to each other. Each of them can be used for certain monomers, initiators or polymerization techniques. Therefore, one should carefully design the synthetic route to prepare the desired functional polymer.
The aim of this review is to provide an insight on the selection of the most suitable CRP technique and click reaction for the synthesis of the desired tailor-made macromolecule. The range of functional initiators and monomers are listed in tables for each CRP technique. Hereby, click reactions performed before (“preclick”) and after (“postclick”) the polymerization initiated by a functional initiator or propagated with a functional monomer are discussed. The special focus will be on the combinations of CRP and click chemistry techniques rather than discussing each of them in details.
2. Overview on click reactions used in combination with controlled radical polymerization techniques
In the following section the click reactions that have been used in combination with CRP are briefly summarized while referring to the original literature (Scheme 1).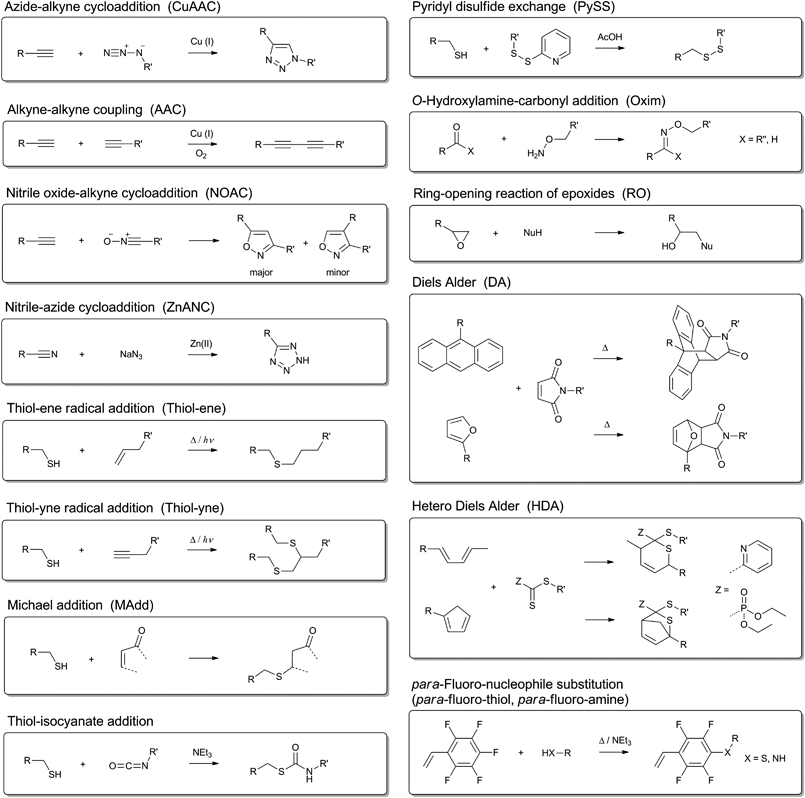 | ||
| Scheme 1 Schematic representation of the click reactions that have been used in combination with controlled radical polymerization techniques. | ||
The most widely applied click reaction has been the copper-catalyzed azide-alkyne 1,3-dipolar cycloaddition (CuAAC) as shown in Scheme 1.40 Sharpless et al. defined this type of cycloaddition as the ideal click reaction and the reaction principle has been employed in various fields of synthetic chemistry, i.e. medicinal chemistry, polymer chemistry, material chemistry, inorganic chemistry and, in particular, organic chemistry.13 This reaction proceeds very rapidly in aqueous medium and even under ambient conditions. The major drawback of this reaction is the necessity of using a copper salt, which requires a purification step following the click reaction.41
Nevertheless, this drawback can be overcome by choosing alkynes with higher reactivity. For instance, cyclooctyne derivatives can undergo strain-promoted azide-alkyne click reactions in biological environments.42 Similarly, electron-deficient alkynes and activated alkynes also provide highly efficient reactions.43 Apparently, alkyne compounds play a crucial role in many different click-like reactions. As an example, Tunca and Hizal et al. demonstrated the synthesis of A2-B2 4-miktoarm star copolymers using the Glaser coupling as an alkyne-alkyne homocoupling reaction (AAC),44 which is a copper-catalyzed reaction that reaches completion at room temperature in three days.45 Recently, Lutz and Heaney et al. reported the cycloaddition of alkynes with nitrile oxides (NOAC)46 in combination with CRP as a potential click reaction.33 This reaction proceeds at room temperature, in polar medium, in the absence of transition metal catalyst, in high yields and is highly regiospecific. Besides alkynes, also nitriles can react with azides in a zinc-catalyzed cycloaddition47 and can be used in combination with CRP.48
Alkynes do not only react with azides49 or its own kind but also react with thiols, known as thiol-yne click reaction.29,30 This represents a very efficient reaction and results in addition of two thiol compounds per alkyne molecule, which can be either added by a base-catalyzed Michael addition or by a photo-initiated Anti-Markovnikov-addition.29,50 Thiol compounds have been in the focus of click reactions in the last couple of years due to their high reactivity towards common functional groups, i.e. alkene, alkyne, isocyanate and pentafluorophenyl.51 The thiol-ene click reaction has been utilized by several groups for the synthesis of dendrimers, the side-chain functionalization of well-defined backbones and for the synthesis of block copolymers.19,28,52 Advantages and disadvantages of this type of click reaction have been discussed in several review articles.10,53 Other thiol-based click reactions can be counted as thiol-maleimide addition (MAdd),28 thiol-isocyanate addition54 and pyridyl disulfide exchange (PySS).36 Thiol-terminated polymers can be easily obtained by RAFT polymerization of a wide range of monomers and a subsequent cleavage of the chain transfer agent. However, hetero Diels–Alder (HDA) reaction using a dedicated chain transfer agent can been employed without a transformation of the RAFT agent to prepare AB block copolymers.55 Commonly, the Diels–Alder reaction (DA) has been employed to construct miktoarm star polymers56 or to engineer self-healing polymers by reversible crosslinking.57
A frequently used reaction for bioconjugation is the oxime formation (Oxim) that has been applied in the conjunction of carbonyl-functionalized proteins and aminoxy-functionalized polymers.58 Furthermore, the ring-opening reaction of epoxides as a well-known spring-loaded reaction13 is used in combination with the CRP of glycidyl methacrylate mostly to introduce other clickable functionalities59 or for attachment on surfaces.60
In practice, some combinations of click reactions and CRP techniques are most commonly applied which will be discussed in detail in the following sections.
3. Strategies towards clicked polymer architectures
There are at least four common ways to combine controlled radical polymerization techniques and click chemistry to construct various clicked polymeric architectures. Thereby, each approach has its inherent drawbacks and amenities. The strategies are summarized in Scheme 2 and will be discussed in a general way in the following.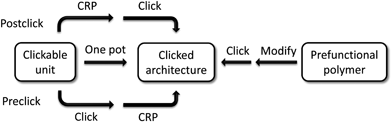 | ||
| Scheme 2 Schematic representation of the strategies towards clicked architectures. | ||
3.1 Postclick strategy
By using functional initiators or monomers with clickable moieties for controlled radical polymerizations, clickable polymers can be achieved to construct various architectures by clicking post to the polymerization.As a limitation, the clickable functionality must not interfere with the polymerization process or has to be protected to gain control over the polymerization and, thus, yielding well-defined polymers with high functional group fidelity up to sufficient monomer conversions. Moreover, most functional monomers or initiators are not commercially available and have to be synthesized prior to the polymerization.
The clickable polymer represents a platform for versatile click functionalization, while retaining the polymer characteristics i.e. chain length, monomer composition and molar mass dispersity. Herein, the approach has a wide scope by means of construction flexibility, in contrast to the preclick approach (see below), since different functional polymers can be prepared from a single batch of a clickable macromolecule. Furthermore, the same molar mass and its distribution allow for a better comparison of changes related to the functionality.
The postclick approach offers high functional group fidelity compared to the postmodification approach. In particular, for the polymerization of clickable monomers each repeating unit of the resultant polymer bears the clickable unit in contrast to a prefunctional homopolymer that have to be modified with the clickable moiety after polymerization. Accordingly, using α-functionalized initiators each chain is monoterminal-functionalized, while end-group functionalization via the postmodification are limited by the yield of the final modification step.
3.2 Preclick strategy
Clicked polymeric architectures can be also obtained by using functional initiators or monomers with clickable moieties that are clicked prior to the polymerization in a so-called “preclick” route. This method is predominantly used if the clickable units interfere with the radical process or the temperature of the polymerization and can not be sufficiently protected.As a limitation, the clicked moiety must be stable under the applied polymerization conditions. It should be mentioned that Diels–Alder adducts tend to undergo retro Diels–Alder reactions at elevated temperatures and, hence, are not in principle amenable for this strategy. However, the stability range of the Diels–Alder adducts is strongly system dependent.61,62 Recently, some literature examples discussed the polymerizability and stability of related clicked initiators and monomers. Maleimide functionalities are often protected by the reversible Diels–Alder reaction with furan to make it more compatible with radical polymerization processes, which was demonstrated by Sanyal and coworkers using a furan-maleimide methacrylate. This monomer was polymerized by a free radical polymerization procedure at 65 °C and deprotected via retro-DA at 125 °C.63 Syrett et al. reported an ATRP reaction with a clicked initiator linked via the maleimide-furan adduct, where the polymerization was successfully performed at 50–60 °C and the retro Diels–Alder reaction occurred at 120 °C.64 By using a clicked initiator with the maleimide-anthracene adduct significantly higher thermal stability was observed upon the retro Diels–Alder reaction. In addition, Barner-Kowollik et al. investigated a hetero Diels–Alder cycloadduct of a dithioester and cyclopentadiene that was rapidly formed at room temperature and cleaved above 80 °C.65 By contrast, the Huisgen 1,3-dipolar cycloaddition of alkynes with azides represents a prominent reaction for this strategy, since the triazole ring is stable under the typically applied polymerization conditions. Furthermore, the bulkiness of the clicked unit should not disturb the polymerization process by causing slow propagation or deactivating of the catalytic system.
In case the clicked monomers can be successfully polymerized via CRP, this procedure provides the highest control over the incorporation of the clicked functionality into polymeric architectures, while having the lowest scope by means of construction flexibility:
On the one hand, the click reaction has been performed with small molecules (monomers or initiators) that can be easily purified and analyzed. Hereon, subsequent polymerization is carried out with less reactive material leading to high functional-group fidelity, whereby no further functionalization is required. However, in order to obtain clicked architectures with different clicked functionalities, the click reaction as well as the polymerization has to be carried out for each clicked architecture. This method has been successfully employed for the preparation of comb-shaped polymers with almost quantitative functionalization of each repeating unit.66,67
3.3 Simultaneous/one pot strategy
In many cases the catalyst used in the ATRP polymerization, i.e. CuBr/PMDETA (N,N,N′,N′′,N′′ pentamethyldiethylene triamine), is the same as for the click reaction that allows a simultaneous/one-pot process of polymerization and click reaction. The combination of the Cu(I)-catalyzed Huisgen cycloaddition and a CRP process allows the one-pot synthesis of a wide range of products, i.e. α-functional- (clickable initiator), grafted- (clickable monomer), star-shaped polymers and polymeric networks, respectively. The simultaneous process means that CRP and click reaction occur at the same time during the polymer synthesis. In contrast to a one-pot process, whereas at first the polymerization and then the click reaction are performed or vice versa (subsequent addition of the second compound). The advantage of this strategy in contrast to the “preclick” or “postclick” way is that for clicked polymeric architectures only one synthesis step and one purification step is required. However, it should be noted that the click reaction generally proceeds much faster than the ATRP and, thus, most click coupling reactions will occur during the initial stages of the polymerization.The first example of this combined route was demonstrated by Haddleton et al. in 2005 using an azide-functionalized ATRP initiator for the polymerization of methyl methacrylate as indicated by a linear relationship of the first order kinetic plot. The efficiency of the click reaction was investigated in the presence of alkyne-functional dyes.68 Another example of simultaneous click and CRP with an unprotected propargyl methacrylate was also reported by Haddleton et al. It was shown that the copper-catalyzed azide-alkyne click reaction proceeds much faster compared to the ATRP.69 The authors could also show that the ratio of rate constants for the polymerization and the click process can be controlled by varying the solvent, the temperature or the concentration of the catalyst. Thus, it was demonstrated that the rate of CuAAC in DMSO is slower than the polymerization, whereas in DMF or toluene the click reaction is faster than the polymerization. The measured PDI values of the synthesized copolymers were found to be below 1.3.69 As a limitation, the clickable functionality must be used without a protection group to allow the Huisgen cycloaddition and, hence, side reactions can occur.
Also the one-pot/tandem process is often used for the preparation of clicked polymeric architectures by using ATRP polymerization and CuAAC. This approach uses the same catalytic system for both the click reaction as well as ATRP, but sequentially. Thereby, both ways are possible: At first the click reaction followed by the polymerization or vice versa (in the manner that the second compound was added later). In contrast, if all components are added at once, the click reaction can be performed at room temperature, while after full conversion, the temperature was raised to initiate the polymerization.70 In all cases well-defined copolymers were obtained. Dubois et al. discussed these different routes and showed that both the preclick as well as the one-pot route give similar results in molar mass and PDI values, whereby the postclick route leads to an increase of the polydispersity index from 1.3 to 1.5.70
3.4 Postmodification strategy
Prefunctional polymers with latent groups prepared by CRP can be modified with clickable moieties to obtain clickable polymers. The modification reaction must be efficient, since it correlates to the fidelity of functional groups in the modified polymer. Mostly, purification steps are necessary, since even efficient modification reactions are not quantitative. In this case, modification as well as clicking are carried out at the polymer, which might complicate in principle the purification and analysis. In particular, incomplete modification is critical for pendant functional polymers, since the unreacted functionalities are attached to the same polymer backbone as the converted ones and, thus, can not be separated as it can be done for endgroup-modified polymers.As an advantage, the starting materials by means of initiators and monomers are in most cases commercially available or can be easily synthesized allowing for large scale experiments. This approach can be used for terminal and pendant functionalized polymers, except for comb-shaped polymers, since electronic and steric effects may hinder full transformation. This method is in particular suitable for clicked architectures where the clickable as well as the clicked moiety interfere with the polymerization process.
It should be noted that in a narrower sense the described reactions are defined as click reactions for the conjunction of small molecules, since they feature among other characteristics a high efficiency and selectivity allowing for the equimolar usage of the click counterparts. However, the conjunction of polymers using these reactions leads not always to full conversion probably due to sterical hindrance or affected diffusion of the polymer chains.53 To drive the reaction to completion, an excess of one clickable polymer can be used. If the click reaction is completed, the typical problems of purification due to the equal solubility behavior of both product and educt polymer can be overcome by adding a click-functionalized resin containing the click counterpart related to the excess one.71,72 The subsequent purification of the desired clicked architecture can be then easily performed by filtration of the clicked resin.
4. Clickable initiators
The use of functional initiators in controlled radical polymerization processes lead to terminal-functionalized polymers in one step. Thereby, two possible routes can be employed using clickable initiators. As illustrated in Scheme 3, click reactions can be performed either after the polymerization (postclick) or prior to the polymerization (preclick). For ATRP initiators only α-clickable polymers are inherently possible in one step. With the ability of the synthesis of initiators/transfer agents for NMP and RAFT with functional groups on both the initiating as well as on the mediating fragment the toolbox expands and ω-clickable polymers as well as α,ω-clickable polymers are accessible in a one step procedure. The advantage of the clickable initiator approach compared to the clickable monomer route is the lower concentration of the clickable unit. Hence, side reactions are reduced and protection is not strictly necessary while leading to a controlled polymerization with a high degree of functionalization of the clickable moiety. In the following, an overview of potential clickable initiators is given and is summarized in Tables 1 to 7. Possible combinations and the restrictions of CRP and click reactions are discussed. Unless otherwise noted, all selected initiators belong to the postclick approach.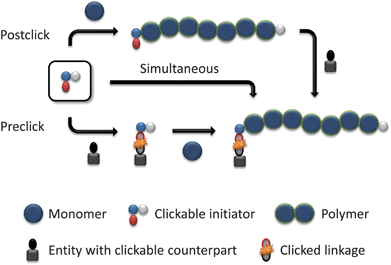 | ||
| Scheme 3 Schematic representation of the strategies using clickable initiators. | ||
| Entry | Structure | Click | CRP | Monomer | Strategy | Ref |
|---|---|---|---|---|---|---|
| (Abbr./Entry) | ||||||
| 1 |
|
CuAAC | ATRP | St, MA, OEGA tBA, NIPAM, tBMA, DEAEMA, HMA | poC | 73,76,78–81,83–89,103,104 |
| AAC | ATRP | St | poC | 44 | ||
| NOAC | ATRP | St | poC | 33 | ||
| 2 |
|
CuAAC | ATRP | NIPAM, EEA | poC | 90,91 |
| 3 |
|
CuAAC | ATRP | NIPAM, 46, 53 | prC | 95 |
| 4 |
|
CuAAC | ATRP | St, tBA, MMA | simult | 96 |
| 5 |
|
CuAAC | ATREP | St, VBA, 50 | simult | 83 |
| 6 |
|
CuAAC | ATRP | St | poC | 97 |
| 7 |
|
CuAAC | ATRP | St | poC | 98 |
| 8 |
|
CuAAC | ATRP | MMA | prC | 99 |
| 9 |
|
CuAAC | ATRP | St, MMA, tBA | poC | 71,94,100 |
| ROP | DMAEMA, NIPAM | |||||
| ATRP | St | prC | 101 | |||
| ROP | ||||||
| 10 |
|
CuAAC | ATRP | tBA | poC | 102 |
| Entry | Structure | Click | CRP | Monomer | Strategy | Ref |
|---|---|---|---|---|---|---|
| 11 |
|
CuAAC | ATRP | DMAEMA, St | poC | 92,106,107 |
| 12 |
|
CuAAC | ATRP | MMA, DEAEMA, HEMA, KSPMA, HMA, NIPAM, DMAEMA, DEAM, St | poC, simult | 68,82,94,109–112 |
| 13 |
|
CuAAC | ATRP | NIPAM | poC | 113 |
| 14 |
|
CuAAC | ATRP | NIPAM | poC | 110 |
| 15 |
|
CuAAC | ATRP | St, nBMA | poC, prC | 114 |
| 16 |
|
CuAAC | ATRP | DEGMA | poC | 104 |
| Entry | Structure | Click | CRP | Monomer (Abbr./Entry) | Strategy | Ref |
|---|---|---|---|---|---|---|
| 17 |
|
DA | ATRP | tBA, MMA | poC | 115,117,120,121,125 |
| prC | 64 | |||||
| MAdd | ATRP | OEGMA, 45, SMA | poC | 66,122,123 | ||
| 18 |
|
MAdd | ATRP | NIPAM | prC | 35,58 |
| 19 |
|
DA | ATRP | St, MMA | poC | 87,89,121,124,125 |
| 20 |
|
DA | ATRP | St | -- | 126 |
| 21 |
|
Thiol-ene | ATRP | St, MMA | poC | 127 |
| 22 |
|
PySS | ATRP | St, tBMA, MMA | poC | 34,128,129 |
| prC | 35,58 | |||||
| 23 |
|
Oxim | ATRP | NIPAM, HEMA, OEGMA | poC | 131,132 |
4.1 Atom transfer radical polymerization (ATRP)
ATRP is the most widely employed CRP technique using the clickable initiator approach because of the easy preparation of functional initiators. Different types of initiators are discussed in the following subsections (Tables 1–3).000 g mol−1, PDI = 1.2–1.3).82 The α-functionalized fluorescent copolymer was clicked onto cotton and both Wang and Merrifield resins using the Huisgen [2+3] cycloaddition. Recently, Tunca and coworkers used the protected initiator (Entry 1) for the preparation of block copolymers of styrene and divinylbenzene to form multiarm star polymers with terminal alkyne groups.87–89 At first, styrene was polymerized to obtain linear alkyne-functional PS (up to Mn = 6000 g mol−1, PDI = 1.1). Subsequently, the prepared PS was used as macroinitiator in the polymerization of divinylbenzene leading to a crosslinked second block that form the core of the multiarm star polymer (up to Mw = 250 000 g mol−1, PDI = 1.2).
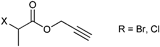
Besides the initiator depicted in Entry 1, its analogue propargyl 2-halopropionate (Entry 2) was also used as clickable initiator for the polymerization of acrylates and NIPAM without any protection.90,91
In most cases of the unprotected initiators, good control over the polymerization was achieved yielding alkyne-functional polymers with low PDI values. In these cases, the undesired chain transfer and termination reactions are suppressed to a negligible amount by decreasing the reaction time84 and keeping the polymerization at low conversions to reduce the termination reactions at the ω-end of the chain. The bromine atom is often substituted in a postmodification reaction with sodium azide to yield heterotelechelic polymers bearing an alkyne and an azide end group, respectively. Furthermore, side reactions involving the alkyne functionality were suppressed by using low alkyne concentrations according to a high monomer-to-initiator ratio92 or by using relatively low temperatures,85,91i.e. 40 °C for the polymerization of tBMA85 or NIPAM.91 In contrast, the polymerization of styrene was conducted for 6 h at 90 °C with the initiator depicted in Entry 1 as nonprotected alkyne initiator revealing significant termination reactions as indicated by SEC measurements.86 It should be noted that direct polymerization throughout the triple bond is hindered, because radical transfer reactions from the styrene or methacrylate radical are suppressed by their low reactivity (Q-values, r-values).
Most of the alkyne-functionalized ATRP initiators are based on the α-halo isobutyrate group. This class of alkyl halides possesses high activation rates due to the suitable radical-stabilization effects of the tertiary carbon and the vicinal ester group resulting in a high initiation efficiency, which is required to control molar masses and molar mass distributions.93 In most of the present cases, this type of initiator is used for ATRP regardless of the polymerized monomer class, e.g. styrenes, acrylates or methacrylates. However, normally the initiator structure is carefully chosen for each given monomer and its reactivity.6,93 Therefore, special focus appear not to be on the optimal initiator structure for control, but on the accessibility of an alkyne-functionalized one. Practically, α-bromoisobutyryl bromide is commercial available and is mostly functionalized via esterificiation reaction with propargyl alcohol.
The most widely employed catalytic system is CuBr and PMDETA again regardless of the initiator or monomer type. Besides, other catalytic systems, e.g. CuCl/tris(2-(dimethylamino)-ethyl)amine (Me6TREN) for the polymerization of NIPAM91,94 or 1,1,4,7,10,10-hexamethyltriethylenetetramine (HMTETA),44 could provide better control over the polymerizations. The catalytic activity of the metal complex and, hence, the ability to control the polymerization can be correlated to the ability to stabilize the Cu(II) oxidation state, thus forming a reducing Cu(I) complex.93 The polymerization can proceed relatively fast even at low temperatures when a catalyst with a high activity is used.6 PMDETA is a good ligand for Cu(I) with moderate activity between the less active bpy (two magnitudes lower in activation rate) and the more active Me6TREN (two magnitudes higher in activation rate) and, therefore, can be used for a wide range of monomers.93
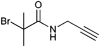
Besides the attachment of propargyl alcohol, the alkyne moiety can also be incorporated onto an ATRP initiator by the reaction of propargyl amine with α-haloisobutyryl halides. An example is shown in Entry 3.95 Hereby, a combination of the pre- and the postclick approach was utilized. The initiator was clicked prior to the polymerization with azide-functionalized dansyl as a fluorescent label. By using this compound as initiator in ATRP different random copolymers of alkyne- or azide-functionalized acrylamides were obtained: Poly(NIPAM-r-propargylacrylamide) (Mn = 20 000 g mol−1, PDI = 1.3) and poly[NIPAM-r-(3-azidopropylacrylamide)] (Mn = 10 000 g mol−1, PDI = 1.2). The pendant clickable polymers were attached onto an azide-functionalized silica particle in an elegant layer-by-layer click approach to yield thermoresponsive microcapsules after removal of the silica template.95
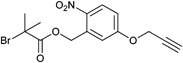
In Entry 4 an initiator is shown where the alkyne is linked to the mediating bromide via an o-nitrobenzyl ester as a photocleavable group.96 The initiator was used in a one-pot click-CRP reaction for the polymerization of either St, tBA or MMA in the presence of azide-functionalized PEO or PS to prepare photocleavable block copolymers as clicked structures with the labile group between the polymer blocks: PEO-b-PS (Mn = 38 000 g mol−1, PDI = 1.2), PEO-b-PtBA (Mn = 63 000 g mol−1, PDI = 1.2), PS-b-PMMA (Mn = 38 000 g mol−1, PDI = 1.2–1.3).
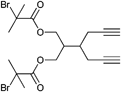
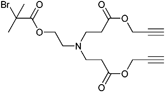
The initiator can bear more than one alkyne functionality. Multifunctional initiators can be used to create two or three dimensional architectures such as star polymers or networks. The initiator depicted in Entry 5 contains two bromoisobutyrate groups as well as two propargyl groups (unprotected alkynes) and is used in atom transfer radical emulsion polymerization in water to synthesize crosslinked nanoparticles of styrene and vinylbenzyl azide.83 An example that can be better analyzed by means of molar mass and its polydispersity index is described for the initiator depicted in Entry 6 bearing both two alkyne and two bromoisobutyrate groups. The polymerization of styrene was carried out for 2 h at 110 °C without the protection of the terminal alkyne groups (Mn = 4 000 g mol−1, PDI = 1.1–1.2), while the subsequent alkyne-azide cycloaddition leads to figure-of-eight-shaped polymers.97
Another alkyne-functionalized initiator (Entry 7) is functionalized with one bromoisobutyrate as well as two propargyl groups.98 Thereby, one alkyne moiety was clicked with an azide-functionalized poly(ethyleneglycol) (PEG) and was subsequently utilized in the polymerization of styrene without protecting the second alkyne moiety that resulted in a “tadpole-shaped” architecture after the second click reaction.98
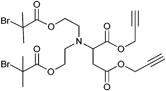
In addition, a trialkyne-functionalized initiator (Entry 8) was synthesized by the substitution reaction of propargyl bromide with pentaerythritol followed by the attachment of α-bromoisobutyryl bromide on the remaining alcohol groups.99 The initiator was used in a preclick approach: First, azido-PS prepared by RAFT (Mn = 2 500 g mol−1, PDI = 1.1) was clicked onto the initiator. Thereby, the molar mass distribution increased to 1.2 due to the presence of small amounts of macroinitiator, where only two PS-chains were attached. Subsequently, the PS-macroinitiator was used in the polymerization of MMA to yield 4-arm stars (Mn = 34 000 g mol−1, PDI = 1.3–1.4). The high polydispersity was assigned to a lack of control in the initial stages of the polymerization.
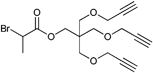
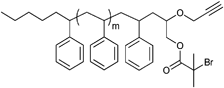
Furthermore, alkyne-functionalized initiators were combined with other polymerization techniques or methods by incorporating, e.g., alcohol groups for ring-opening polymerization (ROP) (Entry 9), whereby the polymerization techniques do not interfere with each other and can be applied simultaneously100 or subsequently.101 Besides the incorporation of α-haloisobutanoyl halides (halide = Cl, Br) into small molecules they can also be incorporated into alcohol-functionalized polymers to form ATRP macroinitiators as shown in Entry 10.102 In this example, polystyrene was synthesized via anionic polymerization and functionalized with propargyl bromide and α-bromoisobutanoyl bromide to provide an alkyne-functionalized ATRP macroinitiator. Polymerization of tBA yielded mid-chain alkyne-functionalized PS-b-PtBA block copolymers (Mn = 15000 g mol−1, PDI = 1.2–1.3).
One can mistrust the requirement of azide-functionalized initiators, since the azide functionality can be easily and efficiently introduced by substitution of the mediating halide with sodium azide. However, the degree of azide functionalization that can be reached with the initiator approach is higher compared to the postmodification route. The reason lies in the nature of controlled radical polymerizations: Termination reactions always occur and, hence, not all polymer chains retain the halide at the ω-terminus which is the bottleneck for the degree of functionalization that can be reached via postmodification. In contrast, using functional initiators, every chain that is initiated by the initiator bears the azide moiety at the α-terminus independently from termination reactions.
The azide moiety is used without protection during polymerization, although some side reactions were described: (i) Cyclization reactions between the azide and the propagating radical that causes low initiator efficiency,68,92 (ii) 1,3-cycloaddition of azides with the double bond of the monomer occurs in the absence of a catalyst at high temperatures and long reaction times, at which the amount decreases in the order of acrylates > acrylamides ≫ methacrylates > styrenes.105
To reduce the side reactions to a negligible amount, short reaction times92,106 and low temperatures92,107 are preferably used. It was shown that the polymerization at room temperature completely suppressed side reactions involving the azide.108 Hence, monomer classes are favored that can be polymerized at moderate temperatures.
In contrast, it was shown that the azide group does not act as an initiating species itself as indicated by controlled polymerizations with an azide-containing initiator.92
The initiator structure depicted in Entry 11 (top) was used for the polymerization of N,N-dimethylamino-2-ethyl methacrylate (DMAEMA)92,106 in THF with CuBr/HMTETA at 60 °C in a controlled way (PDI = 1.1–1.3). However, the initiator efficiency was low (f = 0.4) due to intramolecular cyclization at the early stages of the polymerization involving the azide and the propagating radical.92 Therefore, the “preclick” method was utilized to circumvent these side reactions as the azide-functionalized initiator was “clicked” onto an alkyne-functionalized poly(ε-caprolactone) (PCL). As expected, an increase in the initiation efficiency to f = 0.85 could be observed when using the “clicked” PCL macroinitiator.92 This is a good example where the preclick method is used since the postclick route failed in parts due to side reactions involving the clickable unit.
The initiator structure depicted in Entry 11 (bottom) containing a cleavable p-alkoxybenzyl ester was used in bulk polymerization of styrene at 90 °C, whereby no termination reactions were observed (Mn = 4000 g mol−1, PDI = 1.1–1.2).107
The spacer between the initiating fragment and the azide function seems to have a significant influence on the initiation efficiency in terms of intramolecular cyclization: The initiator depicted in Entry 12 shows, despite a controlled polymerization of MMA (Mn = 6000 g mol−1, PDI = 1.2–1.3), a reduced initiation efficiency for the hexyl spacer (70 to 80%) compared to the propyl spacer (100%).68 Hence, the initiator efficiency can be optimized by choosing an initiator structure by means of a spacer that prevents cyclization in the early stages of the polymerization.
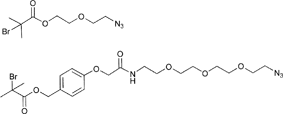
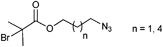
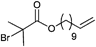
In Table 2, 3-azidopropyl 2-bromoisobutyrate (APBIB) has been the most widely used azide-functionalized initiator (Entry 12, n = 1). Controlled polymerization of St (Mn = 4500 g mol−1, PDI = 1.3),109 NIPAM (Mn = 10000 g mol−1, PDI = 1.1–1.2),110 and DMAEMA (Mn = 10000 g mol−1, PDI = 1.1–1.2) have been reported.111
Moreover, Haddleton and coworkers used the azide-functionalized initiator depicted in Entry 12 for the random copolymerization of MMA and hostasol methacrylate (HMA) (Mn = 8000 g mol−1, PDI = 1.2).82 The α-functionalized fluorescent copolymer was clicked onto cotton and both Wang and Merrifield resins using Huisgen [2+3] cycloaddition.
In addition, Topham and coworkers have polymerized a number of acrylates and methacrylates (MA) in a controlled way using APBIB as initiator:112 2-Aminoethyl methacrylate hydrochloride (Mn = 7000 g mol−1, PDI = 1.1–1.2), 2-(diethylamino)ethyl methacrylate (DEAEMA) (Mn = 21000 g mol−1, PDI = 1.3), DMAEMA (Mn = 7000 g mol−1, PDI = 1.3), 2-hydroxyethyl methacrylate (HEMA) (Mn = 7000 g mol−1, PDI = 1.3), 2-hydroxypropyl methacrylate (HPMA) (Mn = 5000 g mol−1, PDI = 1.2), 2-(methacryloyloxy)ethyl phosphorylcholine (Mn = 15000 g mol−1, PDI = 1.2), glycerol monomethacrylate (Mn = 11000 g mol−1, PDI = 1.2), potassium 3-sulfopropyl methacrylate (KSPMA) (Mn = 20000 g mol−1, PDI = 1.2), and methyl chloride-quaternized 2-(dimethylamino)ethyl methacrylate (Mn = 5000 g mol−1, PDI = 1.2).
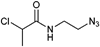
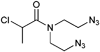
Furthermore, 2-chloropropionamide linked with an ethyl spacer to the azide moiety (Entry 13) was utilized in the controlled polymerization of NIPAM (Mn = 12000 g mol−1, PDI = 1.3).113 The N,N-diazido-2-chloropropionamide (Entry 14) was used in the polymerization of NIPAM to incorporate two azide functionalities on the same chain end (Mn = 10000 g mol−1, PDI = 1.1)110 In this way, 3-arm star polymers can be prepared.
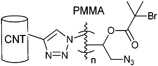
Besides the initiators discussed above, there are also azide-functionalized macroinitiators available. An elegant example of a multi-clickable initiator that is attached to a carbon nanotube (CNT) is shown in Entry 15.114 Poly(glycidyl methacrylate) (PGMA) is functionalized in a ring-opening reaction with sodium azide and subsequently reacted with 2-bromoisobutyryl bromide to yield a multi-clickable polymeric macroinitiator. This multiazide-functionalized polymer was clicked onto a multialkyne-functionalized CNT, whereby the excess of azide functions over the alkyne ones preserve free azides on the surface of the carbon nanotube. This coated CNT was used to click PEG in a grafting-onto approach as well as to polymerize St or nBMA in a grafting-from approach to yield amphiphilic polymer brushes on carbon nanotubes.
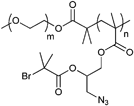
A similar macroinitiator was prepared from a copolymer (PEG-b-PGMA), whereby the poly(glycidyl methacrylate) was functionalized in a ring-opening reaction with sodium azide and subsequently reacted with 2-bromoisobutyryl bromide to yield a multi-clickable polymeric macroinitiator (Entry 16).104 Polymerization of DEGMA yielded azide-functionalized PEO-b-[PGMA-g-(N3)(PDEGMA)] (Mn = 52000 g mol−1, PDI = 1.2). In a grafting-onto approach alkyne-functionalized PDEAEMA was attached via click reaction to obtain coil-rod double hydrophilic diblock copolymers.
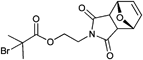
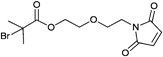
A widely used example is the [4 + 2] cycloaddition of maleimide and anthracene, whereby both moieties can be used as clickable functions attached to common ATRP initiators. The maleimide function must be protected, since it can act as a polymerizable monomer leading to crosslinking and a significant decrease of clickable fidelity after polymerization.122 Therefore, the maleimide is protected prior to the polymerization via Diels–Alder reaction with furan that can be easily cleaved after the polymerization in a retro Diels–Alder reaction by heating the protected polymer. Besides, the maleimide can also undergo Michael addition with thiols as a Michael acceptor (Entry 17, 18).35,58,66,122,123
An often used maleimido initiator is depicted in Entry 17. In general, the maleimide was attached to 2-bromoisobutyrate in a stepwise fashion. At first, maleic anhydride reacts with furan to protect the double bond. Subsequently, the imide was formed with ethanolamine under reflux and as the last step commercially available 2-bromoisobutyryl bromide was reacted.122 This protected initiator was utilized via the postclick approach in the homopolymerization of MMA115,117,120,121 (up to Mn = 3000 g mol−1, PDI = 1.1–1.2), OEGMA (up to Mn = 32000 g mol−1, PDI = 1.2),122 (2,2-dimethyl-1,3-dioxolan-4-yl)methyl methacrylate (up to Mn = 35000 g mol−1, PDI = 1.2),122t-butyl acrylate (tBA)115 (up to Mn = 3000 g mol−1, PDI = 1.2) and the random copolymerization of different methacrylates66,123 containing protected alkyne, ketosol and hostasol or rhodamine B as fluorescent dyes (Mn = 10000 g mol−1, PDI = 1.2). In none of these cases, side reactions were observed or discussed. Furthermore, this initiator was used in preclick approaches by Haddleton and coworkers for the polymerization of MMA at 50 °C (up to 55000 g mol−1, PDI < 1.2).64 Thereby, the alcohol-functionalized maleimide was clicked via DA reaction with an alcohol-functionalized furan or anthracene moiety and was subsequently reacted with 2-bromoisobutyryl bromide to obtain clicked dual-initiators with ATRP-mediating moieties on both click counterparts.
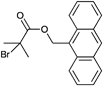
The anthracene-functionalized initiator depicted in Entry 19 was synthesized from commercially available 9-anthracenemethanol and 2-bromoisobutyryl bromide124 and was utilized in the homopolymerization of MMA87,124 (up to Mn = 30000 g mol−1, PDI = 1.1) and styrene121 (Mn = 5000 g mol−1, PDI = 1.1).
Tunca and coworkers used the initiator shown in Entry 19 for the preparation of block copolymers of styrene and divinylbenzene to form multiarm star polymers with terminal anthracene groups.125 At first, styrene was polymerized to obtain linear anthracene-functional PS (up to Mn = 6000 g mol−1, PDI = 1.1). Subsequently, the prepared PS was used as macroinitiator in the polymerization of divinylbenzene leading to a crosslinked second block that form the core of the multiarm star polymer (up to Mw = 75000 g mol−1, PDI = 1.5). Furthermore, anthracene functional PS (prepared with the initiator shown in Entry 19) as well as alkyne functional PS (prepared with the initiator shown in Entry 1) were used as macroinitiators for the ATRP of divinylbenzene yielding multiarm star polymers with terminal alkyne and anthracene groups (Mw = 250000 g mol−1).89 The orthogonality of the two clickable groups were utilized in a sequential double click reaction to selectively attach azide-functionalized PtBA and maleimide-functionalized PMMA.
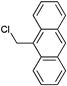
In addition, commercially available 9-chloromethylanthracene was applied as initiator in the polymerization of styrene (Mn = 4500 g mol−1, PDI = 1.2) using CuCl/bpy as catalytic system in THF (Entry 20).126 Although the prepared polymers were not yet applied in a Diels–Alder reaction, the terminal anthracene moiety represents a potential group for this click reaction.
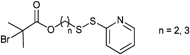
:1). Side reactions could be reduced by using less catalyst130 (catalyst to initiator ratio of 1:0.2) or by changing the catalytic system from CuBr/bpy to CuCl/bpy34 which decreases the amount of free radicals.
An ATRP initiator with a pyridyl disulfide moiety is depicted in Entry 22 and was introduced by Maynard and coworkers.34 It was used for the homopolymerization of MMA (Mn = 10000 g mol−1, PDI = 1.2), N-acetyl-D-glucosamine-functionalized methacrylate128 (Mn = 13000 g mol−1, PDI = 1.1), N-hydroxysuccinimidyl methacrylate129 (Mn = 10000 g mol−1, PDI = 1.3), 2-THP-protected HEMA129 (Mn = 10000 g mol−1, PDI = 1.3), t-butyl methacrylate (tBMA)129 (Mn = 5000 g mol−1, PDI = 1.5), HEMA34 (Mn = 16000 g mol−1, PDI = 1.2–1.3), and St129 (Mn = 13000 g mol−1, PDI = 1.2).
In contrast to these postclick approaches for bioconjugation also the preclick approaches are conducted with retention of the bioactivity.35 As advantages of the grafting-from approach the following issues can be pointed out: (i) The purification of the bioconjugate from catalyst and monomer is simplified compared to the purification from the polymer in the grafting-onto approach and (ii) the placement of the polymer is predetermined facilitating the synthesis and characterization.58 Herein, 2-bromoisobutyrate as an ATRP initiator functionalized with either pyridyl disulfide (Entry 22) or maleimide (Entry 18) was clicked prior to the polymerization onto the free cystein of a protein (Bovine Serum Albumin or T4 lysozyme). Using these protein macroinitiators NIPAM could be polymerized in situ (PDI = 1.3).35,58
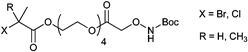
000 g mol−1, PDI = 1.1), HEMA (Mn = 40000 g mol−1, PDI = 1.2), and oligo(ethylene glycol) methacrylate (OEGMA) (Mn = 23000 g mol−1, PDI = 1.2–1.3).131,132
4.2 Reversible addition fragmentation chain transfer (RAFT)
| Entry | Structure | Click | CRP | Monomer | Strategy | Ref |
|---|---|---|---|---|---|---|
| 24 |
|
CuAAC | RAFT | MA, THPA, St, NIPAM | poC | 133–136,143,144 |
| 25 |
|
CuAAC | RAFT | AM, St, MA, 4VP, NIPAM, MMA | poC | 137–140,145,146 |
| prC | 147 | |||||
| 26 |
|
CuAAC | RAFT | St, nBA | prC | 142 |
| Thiol–yne | RAFT | NIPAM | poC | 141 | ||
| 27 |
|
CuAAC | RAFT | VAc, NVP, St, nBA | prC | 142 |
| Entry | Structure | Click | CRP | Monomer | Strategy | Ref |
|---|---|---|---|---|---|---|
| 28 |
|
CuAAC | RAFT | St, VAc, DMA | poC | 99,133,148 |
| 29 |
|
CuAAC | RAFT | St, DMA, NIPAM, nBA, OEGA | poC | 148–152,156 |
| 30 |
|
CuAAC | RAFT | NIPAM, DMA | poC | 105,157,158 |
| 31 |
|
CuAAC | RAFT | VAc | poC, prC | 135,144,153–155 |
| Entry | Structure | Click | CRP | Monomer | Strategy | Ref |
|---|---|---|---|---|---|---|
| 32 |
|
CuAAC | RAFT | HPMAM, MMA, St, OEGA, NIPAM | poC | 159 |
| 33 |
|
PySS | RAFT | nBA, OEGA | poC | 162,36,163 |
| NIPAM | prC | 160,161 | ||||
| 34 |
|
PySS | RAFT | OEGA, St | poC | 164 |
| 35 |
|
HDA | RAFT | St, iBoA | poC | 38,39,55,165–170 |
| 36 |
|
MAdd | RAFT | NIPAM | prC | 171 |
In principle, all types of monomers which can be polymerized by free radical polymerization can also be polymerized by RAFT using the appropriate type of RAFT agents, i.e. dithiobenzoates, trithiocarbonates and xanthates.11,12 AIBN has been the most widely used initiator for RAFT polymerization. In general, clickable moieties are attached to the initiating fragment (R group) of the chain transfer agents. The advantage of the R group compared to the mediating group (Z group) is the high end-group fidelity of the resulting polymer. The related clickable CTAs are depicted in Tables 4 to 6.
The first report on click chemistry and the RAFT process for the preparation of diblock copolymers of styrene and vinyl acetate via polymer-polymer conjugation was provided by Barner-Kowollik and coworkers (Mn = 12 100 g mol−1, PDI = 1.1–1.2).133
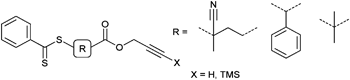
Another combination of click chemistry and the RAFT process was published by Hawker et al. in 2006 showing the facile formation of clickable micelles from block copolymers of protected acrylic acid and styrene polymerized with an alkyne-functionalized RAFT agent (Entry 24). Azide-alkyne cycloaddition is possible with the terminal alkyne-functionalized block copolymers. Protection of the terminal alkyne with the trimethylsilyl group was not necessary in this case.134
Furthermore, a similar approach to well-defined block copolymers was reported using a TMS-protected alkyne dithiobenzoate as shown in Entry 24. Terminal alkyne-functionalized poly(styrene) was synthesized at 60 °C in a controlled way (Mn = 8 200 g mol−1, PDI = 1.1) and it was shown that the molar mass linearly increased with monomer conversion.133 The same RAFT agent was used for the polymerization of an O-methacryloyl mannose monomer resulting in an alkyne-functionalized glycopolymer (Mn = 4 300 g mol−1 and PDI = 1.1–1.2).135 Similarly, an alkyne-functionalized RAFT agent bearing an unprotected alkyne group was used for the polymerization of styrene and NIPAM at 70 to 80 °C. Different homopolymers were synthesized in a molar mass range between 2 700 to 3 700 g mol−1 for poly(styrene) and 4900 to 11000 g mol−1 for poly(NIPAM).136
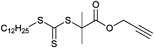
Also a terminal alkyne-functionalized trithiocarbonate (Entry 25) has been used for the RAFT polymerization of different monomers by Brittain.137,138 For example, surface-mediated RAFT polymerization of styrene and methyl acrylate resulted in poly(St-b-MA) with a molar mass of Mn = 34000 g mol−1.137 Furthermore, the same group reported on the modification of silica nanoparticles using the tandem approach of RAFT polymerization of styrene and click chemistry.138
The chain transfer agent as shown in Entry 25 was clicked to azido end-functionalized poly(isobutylene) and was subsequently used as a clicked macro-RAFT agent for the polymerization of NIPAM (Mn = 24 800–53 200 g mol−1, PDI < 1.1). First order kinetic plots for the polymerization of NIPAM were obtained and revealed that this monomer polymerizes in a controlled way.139
The alkyne-functionalized chain transfer agent depicted in Entry 25 was reported by another research group for the polymerization of 4-vinylpyridine (NVP) initiated with AIBN at 80 °C resulting in a polymer with a molar mass of 13 600 g mol−1 with a PDI value of 1.4.140
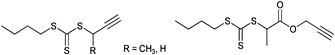
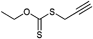
Hyperbranched polymers were prepared by thiol-yne click chemistry by Perrier et al. using the alkyne-terminated transfer agent shown in Entry 26. After a postmodification step to cleave the RAFT agent into a thiol it was clicked (by UV light at room temperature with yields over 95%) to form a styrene hyperbranched polymer.141 Furthermore, a xanthate type of RAFT agent (Entry 27) containing an alkyne group has been used for the polymerization of VAc and NVP by Klumperman.142 In all cases, xanthates were first clicked and then used as functionalized RAFT agents in the polymerization of VAc (Mn = 3 900 g mol−1, PDI = 1.2–1.3), NVP (Mn = 5 400 g mol−1, PDI = 1.1–1.2), St (Mn = 7 500 g mol−1, PDI = 1.1–1.2) and nBA (Mn = 10000 g mol−1, PDI = 1.1).142 Semi-logarithmic kinetic plots indicated controlled polymerizations using these RAFT agents.
It seems that the protection of the alkyne group is not strictly necessary. As shown above, there are examples for both protected and unprotected CTAs demonstrating that polymers with high end-group fidelity (> 90%) can be prepared. The temperature and the ratio between the alkyne terminated RAFT agent and the monomer play an import role to achieve a sufficient control over the polymerization.
The azide moiety is used without protection during the polymerization, although some side reactions were described. To decrease the amount of side reactions, low temperatures and/or low conversions are favored that will be discussed in the following.
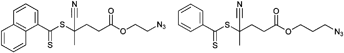
The dithiobenzoate RAFT agents depicted in Entry 28 were used for the bulk polymerization of styrene at 60 °C and provide a good control over the polymerization (Mn = 1900 to 5300 g mol−1, PDI = 1.1143 and Mn = 3200 to 11000 g mol−1, PDI = 1.1133). Moreover, Sumerlin et al. using the initiator listed in Entry 28 for the polymerization of St and DMA resulting in azido functional PS (Mn = 5500 to 12000 g mol−1, PDI = 1.1–1.3) and poly(DMA) (Mn = 10 800 to 21 800 g mol−1, PDI = 1.30).148 A thionaphthoyl RAFT agent containing the azide functionality (Entry 28) was also used for the polymerization of styrene in bulk at 80 °C (Mn = 2 400 g mol−1, PDI = 1.1). A linear relationship in the semi-logarithmic kinetic plot was reported indicating that the concentration of propagating chains are almost constant throughout the reaction.99
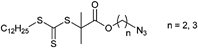
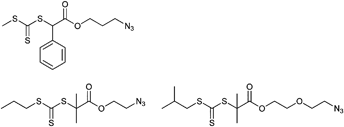
Among the azide-functionalized RAFT agents belonging to the class of the trithiocarbonates, the CTA with a C12-side chain is frequently used for the RAFT polymerization of acrylamides (Entry 29): First order kinetic plots for this CTA indicate a constant concentration of propagating chains.148,149 In addition, Gondi et al. used the initiator depicted in Entry 29 with AIBN for the controlled polymerization of styrene (Mn = 5100–8600 g mol−1, PDI = 1.1–1.2) and DMA (Mn = 5000–10000 g mol−1, PDI = 1.1–1.2).148 Furthermore, the same group reported on the synthesis of poly(DMA-b-NIPAM) using this azido-RAFT agent. In this way, telechelic polymers were synthesized: PNIPAM (Mn = 2700 g mol−1, PDI = 1.1–1.2), PDMA (Mn = 4200 g mol−1, PDI = 1.1) and poly(DMA-b-NIPAM) (Mn = 6000 g mol−1, PDI = 1.15).149 In addition, NIPAM was polymerized with the initiator shown in Entry 29 at 60 °C in a controlled way resulting in azido end-functionalized polymers (Mn = 16 300 g mol−1, PDI = 1.1), which were further used for protein coupling by the copper-catalyzed azide-alkyne cycloaddition.150 The RAFT agent depicted in Entry 29 was also applied for the preparation of 3-miktoarm star polymers by using a combination of RAFT, ring-opening polymerization and click chemistry. After the polymerization at 70 °C of nBA (Mn = 3500 g mol−1, PDI = 1.1), OEGA (Mn = 4800 g mol−1, PDI = 1.1) or NIPAM (Mn = 4600 g mol−1, PDI = 1.1) propargyl diol was clicked to the azide.151 Furthermore, an azide-functionalized CTA (Entry 29) was used for the preparation of hyperbranched polymers. For this purpose, a propagyl acrylate was clicked onto the CTA and subsequently copolymerized with St or NIPAM.152 Perrier et al. used a trithiocarbonate (Entry 30) for the RAFT polymerization of NIPAM (Mn = 2600–10600 g mol−1, PDI = 1.1), but side reactions involving the azido moiety occurred such as 1,3-dipolar cycloaddition with electron-deficient olefins, i.e. NIPAM.105 The cycloadditions to the triazoline or to the pyrazoline (by a second addition of NIPAM) were confirmed by high resolution mass spectrometry.105
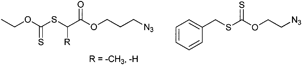
As depicted in Entry 31, an azide-functionalized xanthate has been used for the polymerization of VAc (Mn = 6 800 g mol−1, PDI = 1.15)135 as well as for grafting to an alkyne side-chain functional copolymer.153 A xanthate-terminated dextran was prepared by using click chemistry of an azido RAFT agent shown in Entry 31. The resulting macro-CTA was used for the bulk polymerization of VAc resulting in a block copolymer. The molar mass linearly increased by conversion, although the polydispersity index was increasing.154 A similar xanthate was used for the bulk polymerization of VAc at 80 °C resulting in a broader molar mass distribution at higher monomer conversion (PDI > 1.4).155
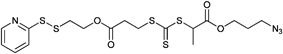
000–14000 g mol−1, PDI = 1.1), NIPAM (Mn = 3200–16200 g mol−1, PDI = 1.12 to 1.14) and OEGA (Mn = 7500–12500 g mol−1, PDI = 1.1).159
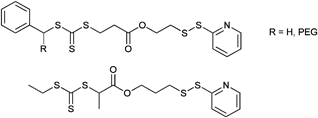
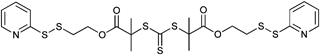
000–34000 g mol−1) were smaller than 1.20 for all samples and for one sample of the block copolymerization with nBA (by increasing the conversion the molar mass distribution broadened). The high end-group fidelity of the pyridyl disulfide groups was indicated by 1H NMR spectroscopy. Furthermore, the same RAFT agent (Entry 33) was used by Davis et al. for the polymerization of NIPAM and OEGA resulting in different molar masses and polydispersity indices:160–163 POEGA (Mn = 15500–23000 g mol−1, PDI = 1.2–1.3) and PNIPAM (Mn = 5100–18000 g mol−1, PDI = 1.2–1.5).163 The PySS end group is often used for the preparation of polymer bioconjugates, e.g. with BSA via the free thiol group. Semilogarithmic kinetic plots are reported for the RAFT polymerization of OEGA and NIPAM with the CTA pictured in Entry 33 (top) by Bulmus and Davis showing a linear relationship between ln[[M]0/[M]t] and reaction time, which indicates a constant level of radical concentration during the polymerization.163 The symmetric trithiocarbonate RAFT agents depicted in Entry 34 were used for the polymerization of OEGA at 70 °C and provide a good control over the polymerization (Mn = 4 600–23 400 g mol−1, PDI = 1.2–1.3). With these homopolymers a chain extension using styrene was performed resulting in block copolymers of type ABA (Mn = 19100–37900 g mol−1, PDI = 1.2–1.3).164
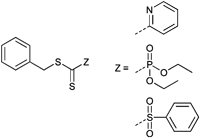
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S double bond (dienophile) was directly used as a clickable group for hetero Diels–Alder reactions with dienes (Scheme 1, HDA). This approach represents a straightforward pathway to block copolymers without an additional synthesis step for a postmodification or any other preparation step. The first study on this approach was reported by Barner-Kowollik, Stenzel and coworkers in 2008. The authors prepared polymer conjugates of PS polymerized by RAFT and a diene-terminated poly(ε-caprolactone). The use of these electron-deficient dithioesters (Entry 35) allow the polymerization of styrene in a controlled manner (Mn = 2200–2800 g mol−1, PDI = 1.1).38 This class of RAFT agents was also used for the polymerizations of St and isobornyl acrylate (iBoA) with different molar masses (stopped at low monomer conversion to ensure high end-group fidelity). All obtained polymers were well-defined and have low polydispersity indices. The prepared polymers were clicked via the HDA with cyclopentadienyl- or 2,4-hexadiene-endcapped polymers.38,39,55,165–170
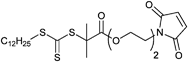
000 g mol−1.171
S double bond (dienophile) was directly used as a clickable group for hetero Diels–Alder reactions with dienes (Scheme 1, HDA). This approach represents a straightforward pathway to block copolymers without an additional synthesis step for a postmodification or any other preparation step. The first study on this approach was reported by Barner-Kowollik, Stenzel and coworkers in 2008. The authors prepared polymer conjugates of PS polymerized by RAFT and a diene-terminated poly(ε-caprolactone). The use of these electron-deficient dithioesters (Entry 35) allow the polymerization of styrene in a controlled manner (Mn = 2200–2800 g mol−1, PDI = 1.1).38 This class of RAFT agents was also used for the polymerizations of St and isobornyl acrylate (iBoA) with different molar masses (stopped at low monomer conversion to ensure high end-group fidelity). All obtained polymers were well-defined and have low polydispersity indices. The prepared polymers were clicked via the HDA with cyclopentadienyl- or 2,4-hexadiene-endcapped polymers.38,39,55,165–170
000 g mol−1.171
4.3 Nitroxide-mediated radical polymerization (NMP)
| Entry | Structure | Click | CRP | Monomer (Abbr./Entry) | Strategy | Ref |
|---|---|---|---|---|---|---|
| 37 |
|
CuAAC | NMP | St | poC | 176 |
| 38 |
|
CuAAC | NMP | St | poC | 179–181 |
| ATRP | MMA | |||||
| AAC | NMP | St | poC | 45 | ||
| ATRP | MMA | |||||
| 39 |
|
CuAAC | NMP | St | poC | 182–184 |
| ROP | ε-CL | |||||
| AAC | NMP | St | poC | 45 | ||
| ROP | ε-CL | |||||
| 40 |
|
CuAAC | NMP | -- | -- | 185 |
| 41 |
|
CuAAC | NMP | -- | -- | 175 |
| 42 |
|
CuAAC | NMP | St, tBA | poC | 174 |
| NMP | St, NIPAM, nBA | prC | 176,188 | |||
| 43 |
|
DA | NMP | St | poC | 118 |
| ATRP | tBA | |||||
| NMP | St | prC | 119,190 | |||
| ATRP | tBA |
For nitroxide-mediated polymerizations very few examples of clickable initiators were described up to now. In comparison to ATRP and RAFT polymerizations, usually higher reaction temperatures are necessary for NMP. This decreases the number of suitable click functionalities that can be used during the polymerization without exceeding acceptable amounts of side reactions.
It was shown that unimolecular initiators such as phenylethyl-alkylated 2,2,6,6-tetramethylpiperidinylnitroxide (TEMPO) and 2,2,5-trimethyl-4-phenyl-3-azahexane-3-nitroxide (TIPNO) can be functionalized without influencing the control over the polymerization.172–174 Thereby, the functionalization can be performed in principle at the initiating as well as at the mediating fragment of the alkoxyamine. Until now, clickable moieties are only attached to the initiating fragment. The advantage of this side compared to the mediating side is the high end-group fidelity of the resulting polymer. This is caused by the nature of the NMP process: The incorporation of a functional group at the initiating chain end is done in one step, whereas the incorporation of a certain functionality at the mediating chain end contains many reaction steps until the final polymer is formed, which increases the probability of side reactions.
The general synthetic strategy towards a functional unimolecular initiator is the radical coupling reaction of TIPNO or of the commercially available TEMPO with a functionalized vinyl compound activated by a manganese(III) salen complex (Jacobsen's reagent).175 The related clickable initiators for NMP are shown in Table 7.
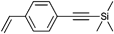
The alkyne was protected with the TMS group to reduce the possible side reactions under polymerization conditions: (i) chain transfer by hydrogen abstraction from the alkyne as well as polymerization along the triple bond leading to crosslinking76,77,176,177 and (ii) addition of nitroxide radicals to the triple bond.178 It can be noted that even a small loss of nitroxide during polymerization has a large impact on the controlled character of the polymerization, since it shifts the equilibrium towards the free propagating radical resulting in a significant increase in termination and transfer reactions in particular at higher conversions.
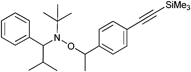
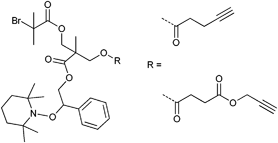
The protected initiator (Entry 37) was used in the homopolymerization of styrene resulting in α-functionalized clickable PS (Mn = 24000 g mol−1, PDI = 1.1) after deprotection with tetrabutylammonium fluoride (TBAF). In contrast to ATRP, no uncontrolled deprotection of the TMS-alkyne was noticed during the polymerization. Furthermore, a combination of ATRP and NMP with the same initiator is possible as demonstrated by Tunca and Hizal et al.45,179–181 Although both techniques are radical polymerizations and, hence, might interfere with each other, they can be applied subsequently: (i) NMP of styrene can be conducted without affecting the ATRP initiator in the absence of any metal catalyst45,180,181 and (ii) ATRP of methacrylate can be conducted at lower temperature (60 °C) at which the nitroxide is still inactive and not able to mediate radical propagation.179 Entry 38 shows such an alkyne-functionalized multifunctional initiator for ATRP and NMP.45,179–181 The structure contains (i) 2-bromoisobutyrate as mediator for the ATRP polymerization of MMA, (ii) TEMPO as mediator for NMP polymerization of styrene and an unprotected alkyne, whereas the fragments are linked via ester groups.
The unprotected alkyne is thermally stable up to 125 °C and the TEMPO-mediated polymerization of styrene seems not to interfere with the unprotected alkyne. Apparently, even after 17 h at 125 °C the polymerization was controlled and no significant loss of alkyne was observed for the resulting PS-macroinitiator (Mn = 10000 g mol−1, PDI = 1.2) as judged by 1H NMR spectroscopy and SEC measurements of the subsequent clicked structure.181 By contrast, the ATRP of MMA was kept short in every case (30 min at 60 °C) to prevent significant amounts of side reaction with the catalytic system resulting in PMMA macroinitiator (Mn = 6000 g mol−1, PDI = 1.2).179 However, the amount of side reactions is negligible in most cases due to the low concentration of alkyne-containing initiator.
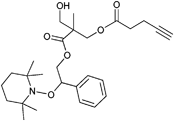
Furthermore, alkyne-functionalized NMP initiators are combined with other controlled polymerization techniques by the incorporation of, e.g., alcohol groups for the ring-opening polymerization of ε-caprolactone (Entry 39). These polymerization techniques do not interfere with each other and can be applied simultaneously182,183 or subsequently.45,184 For the consecutive procedure, first the ROP of ε-CL with the initiator depicted in Entry 39 was conducted at 110 °C for 2 h using Sn(Oct)2 as catalyst (Mn = 4000 g mol−1, PDI = 1.1) followed by NMP of styrene at 125 °C for 15 h (Mn = 19000 g mol−1, PDI = 1.3).45,184 On the other hand, in a simultaneous one-pot approach St and ε-CL were heated with Sn(Oct)2 for 22 h at 120 °C (Mn = 12000 g mol−1, PDI = 1.1).182 For all examples, copolymers of PCL-b-PS with an alkyne as clickable function at the junction point were achieved, whereby the alkyne was not protected even in the polymerization at 120 °C for 22 h. Hence, the alkyne must be stable under the applied polymerization conditions of NMP and ROP.
In addition, since both polymerization techniques do not interfere with the azide-alkyne cycloaddition, 3-miktoarm star terpolymers can be constructed by conducting ROP of ε-CL (with Sn(Oct)2), NMP of St (with initiator shown in Entry 39) and either simultaneously (one-pot/one-step) or subsequently (one-pot/two-step) clicking an azide-functionalized polymer with CuBr/PMDETA as catalyst.183 Thereby, 3-arm stars of PEG-PCL-PS (Mn = 14000 g mol−1, PDI = 1.3) and PMMA-PCL-PS (Mn = 14 500 g mol−1, PDI = 1.2) with the one-pot/one-step technique and stars of PtBA-PCL-PS (Mn = 16000 g mol−1, PDI = 1.1) and PEG-PCL-PS (Mn = 15000 g mol−1, PDI = 1.1) in the one pot/two step approach could be synthesized, respectively.183
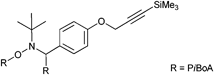
Another type of an alkyne-functionalized macroinitiator (Mn = 7700 g mol−1, PDI = 1.1–1.2) is depicted in Entry 40, whereas PiBoA is attached on both the initiating and the mediating fragment of the alkoxyamine.185 The macroinitiator was obtained by a nitrone-mediated radical coupling reaction of activated ATRP-made PiBoA (Mn = 4300 g mol−1, PDI = 1.2) in the presence of an alkyne-functionalized nitrone. To the best of our knowledge, this macroalkoxyamine had not yet been used as initiator in NMP. Nevertheless, it seems to be a potential candidate, since in a similar approach polystyrene midchain-functionalized with a parent alkoxyamine (not including the alkyne moiety) was used as initiator in NMP of St, n-BA and NIPAM for the chain extension towards ABA triblock copolymers.186,187
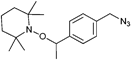
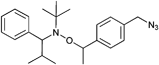
In a similar approach the p-(azidomethyl)phenylethyl-TIPNO was synthesized, whereby the p-chloromethyl styrene was functionalized with the azide prior to the radical coupling with TIPNO (Entry 42). The initiator was used in the polymerization of styrene (Mn = 9000 g mol−1, PDI = 1.1 after 3 h, 120 °C) and n-butylacrylate (Mn = 6 500 g mol−1, PDI = 1.2–1.3 after 13 h at 120 °C), whereby the polymerization was controlled and no side reactions of the azide were discussed.174
In contrast, Voit et al. discussed that the polymerization of styrene with the azido-functionalized initiator depicted in Entry 42 failed due to the side reactions that were assigned to the cyclization of the azide with the vinylic double bond of the monomer.176 Since the azide group is not thermally stable,49 either postmodification or preclick approaches could be successfully utilized to prepare the α-functionalized polymers. In the postmodification, p-(chloromethyl)phenylethyl-TIPNO was used in the polymerization of styrene and the chloro group was subsequently converted with sodium azide into azide (N3-PS: Mn = 8000 g mol−1, PDI = 1.2). Following the preclick approach, an alkyne-functionalized moiety (Cbz-protected adenine derivative) was clicked onto the azide prior to the polymerization of styrene resulting in α-functionalized PS (Mn = 52000 g mol−1, PDI = 1.2).176
To study the steric and electronic influence of the triazole moiety for the initiation quality of the alkoxyamine shown in Entry 42, two different alkoxyamines were synthesized starting from 4-(chloromethyl)phenylethyl-TIPNO, whereby the chloro group was substituted either by azide or by 4-azidobenzoate to vary the distance of the azide to the alkoxyamine skeleton.188 Polymerization of NIPAM using these azido-functionalized alkoxyamines as initiators failed in both cases. Therefore, the azido group was functionalized via 1,3 dipolar cycloaddition with either a 1,2-dihydroxyalkyl moiety, a barbituric acid moiety or a phenyl moiety. The initiators where the triazole was directly bound to the alkoxyamine group showed poor initiation. In contrast, the alkoxyamine with a rigid spacer revealed good control over the polymerization of NIPAM (Mn = 5000 g mol−1, PDI = 1.2) and n-BA (Mn = 6000 g mol−1, PDI = 1.2). The poor initiation efficiency in the first case was partly ascribed to an electronic influence but mostly to intramolecular hydrogen bonding between the propagating radical and the barbituric acid that hindered propagation. This was sterically prevented with the rigid spacer in the second case. In these preclick cases the azide moiety of the initiator described in Entry 42 opens the field towards versatile functionalized initiators via the facile incorporation of functional groups.
The anthracene-functionalized mikto-initiator depicted in Entry 43 was only used in the preclick approach, whereby the Diels–Alder reaction was conducted prior to the NMP of styrene followed by ATRP of tBA. With this strategy, different block copolymers were obtained: (i) H-shaped terpolymers119 (PS)(PtBA)-PEO-(PtBA)(PS) (Mn = 18000 g mol−1, PDI = 1.3) and (PS)(PtBA)-PPO-(PtBA)(PS) (Mn = 31000 g mol−1, PDI = 1.3) and (ii) 3-miktoarm star polymer190 PEG-PS-PtBA (Mn = 18000 g mol−1, PDI = 1.3). The protected maleimide-functionalized miktoinitiator described in Entry 43 was initially used in the ATRP of tBA (Mn = 4 000 g mol−1, PDI = 1.3) and subsequently “clicked” with anthracene-functionalized PCL prior to the NMP of styrene to obtain a 3-miktoarm terpolymer (PCL-PtBA-PS) (Mn = 40000 g mol−1, PDI = 1.7).118 The high polydispersity index was ascribed to a loss of TEMPO during the Diels–Alder reaction at 100 °C, indicated by a shoulder in the SEC trace at lower molar masses.
The preclick approach for the anthracene or maleimide moiety in combination with NMP should be used, since the maleimide and anthracene moieties cause side reactions under the polymerization conditions of NMP. Hence, to the best of our knowledge no example for the postclick approach is yet reported.
5. Clickable monomers
Clickable monomers can be used to synthesize pendant functionalized polymers (Scheme 4) that can be easily modified in a grafting-onto approach via click chemistry. Thereby, the clickable monomer can be homopolymerized or copolymerized to obtain versatile random-, block- or comb polymers. Most widely used in controlled radical polymerization processes are MMA and St derivates.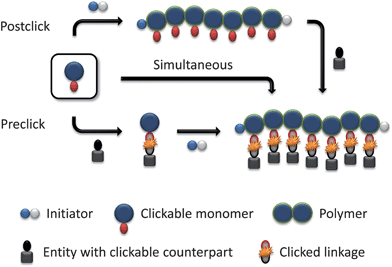 | ||
| Scheme 4 Schematic representation of the strategies via clickable monomers. | ||
The polymerization of these click-functionalized monomers represents often a synthetic challenge, because the clickable unit as a reactive group is frequently in conflict with the radical polymerization conditions. In contrast to the initiator approach a higher amount of side reactions involving the clickable functionality is expected, which is caused by the higher concentration of the monomer used during the polymerization process compared to the initiator. This fact is in particularly pronounced for bulk polymerizations or for side reactions involving besides the clickable unit other parts of the monomer, e.g. the vinyl group of azide-containing monomers, where cycloaddition between the double bond and the azide can occur. To circumvent such side reactions, either the clickable unit has to be protected, or polymerization time or temperature have to be reduced.
However, the degree of functionalization is much higher in this approach compared to the initiator one. Clickable initiators would provide polymers only with terminal functionalities, which would be one or two for linear polymers and equal the number of arms for star-shaped polymers. In contrast, homopolymerization of a clickable monomer would yield a polymer with functionalities as many as the number of repeating units. The degree of functionality can be decreased by copolymerization, which also decreases possible side reactions that are caused by the clickable monomers. In this section we discuss clickable monomers according to the functional groups, i.e. alkyne, azide, diene, thiol, para-fluoro and others. An overview over the clickable and clicked monomers is given in Tables 8–10.
| Entry | Structure | Click | Strategy | CRP | Initiator/CTA (Abbr./Entry) | Comonomers | Ref |
|---|---|---|---|---|---|---|---|
| 44 |
|
CuAAC | poC | RAFT | MCPMDB | THPA, St | 194,203 |
| NMP | PhEt-TIPNO | St, DMAM, HEMA, GMA, tBOSt, tBA, THPA | 60,193–195 | ||||
| 45 |
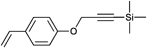
|
CuAAC | poC | NMP | PhEt-TIPNO | St, tBOSt, AcOSt | 195,201 |
| RAFT | DDAT | St | 201 | ||||
| 46 |
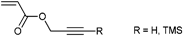
|
CuAAC | poC | NMP | PhEt-TIPNO | tBA | 193 |
| RAFT | BPIT | AA | 205,206 | ||||
| prC | RAFT | 28 | NIPAM | 156 | |||
| 47 |
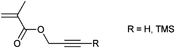
|
CuAAC | poC | ATRP | 15, TosCl, BMP | ε-CL, MMA, SMA, mPEG | 3,123,192,209 |
| RAFT | CPDB,CPADB,CDB,CBDB, DDAT, MCPMDB | OEGMA, MMA, GMA | 153,191,199,200,207,208 | ||||
| simult | ATRP | BMPABE | -- | 69 | |||
| prC | RAFT | CPDB | MMA | 214 | |||
| ATRP | 17 | -- | 66 | ||||
| 48 |
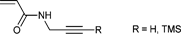
|
CuAAC | poC | ATRP | 3 | NIPAM | 95 |
| NMP | PhEt-TIPNO | HEMA, DMAM | 193 | ||||
| prC | RAFT | CPADB | NIPAM | 215 | |||
| 49 |
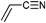
|
ZnANC | poC | ATRP | BPN, PEB | St | 48 |
| NMP | BPO, TEMPO | St | 210 | ||||
| 50 |

|
CuAAC | poC | ATRP | EBiB | MMA | 213 |
| prC | RAFT | MPPCTTA | St | 67 | |||
| 51 |
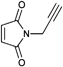
|
CuAAC | poC | ATRP | PEB | St | 216 |
5.1 Alkyne-containing monomers
The synthesis of alkyne-functionalized monomers is straightforward, whereby most synthetic routes involve the esterification of (meth)acryloyl chloride with propargyl alcohol or propargyl amine for (meth)acrylates3,66,153,191,192 and acrylamides,95,193 respectively. The propargyl derivatives can be protected by the reaction with trimethylsilyl chloride and 1,8-diazabicyclo[5.4.0]undec-7-ene catalyzed by silver chloride. The synthesis of alkyne-functionalized styrene is often accomplished via the Sonogashira reaction of 4-bromostyrene with (trimethylsilyl)acetylene.60,193–195The protection of acetylene-functionalized monomers by the alkylsilyl group is of prime importance, because the terminal alkyne is known to be chemically196 and thermally197 not stable under the polymerization conditions required for the CRP techniques.194 However, some researchers have used unprotected alkynes and indeed demonstrated that the terminal alkyne undergoes side reactions such as (i) radical addition to the triple bond,193 (ii) chain transfer,198 (iii) complexation of the terminal triple bond to copper-based ATRP catalysts and insertion reactions leading to insoluble crosslinked networks.77,193,199,200 In one of these reports, Matyjaszewski and coworkers reported that the ATRP of unprotected propargyl methacrylate was hardly controllable (PDI values > 3), due to the involvement of the acetylene moiety during the catalyzed radical process.77
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C-TMS) (Entry 44) in nitroxide-mediated polymerizations for the preparation of co- and terpolymers.193 By utilizing the PhEt-TIPNO alkoxyamine as unimolecular initiator, poly(St-co-St-CCH) (Mn = 47000 g mol−1, PDI = 1.2), poly(St-CCH-co-DMAM) (Mn = 40000 g mol−1, PDI = 1.2) and poly(St-r-St-CCH-r-HEMA) (Mn = 32000 g mol−1, PDI = 1.2) could be prepared in a controlled manner, whereby the alkyne monomer was incorporated into the polymers up to 10%. Without TMS-protection, significant amounts of crosslinked polymers at a higher content of alkyne-functionalized monomer or at high conversion were observed. The efficiency and the orthogonality of the alkyne-azide click reaction were proven by one-pot functionalization either in a cascade or in a simultaneous approach. In related work, Voit et al. synthesized poly(St-r-St-CCH-r-GMA) (Mn = 30000 g mol−1, PDI = 1.3) random terpolymers via NMP using the TMS-protected alkyne monomer. The deprotection with TBAF was performed without affecting the glycidyl moiety resulting in a pendant functionalized terpolymer bearing two orthogonal clickable moieties.60
C-TMS) (Entry 44) in nitroxide-mediated polymerizations for the preparation of co- and terpolymers.193 By utilizing the PhEt-TIPNO alkoxyamine as unimolecular initiator, poly(St-co-St-CCH) (Mn = 47000 g mol−1, PDI = 1.2), poly(St-CCH-co-DMAM) (Mn = 40000 g mol−1, PDI = 1.2) and poly(St-r-St-CCH-r-HEMA) (Mn = 32000 g mol−1, PDI = 1.2) could be prepared in a controlled manner, whereby the alkyne monomer was incorporated into the polymers up to 10%. Without TMS-protection, significant amounts of crosslinked polymers at a higher content of alkyne-functionalized monomer or at high conversion were observed. The efficiency and the orthogonality of the alkyne-azide click reaction were proven by one-pot functionalization either in a cascade or in a simultaneous approach. In related work, Voit et al. synthesized poly(St-r-St-CCH-r-GMA) (Mn = 30000 g mol−1, PDI = 1.3) random terpolymers via NMP using the TMS-protected alkyne monomer. The deprotection with TBAF was performed without affecting the glycidyl moiety resulting in a pendant functionalized terpolymer bearing two orthogonal clickable moieties.60
Furthermore, St-CCH was used for the preparation of poly(tBOSt-b-[St-co-St-CCH]) (Mn = 60000 g mol−1, PDI = 1.3–1.4) and poly(pHSt-b-[St-co-St-CCH]) (Mn = 26000 g mol−1, PDI = 1.2) diblock terpolymers which were applied in block copolymer lithography.195,201
In addition, St-CC-TMS was used in the synthesis of amphiphilic diblock terpolymers consisting of a hydrophilic poly(acrylic acid) block and a hydrophobic copolymer poly(St-co-St-CCH).194 Since acrylic acid can not be directly polymerized in a sufficiently controlled way with NMP202 – due to decomposition of the nitroxide under acidic condition – one can use the protection/deprotection strategy. First, a PtBA-macroinitiator was applied in the nitroxide-mediated polymerization of St and St-CC-TMS leading to poly(tBA-b-[St-co-StCCTMS]) (Mn = 32000 g mol−1, PDI = 1.2–1.3). However, deprotection of the PtBA block to PAA leads to a significant loss of alkyne functionality even of the protected one. Therefore, tetrahydropyran acrylate (THPA) was used which can be deprotected under milder conditions, but this compound was not stable under the temperature required for NMP. The P(THPA) macroinitiator could be polymerized via RAFT at 70 °C and was used in the copolymerization of styrene and 4-(trimethylsilylethynyl)styrene. Following the deprotection amphiphilic block copolymers PAA-b-[PS-co-PSCCH] (Mn = 16000 g mol−1, PDI = 1.2) were obtained that are capable to form micelles with a clickable hydrophobic core.194,203
In the discussed cases protected St-CCH was incorporated up to 20% in a statistical copolymerization with styrene. This might be sufficient for the specific attachment of functional groups by clicking, but for the tailoring of macroscopic properties homopolymers with pendant alkyne groups seem to be more promising. Voit and coworkers showed that by increasing the amount of protected 4-ethynylstyrene the control of nitroxide-mediated polymerization with PhEt-TIPNO as unimolecular initiator is lost indicated by polydispersity indices around 1.9 and large differences of the calculated molar masses to the observed ones for the homopolymer of poly(St-CC-TMS).195 The authors assigned the loss of control to a shift of the active-dormant species equilibrium towards the active side caused by sterical hindrance and also to the recombination of the nitroxide and the TMS group of the propagating 4-(trimethylsilylethynyl) styrene radical. This assumption is supported by the fact that a controlled polymerization is obtained if an excess of free nitroxide is added to the polymerization medium. This shifts the equilibrium back to the dormant side and results in the synthesis of well-defined poly(St-CC-TMS) (Mn = 3 500 g mol−1, PDI = 1.2).195
To circumvent the sterical hindrance during the polymerization as well as to provide enhanced accessibility to the alkyne for the postmodification, 4-(3′-trimethylsilyl-ethynylmethoxy)styrene (St-OMe-CCTMS) containing a methoxy group as a flexible spacer was investigated (Entry 45). The synthesis of the monomer was performed by a substitution reaction of 4-hydroxystyrene with propargyl bromide and subsequent protection with TMS. The homopolymerization proceeded in a controlled manner without the necessity of any free nitroxide (poly(St-OMe-CCTMS): Mn = 6000 g mol−1, PDI = 1.2–1.3). Besides, the TMS group is labile under basic and acidic conditions prohibiting the use of acetic acid as a polymerization enhancer. Moreover, the TMS group is thermally labile and a partial loss of the protecting group was observed for the reaction at 120 °C. Unfortunately, the more stable t-butyldimethyl-silyl (TBDMS) or triisopropylsilyl (TIPS) protected monomers could not be synthesized. With St-OMe-CCTMS as monomer in hand the following diblock copolymers were synthesized: poly(tBOSt-b-St-OMe-CCH) (Mn = 52000 g mol−1, PDI = 1.2), poly(AcOSt-b-St-OMe-CCH) (Mn = 21000 g mol−1, PDI = 1.4) as well as poly(pHSt-b-St-OMe-CCH) (Mn = 16000 g mol−1, PDI = 1.2).195,201
Not only nitroxide-mediated polymerization but also the RAFT polymerization technique was used for the preparation of diblock copolymers poly(St-b-St-OMe-CCH) with a trithiocarbonate RAFT agent.201 Although alkyne-functionalized ATRP initiators are widely used for the polymerization of styrene, no alkyne-functionalized styrene derivative was polymerized via ATRP so far. A reason for that might be the higher polymerization temperature for the ATRP of styrene compared to other monomer classes promoting side reactions. It can be noted that the partial loss of the trimethylsilyl-protecting group at elevated temperatures leads to a higher amount of terminal acetylene compared to the initiator approach, which might cause a significant loss of control due to interference with the copper catalyst.
The unprotected propargyl acrylate depicted in Entry 46 was copolymerized with acrylic acid at 60 °C by Caruso et al. using the RAFT method with a trithiocarbonate CTA resulting in a broad molar mass distribution (Mw = 86000 g mol−1 and a PDI value of 2.2).205 In addition, a similar copolymer was prepared using a trithiocarbonate CTA yielding a copolymer with a molar mass of Mn = 53000 g mol−1 and a polydispersity index of 1.9. The broad mass distribution is attributed to branching of the unprotected alkyne-functionalized monomer.206
Furthermore, TMS-protected propargyl acrylate was randomly copolymerized with tBA using PhEt-TIPNO as initiator for NMP by Malkoch and coworkers, but no detailed discussion was provided regarding the obtained molar masses and polydispersity of the isolated polymers.193
000 g mol−1 are achievable.153 The polymerizations were carried out for 3 to 16 h with protection of the terminal alkyne group resulting in well-defined polymers (PDI < 1.3). Also some block and random copolymers were prepared with different comonomers (MMA, OEGMA).208 A kinetic study for propargyl methacrylate (Entry 47), including semilogarithmic kinetic plots of the RAFT polymerization of the propargyl methacrylate and the silyl-protected monomer was reported by Barner-Kowollik and coworkers. The authors demonstrated that the protected monomer polymerizes at a much lower rate than the nonprotected monomer.153,191 Another RAFT copolymerization of the unprotected methacrylate (Entry 47) and MMA or GMA showed that the PDI values increase (PDI = 1.6–2.0, Mn = 15000–25000 g mol−1) and that under these polymerization conditions side reactions such as transfer and insertion reactions occur.199
TMS-protected propargyl methacrylate was also polymerized by ATRP. The first contribution was reported by Haddleton and coworkers using CuBr/N-ethyl-2-pyridylmethanimine as catalytic system. Kinetic studies indicated a living process and SEC measurements of the resulting polymers revealed a good control over the polymerization (PDI < 1.3).3 This monomer is also used for the synthesis of block copolymers with poly(ε-caprolactone) as macroinitiator. A block copolymer (Mn = 14 100 g mol−1, PDI = 1.2) was obtained after the deprotection with TBAF and followed by the alkyne-azide cycloaddition leading to a functional graft copolymer.192
Drockenmuller et al. reported an in situ approach of ATRP polymerization and copper-catalyzed azide-alkyne click reaction using propagyl methacrylate as clickable monomer. The resulting functionalized copolymers revealed a broader molar mass distribution (PDI = 1.3–2.1) due to the use of unprotected alkyne.209
900–19600 g mol−1, PDI = 1.2–1.3).95
Additionally, the protected acrylamide was copolymerized with (N,N-dimethyl)acrylamide and TMS-protected 2-(hydroxyethyl)methacrylate via NMP using PhEt-TIPNO as initiator to prepare water-soluble random terpolymers.193
Acrylonitrile was polymerized via ATRP using 2-bromopropionitrile as initiator and CuBr/bpy as catalytic system by Du Prez and Matyjaszewski. The initiator contains the monomer group as initiating fragment that posses equal radical reactivity as the monomer itself providing fast initiation.48 Thereby, poly(acrylonitrile) PAN (Mn = 40000 g mol−1, PDI = 1.1) as well as poly(AN-b-St) (PDI = 1.1) and poly(AN-r-St) (Mn = 8500 g mol−1, PDI = 1.1) were prepared. For the block copolymer, first a PAN-macroinitiator was prepared followed by the polymerization of styrene as the second block to ensure high initiating rates and, hence, a narrow molar mass distribution.48 In addition, acrylonitrile was polymerized via NMP using TEMPO/dibenzoylperoxide (BPO) as bimolecular initiator for the polymerization of the random copolymer poly(St-r-AN) (Mn = 10000 g mol−1, PDI = 1.3–1.4) and the diblock polymer poly(St-b-[St-r-AN]) (Mn = 87000 g mol−1, PDI = 1.2) that was initiated with a PS-macroinitiator.210 Thereby the control increases with the content of acrylonitrile in the acrylonitrile/styrene feed.
Regarding possible postmodifications, it should be noted that the pendant nitrile group in polymeric materials is up to now efficiently modified only to the corresponding tetrazole ring using the reaction with sodium azide and zinc chloride as catalyst in DMF 120 °C for 40–50 h.48,210 Herewith, the nitrile-azide cycloaddition is used to modify macroscopic properties rather than to place functional groups or to attach polymeric side chains.
The reason for this limitation lies within the nature of the ring formation of the tetrazoles: To allow an efficient ring formation under mild conditions, the azide should not be sterically hindered (which limits the use of polymeric azides), while the nitrile group has to be electron-poor (e.g. tosylnitrile), which is not sufficiently fulfilled for the nitrile group along the backbone of (poly)acrylonitrile.49,211,212
However, with the efficient modification to the corresponding tetrazole ring the macroscopic properties of the prepared diblock copolymers changes: (i) solubility and swellability in protic solvents increase, (ii) the morphology changes, since the tetrazole formation increases the incompatibility between the blocks210 and (iii) the temperature stability for the tetrazole-modified material significantly decreases (by 60–120 °C) compared to the nitrile-based polymer.48
000–12000 g mol−1, PDI = 1.1–1.5).213
Alkyne-functionalized maleimide (Entry 51) will be discussed in Section 6.1.2.7.
5.2 Azide-containing monomers
| Entry | Structure | Click | Strategy | CRP | Initiator/CTA (Abbr./Entry) | Comonomers | Ref |
|---|---|---|---|---|---|---|---|
| 52 |
|
CuAAC | simult | ATREP | 5 | St | 83 |
| 53 |
|
CuAAC | poC | ATRP | EBiB, TosCl, | MMA, DMAEMA, tBMA | 77,90,209,220,221 |
| RAFT | CDB, CBDN, CPADB | MMA | 108,207,219,221 | ||||
| 54 |
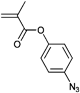
|
CuAAC | poC | RAFT | BICDT, CPDB | MMA, MA, St | 217 |
| 55 |
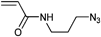
|
CuAAC | poC | ATRP | 3 | NIPAM | 95 |
| RAFT | CPADB | NIPAM, DMA | 218 |
| Entry | Structure | Click | Strategy | CRP | Initiator/CTA | Comonomers | Ref |
|---|---|---|---|---|---|---|---|
| 56 |
|
DA | poC | RAFT | BDAT | St | 228 |
| 57 |
|
DA | poC | ATRP | EBiB, BBiBE | MMA, EHA | 57,229–232 |
| 58 |
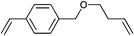
|
Thiol ene | poC | RAFT | MCPMDB | St | 127 |
| 59 |
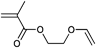
|
Thiol ene | poC | RAFT | CPADB | -- | 234 |
| 60 |
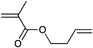
|
Thiol ene | poC | ATRP | EBiB | MMA | 127 |
| 61 |
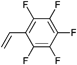
|
Thiol para-fluoro, Amine para-fluoro | poC | NMP | Blocbuilder®, PhEt-TIPNO | St | 31,32 |
| 62 |
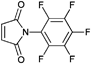
|
Thiol para-fluoro | poC | ATRP | PEB | St | 216 |
| 63 |
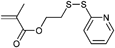
|
PySS | poC | RAFT | CPADB | HPMAM | 235,236 |
| 64 |
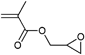
|
RO | poC | NMP | TIPNO | St, MA | 9,60 |
| RO | poC | ATRP | EBiB | tBMA, MMA | 59,114,244 |
Azide-containing monomers based on (meth)acrylates or (meth)acrylamides are prepared by an esterification of 2-azidoethanol or 3-azidopropylamine with a (meth)acryloyl chloride or an activated acid of (meth)acrylic acid.77,90,108,217–220 In addition, the route starting from the 2-hydroxyethyl methacrylate to the corresponding azide via the Mitsunobu reaction is described.221 In the case of azide-functionalized styrene, the alkyne functionality is incorporated via substitution reaction of 4-vinylbenzyl chloride with sodium azide.83
The used temperature represents an important criterion for the controlled polymerization of these monomers. The instability of the azide group at elevated temperatures is described, resulting in (i) the decomposition with evolution of nitrogen to form reactive nitrenes that undergo insertion reactions49,217,222–225 or (ii) cycloaddition with the monomer to form triazolines.49,105,108,226,227 Both side reactions result in the formation of a crosslinked polymer. The higher concentration of azide groups in the polymerization solution (1–2 M or bulk) compared to the initiator approach constrains the polymerization conditions of CRP to lower temperatures to gain sufficient control over the polymerization.77,90,95,108,217,219,220 Nonetheless, there are also two reports published using normal ATRP conditions (65–70 °C) claiming that the polymerization proceeded in a controlled manner.207,218 Regarding the orthogonality of ATRP and azide-containing monomers, it is known for azides to undergo the Staudinger reaction, i.e. reduction to amines, in the presence of phosphines that are typically used as ligand for iron complexes as the “new generation” catalytic systems in ATRP.6,15,49
500 g mol−1, PDI = 1.1).108 Benicewicz and coworkers also reported that the RAFT polymerization of 6-azidohexyl methacrylate showed a linear pseudo-first-order kinetic plot at 30 °C. It was noted that with higher monomer conversion (25%) a high molar mass shoulder was observed by SEC measurements.219 2-Azidoethyl methacrylate (Entry 53) was used for the copolymerization with MMA via ATRP or RAFT. The copolymer prepared by ATRP had a molar mass of Mn = 5400 g mol−1 and a PDI value of 1.2, whereas the copolymer prepared by RAFT had a molar mass of Mn = 7100 g mol−1 and a PDI value of 1.4. These copolymers were used for further functionalization with cyclooctyne derivatives (copper-free clicking).221 The 4-azidophenyl methacrylate (Entry 54) can be copolymerized with different monomers in a controlled manner at room temperature by using a carbodithioate or a dithiobenzoate as initiator. Methyl acrylate, methyl methacrylate and styrene have been used as comonomers. The resulting copolymers were well-defined (PDI < 1.3) and in a molar mass (Mn) range between 3000 and 16000 g mol−1.217
With the azide-functionalized monomer depicted in Entry 53 also a tandem click chemistry/ATRP procedure was applied by Drockenmuller et al. using CuBr/bpy as the catalytic system and TosCl as the initiator. Different copolymers with MMA were prepared (Mn < 22400 g mol−1, PDI = 1.5).209
500–13300 g mol−1, PDI = 1.2).95 In another study, the authors polymerized 3-azidopropylacrylamide by RAFT with DMAM and NIPAM, where 4-cyanopentanoic acid dithiobenzoate was used as CTA and 4,4′-azobis(4-cyanopentanoic acid) as initiator to yield random copolymers (Mn = 14700 g mol−1, PDI = 1.2).218
5.3 Monomers for Diels–Alder reactions
There are only a few examples for clickable monomers (dienophile or diene containing ones) that are suitable for Diels–Alder reactions. The furfuryl methacrylate is commercially available and therefore accessible for the controlled polymerization without further functionalization.000–6000 g mol−1, PDI = 1.2–1.4).228
000 g mol−1, PDI = 1.3) were obtained.57,229–231 Furthermore, FMA was successfully used in the preparation of block copolymers with 2-ethylhexyl acrylate (EHA) and 1,2-bis(bromoisobutyryloxy)ethane (BBiBE) as initiator.232 The ATRP reaction was conducted at 90 °C using CuCl/HMTETA as catalytic system. The obtained PFMA-b-PEHA-b-PFMA (up to Mn = 51000 g mol−1, PDI = 1.3) was crosslinked with a bismaleimide to yield materials with self-healing properties.
5.4 Monomers for thiol-ene clicking
In the last years, the thiol-ene click reaction has attracted significant attention in the field of polymer science. These robust and efficient reactions have enormous advantages for the construction of polymeric structures. Recently, several reviews summarized the power of thiol-ene chemistry.19,28,53,233Since thiol groups readily undergo side reactions under radical polymerization conditions, preferably ene-functionalized monomers were synthesized and used as clickable monomers. (Meth)acrylates containing a terminal double bond are prepared by an esterification of an alcohol (e.g. ethylene glycol vinyl ether or 3-butene-1-ol) with an acid chloride or an anhydride of (meth)acrylate acid.127,234 Styrenes containing a terminal double bond are synthesized by substitution of a chloride group (e.g. from 4-vinylbenzyl chloride, using 3-butene-1-ol).127 The controlled polymerization of this class of monomers is easily possible due to the rather low reactivity of the unconjugated alkene group. Thus, no or less crosslinking occurs during the polymerization.127
000 g mol−1 and a PDI value of 1.1.127
000 g mol−1, PDI = 1.2).127
5.5 Monomers for para-fluoro substitution
000 g mol−1, PDI = 1.1) and diblock copolymers poly(PFS-b-PS) (Mn = 17000 g mol−1, PDI = 1.2) could be prepared, where either a PFS- or a PS-macroinitiator was used.32 Terpyridine-functionalized PhEt-TIPNO alkoxyamine as unimolecular initiator was also used for the NMP of PFS at 120 °C to yield poly(PFS) (Mn = 4500 g mol−1, PDI = 1.1) and poly(PFS-b-PS) (Mn = 10000 g mol−1, PDI = 1.2).31
The pentafluorophenyl-functionalized maleimide (Entry 62) will be discussed in Section 6.1.2.7.
5.6 Monomers for pyridyl disulfide exchange
In entry 63 a monomer is shown, where the pyridyl disulfide moiety is linked to methacrylate. The PySS group can be exchanged in a postmodification or premodification step by thiol functional compounds under mild reaction conditions, because of the facile leaving character of the 2-pyridinethione. The release of 2-pyridinethione allows the monitoring of the PySS exchange by UV/vis spectroscopy. This procedure is often used for the preparation of polymer bioconjugates (e.g. with oligopeptide) or for anticancer drugs, such as doxorubicin linked via the free thiol group.235,236The monomer was used in RAFT polymerization with CPADB as CTA. These polymers were used as macro CTAs for the preparation of block copolymer of HPMAM resulting in different block segments with different molar mass (Mn = 13400–49000 g mol−1, PDI = 1.2–1.3). These block copolymers were further crosslinked as micelles.235
Moreover, homopolymers of the PySS monomer were prepared by using the RAFT method by Bulmus et al. Different semilogarithmic plots are examined for this monomer indicating the controlled character of the polymerization. Numerous homopolymers were synthesized as basis for further biofunctionalization (Mn = 8000–12600 g mol−1, PDI = 1.1–1.4).236
5.7 Monomers for ring-opening reactions
Ring-opening reaction of strained heterocycles are considered as a click reaction based on the spring-loaded character towards nucleophiles by Sharpless and coworkers.13 For this purpose, glycidyl methacrylate (GMA) is a commercially available monomer, and thus can be used as a clickable monomer without further modification (Entry 64). CRP of GMA has been well studied by NMP,9 RAFT237 and ATRP.238–243 For ATRP of GMA it is important to note that the epoxide might react with free ligand, which leads to unwanted branching. Thus, the catalyst has to be preformed before the addition of the monomer.59 Moreover, any strong nucleophile should be avoided during polymerization. Apart from that, the epoxide is stable under the polymerization conditions even at elevated temperatures typically required for NMP.With the “boom” of click chemistry in polymer science, the ring opening of epoxides with nucleophiles as a click-type reaction experienced also a revival in the last few years, but rather to introduce azides or alkynes than as a click reaction itself (Scheme 1, Entry 89). Why modifying towards another clickable functionality although the ring-opening reaction is an efficient click reaction itself? This is caused by the poor selectivity of epoxides. In contrast, the orthogonality is strongly increased for, e.g., the azide-alkyne click reaction allowing the orthogonal functionalization in one-pot, simultaneous or cascade reactions.193
Various polymers containing glycidyl methacrylate were synthesized as basis for further click reactions: Poly(St-r-(CC-CH2-St)-r-GMA) was prepared via NMP with TIPNO as mediating nitroxide (Mn = 30000 g mol−1, PDI = 1.3).60 By ATRP the homopolymer as well as the random copolymers were synthesized (Entry 64): poly(GMA) with Mn = 27000 g mol−1 and PDI = 1.3,114 poly(GMA-r-tBMA) with Mn = 8000 g mol−1 and PDI = 1.2244 as well as poly(GMA-r-MMA) with Mn = 20000 g mol−1 and PDI = 1.2 to 1.5,59 whereby the polydispersity index for the latter increases by increasing the fraction of GMA used in the feed.
5.8 Clicked monomers
Clicked monomers are formed by the azide–alkyne 1,3-dipolar cycloaddition and contain the triazole ring which is linked to the polymerizable vinyl group. There are only few examples of clicked monomers. Hawker et al. have described the RAFT polymerization of a series of 4-vinyl-1,2,3-triazoles (Entry 50). Different (co)polymers were prepared in a controlled manner using a dithioester.67 Sumerlin et al. used propargyl acrylate (Entry 46) that was clicked onto an azide-containing trithiocarbonate (Entry 29) for the RAFT polymerization of branched poly(N-isopropylacrylamide) (PDI = 1.5–2.1).156 Also the chain extension of this macro-CTA with DMA was performed. 1-(3′-Aminopropyl)-4-acrylamido-1,2,3-triazole hydrochloride (Entry 48) was polymerized by the RAFT using a poly(NIPAM) macro-RAFT agent resulting in block copolymers (Mn = 19600 g mol−1, PDI = 1.2).215 Propargyl methacrylate (Entry 47) was clicked by a Cu-catalyzed 1,3-dipolar cycloaddition with 3-azido-7-diethylaminochromen-2-one and subsequently copolymerized with MMA by RAFT using 2-cyanoprop-2-yl dithiobenzoate (CPDB) as chain transfer agent (Mn = 10200 g mol−1, PDI = 1.2).214
6. Postmodification
Postmodification reactions are used to transform latent functional groups into clickable units, whereby the polymer can be modified either at pendant or at terminal positions (Scheme 5). For the terminal modification the initiating side, the mediating side or both can be modified. An overview of the end-group modification approaches are depicted in Tables 11 and 12 and the side-group modifications are shown in Table 13, which will be discussed in the following.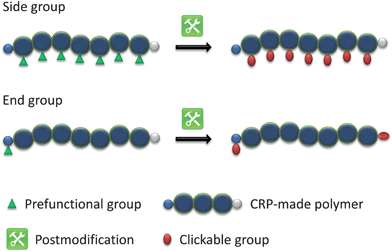 | ||
| Scheme 5 Schematic representation of the postmodification strategies. | ||
| Entry | Modification | Click | CRP | Type | Monomer | Ref |
|---|---|---|---|---|---|---|
| 65 |

|
CuAAC | ATRP | Nucleophilic substitution | St, tBA, MA, OEGA, iBoA, MMA, GMA, OEGMA | 59,78–80,90,97,98,102,114,251–261 |
| 66 |
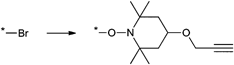
|
CuAAC | ATRP | Nitroxide radical coupling | St | 263 |
| 67 |
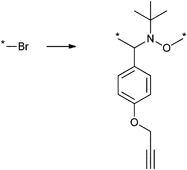
|
CuAAC | ATRP | Nitrone-mediated radical coupling | iBoA | 185 |
| 68 |

|
DA | ATRP | Nucleophilic substitution | St, MMA, MA, iBoA | 39,266 |
| 69 |

|
Thiol–ene | RAFT | Aminolysis | NIPAM, OPA, EA, HEA | 116,245,267,270,275–278 |
| Thiol–yne | RAFT | Aminolysis | St | 141 | ||
| MAdd | RAFT | Aminolysis | NIPAM | 116,273 | ||
| 70 |
|
Thiol–ene | RAFT | Aminolysis | DEAEMA, BA, NIPAM, MMA, HPMAM, OEGA | 50,267–271 |
| Thiol–yne | RAFT | Aminolysis | NIPAM | 50 | ||
| Thiol-Isocyanate | RAFT | Aminolysis | DEAEMA | 54 | ||
| MAdd | RAFT | Aminolysis | DEAEMA, NIPAM, MMA | 50,233,271,272,274 | ||
| 71 |
|
CuAAC | RAFT | Aminolysis, Substitution | MMA, DEGMA, LMA, St, NIPAM | 280 |
| 72 |
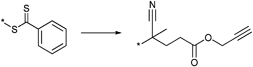
|
CuAAC | RAFT | Radical exchange | St | 143 |
| 73 |
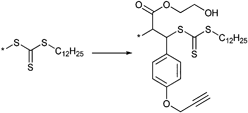
|
CuAAC | RAFT | Radical insertion | St, MA | 281 |
| Entry | Modification | Click | CRP | Type | Monomer | Ref |
|---|---|---|---|---|---|---|
| 74 |
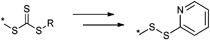
|
PySS | RAFT | Aminolysis, Substitution | NIPAM | 267 |
| 75 |
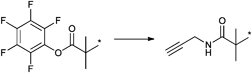
|
CuAAC | RAFT | Ester cleavage | OPA | 245 |
| 76 |

|
CuAAC | NMP | Oxidative cleavage | St | 284 |
| 77 |
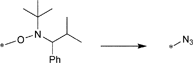
|
CuAAC | NMP | Radical exchange | St | 247 |
| 78 |
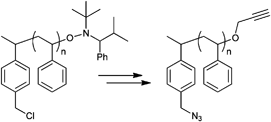
|
CuAAC | NMP | i) Substitution | St | 247 |
| ii) Oxidative cleavage | ||||||
| 79 |
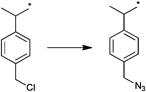
|
CuAAC | NMP | Substitution | St, tBA | 134,176,247 |
| 80 |
|
CuAAC | NMP | Substitution | St | 71,72 |
| ROP | ε-CL | |||||
| ATRP | tBA | |||||
| 81 |
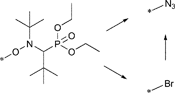
|
CuAAC | NMP | Radical exchange, Substitution | 285 |
| Entry | Modification | Click | CRP | Type | Comonomers | Initiator | Ref |
|---|---|---|---|---|---|---|---|
| 82 |
|
CuAAC | ATRP | Quaternization | DMAEMA, DEGMA, DEAEMA | EBiB | 288 |
| 83 |
|
CuAAC | ATRP | Esterification | — | EBiB | 289 |
| 84 |
|
CuAAC | NMP | i) Hydrolysis | St | PhEt-TIPNO | 291 |
| ii) Substitution | |||||||
| 85 |
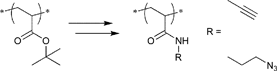
|
CuAAC | NMP | i) Acidolysis | St | PhEt-TIPNO | 292,295 |
| ii) Amidation | |||||||
| 86 |
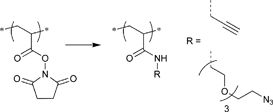
|
CuAAC | NMP | Amidation | St, tBA, AM | PhEt-TIPNO | 193 |
| 87 |
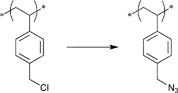
|
CuAAC | NMP | Substitution | St, tBA | PhEt-TEMPO | 117,292–294 |
| 88 |
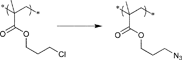
|
CuAAC | RAFT | Substitution | AA | BPIT, PEDT | 205,206 |
| 89 |
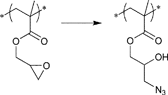
|
CuAAC | ATRP | Ring opening | MMA, tBMA | EBiB | 59,104,114,244,290 |
| 90 |
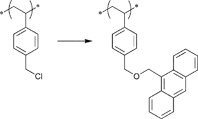
|
DA | NMP | Etherification | St | PhEt-TEMPO | 117,120 |
6.1 End-group modification
Linear polymers prepared by controlled radical processes contain two functional end groups, one on the initiating end and the other one on the mediating end. Therefore, a polymer can be further functionalized on the α- and/or the ω-terminus. In particularly for the ω-terminus, unavoidable radical termination reactions during the polymerization such as recombination as well as disproportionation occur and, thus, parts of its functionality are lost to a certain amount. Assuming an efficient postmodification reaction, these side reactions for the prefunctional polymer can be considered as the bottleneck for the degree of functionalization that can be reached for ω-functionalized polymers via the postmodification strategy.6.1.1.1 RAFT – Modification towards alkyne functionality. An α-endgroup modification of the initiating group was reported by Bertozzi et al., where a pentafluorophenyl ester as a labile ester group (Entry 75) was attached to the initiating fragment. After polymerization the labile ester was cleaved with diisopropylethylamine in the presence of propargyl amine resulting in an α-terminated alkyne polymer. Apparently, the trithiocarbonate was not attacked by the amine and, therefore, a defined polymer could be obtained.245
6.1.1.2 NMP – Modification towards azide functionality. Despite the versatility of functional groups that can be introduced as an initiating fragment on the α-functionalized polymers prepared by NMP with functional alkoxyamines,175 there is only one synthetic route reported for the transformation into a clickable moiety: The modification of the benzylic chloro group into an azide via a nucleophilic substitution with sodium azide (Entry 78, 79).
Initially, Hawker et al. described the postmodification from chloride into azide α-functionalized polystyrene in 2004.246 Thereby, the sodium azide was activated by the addition of catalytic amounts of 18-crown-6 ether while an excess of sodium azide was used. This postmodification was also utilized by Voit et al. using acetone as solvent and three equivalence of sodium azide at room temperature to achieve full conversion after several hours.176 In 2008, Braslau et al. obtained azide-functionalized polystyrene by the reaction of the chloride counterpart in DMF with sodium azide (3 eq.) at 50 °C in the absence of crown ethers to almost full conversion.247 Furthermore, the transformation was applied by O'Reilly et al. for the preparation of azido-functionalized poly(acrylic acid-b-styrene) (N3CH2-PhEt-PAA-b-PS) as clickable amphiphilic diblock copolymers. The postmodification of the benzylic chloro group attached to the PAA block was conducted in water at room temperature using a 5-fold excess of sodium azide to yield clickable micelles in water with azide groups on the outer shell.134
6.1.2.1 ATRP – Modification towards azide functionality. The most prominent postmodification reaction towards a clickable functionality of ATRP-prepared polymers is the substitution of halides such as bromide against azide, since the halide is inherently present at the ω-chain end after the ATRP process (Entry 65). The reaction is very efficient with sodium azide in DMF at room temperature, allowing for a specific transformation in the presence of distinct labile groups. In this context, it is important to note that Matyjaszewski and coworkers developed the method for this nucleophilic substitution in 1997/1998 for polystyrenes and polyacrylates to allow an easy access to amines over the azide via a reduction248 or via the Staudinger process.249 The authors also studied the rate constant for the substitution of selected model halogen compounds with sodium azide.250
Up to now a lot of different polymers were postmodified via this azidation method. Poly(styrenes), poly(acrylates), poly(methacrylates) and their copolymers at different molar masses have been reported.59,78–80,90,97,98,102,114,251–261 It should be noted that the azidation of poly(methacrylates) is significantly slower compared to poly(acrylates) and poly(styrenes) due to the tertiary bromine.262 The modification can be used, e.g., in combination with multifunctional initiators bearing functional termini for NMP or ROP (Entry 80). The multifunctional initiator was used in NMP of styrene, ROP of ε-caprolactone as well as ATRP of tBA followed by the quantitative substitution of the bromine against azide, while the molar masses and the polydispersity indices retained.71,72
6.1.2.2 ATRP – Modification towards alkyne functionality. Only a few examples are described for polymers prepared by ATRP dealing with the exchange of the bromine end group against alkyne-functionalized groups.185,263 Low molar masses and conversions are targeted by preparing the polymers to assure a high end-group fidelity of the ω-terminating bromide.
Monteiro and coworkers used the high reactivity of PS towards cleavage of the bromine end group in the presence of Cu(I)Br, Me6TREN and DMSO promoting a single electron transfer (SET) via the formation of nascent Cu(0).263 The emerging carbon-centered radical was trapped by an alkyne-functionalized nitroxide (2,2,6,6-tetramethyl-4-(prop-2-ynyloxy)piperidin-1-yloxyl)264 to result in alkyne-terminated PS with near quantitative yields within 10 min at room temperature (Entry 66). A unique feature of the nitroxide radical coupling (NRC) as a selective and highly efficient reaction is the reversible formation of the C–O bond of the alkoxyamine that undergoes homolytical cleavage upon heating. In this fashion, the coupled nitroxide can be substituted by an excess of other functional nitroxides (10-fold excess) at elevated temperatures.
Recently, Barner-Kowollik and coworkers established a new approach towards midchain-functionalized polymers by using nitrones in a dual radical capturing process.185,265 Nitrones can rapidly react with carbon-centered radicals to form nitroxides that further trap radicals to form alkoxyamines. As such, nitrones can be used for efficient polymer conjugation by mediating the radical coupling reactions of macroradicals. The authors activated ATRP-made PiBoA with Cu(0)/PMDETA in toluene at 60 °C in the presence of an alkyne-functionalized nitrone (2-fold excess), while the alkyne was protected with TMS.185 In this vein, midchain alkyne-functionalized polymers as depicted in Entry 67 can be prepared in high yields as indicated by 1H NMR spectroscopy (∼90%). The midchain functional PiBoA was clicked after deprotection with either PS-N3 or PiBoA-N3 to form 3-arm star polymers.
As an additional feature, the prepared polymers bear an alkoxyamine functionality in the backbone that could in principle be used as a macroinitiator in a nitroxide-mediated polymerization for the insertion of another polymer block towards triblock copolymers. Hence, the present approach can also be considered as a preparation of clickable or clicked macroinitiators for NMP (Entry 40; Section 4.3).
6.1.2.3 ATRP – Modification toward diene functionality for Diels–Alder reactions. In Entry 68 a nucleophilic substitution of a bromo-terminated polymer with cyclopentadienyl as reported by Barner-Kowollik is described. This strategy allows the direct access to dienes which can be used for catalyst-free click reactions, i.e. Diels–Alder reaction. The reaction was performed with sodium cyclopentadienide (NaCp) or nickelocene (NiCp2) as the substituting agent at ambient temperature. It should be mentioned that side reactions can occur during the substitution with the more reactive NaCp, in particular for PMMA-Br, PiBoA-Br and PMA-Br, but not for PS-Br. It could be shown that the use of NiCp2 using tributylphosphine and sodium iodide as promoters eliminates these side reactions. In this manner, cyclopentadienyl-functionalized polymers of PMMA, PiBoA, PMA and PS are reported (PDI < 1.3).39,266
6.1.2.4 RAFT – Modification towards thiol functionality. In recent years, an alternative click strategy has been established in polymer research for the design of complex macromolecular architectures. This strategy is based on the special chemical nature of thiol compounds that can be used for radical coupling processes in thiol-ene and thiol-yne reactions, as well as for nucleophilic addition reactions such as thiol-isocyanate addition or Michael addition. The radical-mediated addition of a thiol to an yne is a “sister” reaction to the radical thiol-ene reaction, whereby two thiols can be added (two-step process). All these reactions can be performed under mild conditions.
The ω-end of the polymer chains prepared by the RAFT polymerization can be easily modified to generate a reactive thiol group. In most of the reports a trithiocarbonate (Entry 69) or a dithiobenzoate (Entry 70) have been used as chain transfer agents for RAFT. These group can be easily modified into thiol groups. There are two synthetic pathways towards thiols: (i) Aminolysis with a primary amine50,54,141,245,267–275 and (ii) reduction with NaBH4.276–278 Care should be taken in this postmodification to prevent the coupling of two chains to form a disulfide bridge, which are often visible by a shoulder at lower elution volumes in the SEC measurements. However, coupling of thiols can be easily reversed by addition of a reducing agent, e.g. phosphine derivatives.50,54,116,268,271,273,276 Limitations of the thiol-ene reaction for polymer-polymer conjugation were recently described by Du Prez et al.53
In some cases, further purification of the ω-thiol functional polymers is necessary and dialysis or precipitation are the preferred techniques. A calorimetric method for the determination of the free thiol concentration was developed by Ellman.279 Ellman's reagent converts a thiol into a 5,5′-dithiobis(2-nitrobenzoic acid) derivative, which has a strong absorption and therefore the degree of functionalization can be determined.267,269,270,273,275 Alternatively, this can be indirectly estimated by clicking a fluorescence dye (e.g. pyrene).274,276 The degree of functionalization can be varied from 60% up to 99.5%. Often short polymer chains are used for the modification to provide high end-group fidelity on the mediating side (Mn < 10000 g mol−1). Different polymers were used for the modification towards thiols, i.e. acrylate-, methacrylate- and acrylamide derivatives.
An elegant example for the orthogonality of the Michael addition as nucleophilic thiol-ene reaction to the radical thiol-ene was recently described by Lowe et al.50 By using the fact that the nucleophilic reaction of the thiols is selective for double bonds conjugated with electron-withdrawing groups (e.g. α,β-unsaturated carbonyl compounds), a consecutive reaction of a heterofunctional polymer with different thiols, first reacted via Michael addition and followed by the radical thiol-ene or thiol-yne reaction could be shown. The reverse case leads to the loss of orthogonality due to the unselective nature of the radical coupling.
6.1.2.5 RAFT – Modification towards alkyne functionality. An elegant approach to generate ω-end group functionalized triple bonds, which can be used for the Cu(I)-catalyzed cycloaddition, was reported by Theato and coworkers using butynyl methane thiosulfonate (Entry 71). Five different acetylene-terminated polymers (MMA, DEGMA, LMA, St, NIPAM) could be clicked to an azide.280 Another approach towards alkyne-terminated polymer chains is the cleavage of the RAFT agent by a radical process (Entry 72).143 Thereby, an excess of alkyne-modified initiator is used, which decomposes while generating radicals that react with the C = S of the thiocarbonylthio moiety in the polymer chain. Higher temperatures (80 °C) and a large excess of the initiator are necessary and it should be taken into account that side reactions during end-group modification can occur by means of recombination during insertion.143 ABCD 4-miktoarm star polymers could be prepared by RAFT polymerization of styrene followed by an insertion reaction applying a large excess of 2-hydroxyethyl-3-(4-(prop-2-ynyloxy)phenyl) acrylate to stop the polymerization at 110 °C. Hence, a unprotected triple bond attached at the end of the polymer chain could be obtained (Entry 73). The reaction is not very efficient, because a large amount of unreacted macroinitiator is present after the diblock copolymerization.281
6.1.2.6 RAFT – Modification towards pyridyl disulfide functionality. Chemical modification of the ω-endgroup by aminolysis in the presence of 2,2′-dithiodipyridine was reported to generate a pyridyl-disulfide-terminated polymer chain (Entry 74). The pyridyl disulfide end groups allowed straightforward conjugation with oligonucleotides and peptides.267
6.1.2.7 NMP – Conventional modification and possibilities. Only a few examples are described dealing with the exchange of the nitroxide moiety on the polymer. The first study was conducted by Rizzardo, reducing the TEMPO to a hydroxy group with a mixture of zinc/acetic acid.282 A radical-cleavage approach towards more versatile functional end groups was presented by Hawker and coworkers. Thereby, the propagating radical was trapped at the polymerization temperature by N-functionalized maleimides as non-self-polymerizing monomers, while the TIPNO nitroxide decomposed at elevated temperatures resulting in the maleimide-functionalized polymer.283 The transformation yield for polystyrene, polyisoprene and poly(n-butylacrylate) was typically 90 to 95%, while no changes in molar masses and the polydispersity indices were observed.
This approach offers the possibility to introduce clickable units at the ω-terminus by using functionalized maleimides. Following this strategy, Lutz and coworkers synthesized a library of functional maleimides and used them in the copolymerization with styrene via ATRP, while the maleimide was incorporated at a specific place in the polymer chain.216 Among those, N-pentafluorophenyl maleimide or N-propargyl maleimide could be incorporated into the polymer, while the latter one had to be protected due to pronounced side reactions (Entry 51, 62). In principle, clickable ω-functionalized polymers should be accessible using these monomers in the radical-cleavage procedure by Hawker as well.
6.1.2.8 NMP – Modification towards alkyne functionality. In contrast to the thermal cleavage of the alkoxyamine-terminated polymer, oxidative cleavage under much milder thermal conditions can be achieved by single electron oxidation with ceric ammonium nitrate (CAN).247,284 After the oxidation the alkoxamine cleaves heterolytically into a nitroxide and the cation-terminated polymer that can be trapped by nucleophiles. Braslau and coworkers treated TIPNO-terminated polystyrene with CAN and propargyl alcohol as nucleophile at room temperature under anhydrous conditions to obtain alkyne-functionalized PS (Entry 76) with an end-group fidelity of 65% as determined by UV-vis experiments.247 Investigating PhEt-TEMPO as a model compound for TEMPO-terminated polymers, a heterolytic cleavage similar to the TIPNO counterpart was obtained. It could be shown that this method can also be used for TEMPO-terminated polymers. In contrast, polyacrylates terminated with TIPNO or TEMPO also undergo oxidative cleavage, but the cation-terminated polymer interferes with CAN by forming nitrate ester and prohibit further attachment of functional groups via the addition of nucleophiles.284
The modification of polystyrene that was prepared by NMP with commercially available β-phosphonylated alkoxyamine BlocBuilder® cannot be performed via the described oxidative cleavage with CAN due to the electronic and steric nature of SG1.285
6.1.2.9 NMP – Modification towards azide functionality. Terminal azide-functionalized polystyrene could be obtained starting from the nitroxide-terminated polymer through the reaction with ethanesulfonyl azide (EtSO2N3) that was introduced by Renaud and Ollivier286 as an azidation method for carbon radicals. Braslau and coworkers showed that polystyrene prepared by NMP reacts in the presence of an excess of EtSO2N3 in N-methyl-2-pyrrolidinone at 120 °C to the azido-functionalized PS (Entry 77), although the azidation was incomplete as judged by labelling experiments with an alkyne-functionalized dye analyzed via UV-vis experiments.247 Using PhEt-TIPNO as a model compound, the treatment with EtSO2N3 led to less than 30% of the desired transformation.247 This was explained by the weak electrophilic character of the styryl radical, since only electron-rich radicals can efficiently add onto EtSO2N3.287 This also explains why polyacrylates could not be modified with this method.
The azidation reaction of nitroxide-terminated polystyrene was further studied by Bertin and coworkers for polystyrenes prepared with the commercial available BlocBuilder®.285 Thereby, a one-step as well as a two-step approach towards the terminal azido-functionalized polymer were performed (Entry 81). In the one-step approach EtSO2N3 was used under optimized reaction conditions using a large excess (50 eq.) at 90 °C. In the two-step approach the alkoxyamine was reacted at 75 °C in a radical exchange reaction with 2-bromoisobutyrate as solvent as well as the bromination agent to obtain the exchange of the nitroxide by bromine. Furthermore, this bromo-functionalized polymer was reacted with sodium azide following the well-known ATRP postmodification procedure at room temperature in DMF. For both approaches the azide-functionalization degree was around 70%.
6.1.3.1 NMP. Since the azide group is stable against the mild conditions of the oxidative exchange of the nitroxide against propargyl alcohol by ceric ammonium nitrate (Section 4.1.1), alkyne-azide-functionalized heterotelechelic polystyrene could be efficiently synthesized in a two step synthesis as demonstrated by Braslau et al. (Entry 78).247 At first the chloro group of the α-functionalized polystyrene prepared by NMP was transformed into the azide followed by the oxidative cleavage reaction with CAN at room temperature and the in situ nucleophilic addition of propargyl alcohol.
6.2 Side-group modification
In contrast to the terminal functionalization, the modification of pendant groups is more influenced by steric or electronic effects, in particular for homopolymers, where every repeating unit has to be modified. Nonetheless, this method represents a convenient alternative route to multiple click-functionalized polymers, if the direct polymerization of these monomers is relatively more difficult and/or side reactions occur during the polymerization.Modification towards alkyne functionality. In Entry 82 the alkyne moiety could be selectively introduced by quaternization of the amine of 2-(dimethylamino)ethyl methacrylate (DMAEMA) with propargyl bromide at room temperature (Menschutkin reaction).288 Due to sterical hindrance, the DMAEMA is more reactive towards quaternization than DEAEMA allowing for the selective modification of a terpolymer containing DEGMA, DMAEMA and DEAEMA.288 The purification of the quaternized copolymer was achieved simply by precipitation. The extent of quaternization of the DMAEMA units was evaluated by 1H NMR spectroscopy to be 35%.
Alkynyl side groups were introduced into polymeric backbones of a linear poly(2-hydroxyethyl methacrylate) by an esterification reaction between the hydroxyl groups and 4-pentynoic acid that was activated by N,N′-dicyclohexylcarbodiimide (Entry 83). The degree of functionalization was estimated by 1H NMR spectroscopy to be close to 100%. No change of the apparent molar masses and PDI values occurred.289
Modification towards azide functionality. An efficient and convenient synthesis route to azides is the ring-opening reaction of epoxides as shown in Entry 89. The oxirane ring is often opened with sodium azide in the presence of ammonium chloride to yield the corresponding 1-hydroxy-2-azido compounds. The reactions can be followed by IR spectroscopy of the characteristic vibration bands of the azide (2104 cm−1) and the epoxide ring (909 cm−1). This click reaction was applied for the preparation of copolymers with multiple azide groups, whereby different methacrylates are used, resulting in defined copolymers.59,104,114,244,290
Modification towards azide functionality. In Entry 88, a nucleophilic substitution of a pendant chloro group against azide was performed for a poly(3-chloropropyl acrylate-co-acrylic acid). The reaction was conducted with sodium azide at 80 °C and the final product was subsequently dialyzed. High molar mass copolymers were obtained: Mn = 86
000 and 135000 g mol−1 with polydispersity indices of 2.2 and 1.4, respectively.205,206
Modification towards either alkyne or azide functionality. A substitution reaction was carried out as pendant postmodification for the random copolymer poly[styrene-r-(4-acetoxystyrene)] (Entry 84).291 The acetyl group was used as a protection for the phenolic hydroxy group that cannot be polymerized directly, since it acts as an radical scavenger. Deprotection was accomplished by using hydrazine monohydrate as a base for hydrolysis at room temperature to yield poly[styrene-r-(4-hydroxystyrene)]. The hydroxy group was further reacted with propargyl bromide under basic conditions in a Williamson ether synthesis to yield the clickable styrenic copolymer while retaining molar mass and polydispersity indices of the protected copolymer.291
Another postmodification strategy towards clickable side groups is the carbodiimide-mediated condensation which was demonstrated for the block copolymer poly(tBA-b-St) by Wooley and Hawker (Entry 85).292 First, the t-butyl group, acting as a protecting group, was cleaved using trifluoroacetic acid at room temperature. The resulting amphiphilic diblock polymer poly(acrylic acid-b-styrene] was assembled into micelles in water and partly functionalized with either 3-azidopropylamine or propargyl amine in a condensation reaction at room temperature using 1-[3′-(dimethylamino)propyl]-3-ethylcarbodiimide methiodide to activate the acid. The residual acid groups were crosslinked to obtain nanoparticles with azides or alkyne moieties on the outer shell.
Another postmodification strategy uses succinimide-functionalized acrylates as active ester for the condensation with amines under mild conditions.193 The active ester is stable under the applied polymerization conditions and can be polymerized in a controlled way, while in contrast to alkyl acrylates no deprotection for further modification is required. Hereon, Malkoch et al. showed the preparation of random copolymers of N-(acryloyloxy)succinimide with either styrene, t-butylacrylate or acrylamide followed by a successful condensation reaction with propargyl amine or 1-amino-11-azido-3,6,9-trioxoundecane at 50 °C (Entry 86). In addition, the amidation reaction does not interfere with the click reaction and can be used in a one-pot cascade reaction or simultaneously.193
A facile approach towards pendant azide-functionalized polymers via postmodification is the reaction of benzylic chloride with sodium azide in a nucleophilic substitution reaction (Entry 87). The reaction is efficient at room temperature in DMF. Following this procedure, a random copolymer consisting of 4-(chloromethyl)styrene and styrene was transformed with sodium azide.293 Furthermore, a random terpolymer consisting of 4-(chloromethyl)styrene, styrene as well as 4′-(anthracene-methyloxymethyl)styrene was efficiently modified to the pendant azido- and anthracene-containing derivative.117 O'Reilly et al. modified the amphiphilic diblock copolymer poly[(acrylic acid)-b-styrene-r-4-chloromethylstyrene] via the described procedure to obtain, after crosslinking, nanoparticles with azide-functionalized cores.292 In 2009, Ting et al. transformed homopolymers of 4-(chloromethyl)styrene as well as random and block copolymers with styrene at 60 °C in a mixture of DMSO/THF to the pendant azide-functionalized polymers.294
Modification towards diene functionality for Diels–Alder reaction. The pendant benzylic chloro group in 4-(chloromethyl)styrene containing polymers were also modified by the etherification reaction with 9-(oxymethyl)anthracene (Entry 90). Thereby, an excess of 9-(oxymethyl)anthracene was activated with sodium hydride to react with poly(styrene-r-4-chloromethylstyrene) to the anthracene-functionalized polymer at room temperature,117 although the conversion only went to completion by using higher temperatures.120
7. Applications of clickable polymers
The concept of click chemistry combined with the concept of controlled radical polymerizations represent an ideal pair for the preparation of tailor-made macromolecular architectures.The striking advantage of this combination can be clearly seen by the variety of clicked architectures that become possible by using clickable polymers as building blocks (Schemes 6 and 7). The schemes should provide an overview over the clickable and clicked architectures, while assigning assorted references. The origin for the large variety of architectures is the overcoming of limitations inherent for other techniques, which allows the design of new architectures prepared in high yields and with a wide range of accessible molar masses and constitutions. In this way, some block combinations become possible, which were not directly polymerizable due to disparate reactivities. An example for such an architecture is the block copolymer of styrene and ethylene glycol (EG) using the 1,3-dipolar cycloaddition of azides and acetylene of the respective end groups.
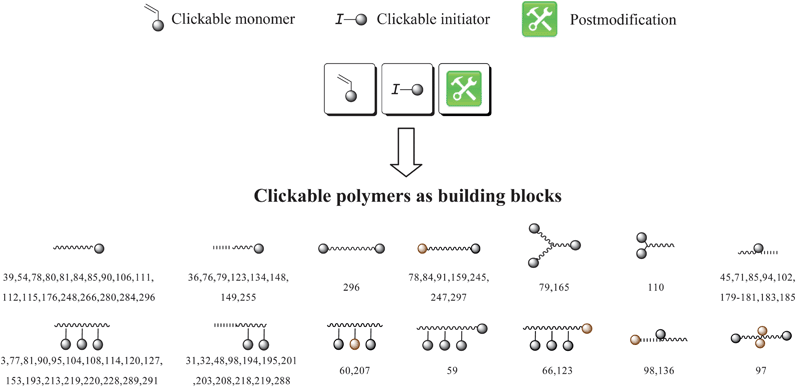 | ||
Scheme 6 Schematic representation of clickable polymers:  = polymer chain; = polymer chain;  , ,  = orthogonal click functionalities, = orthogonal click functionalities,  = block segments. = block segments. | ||
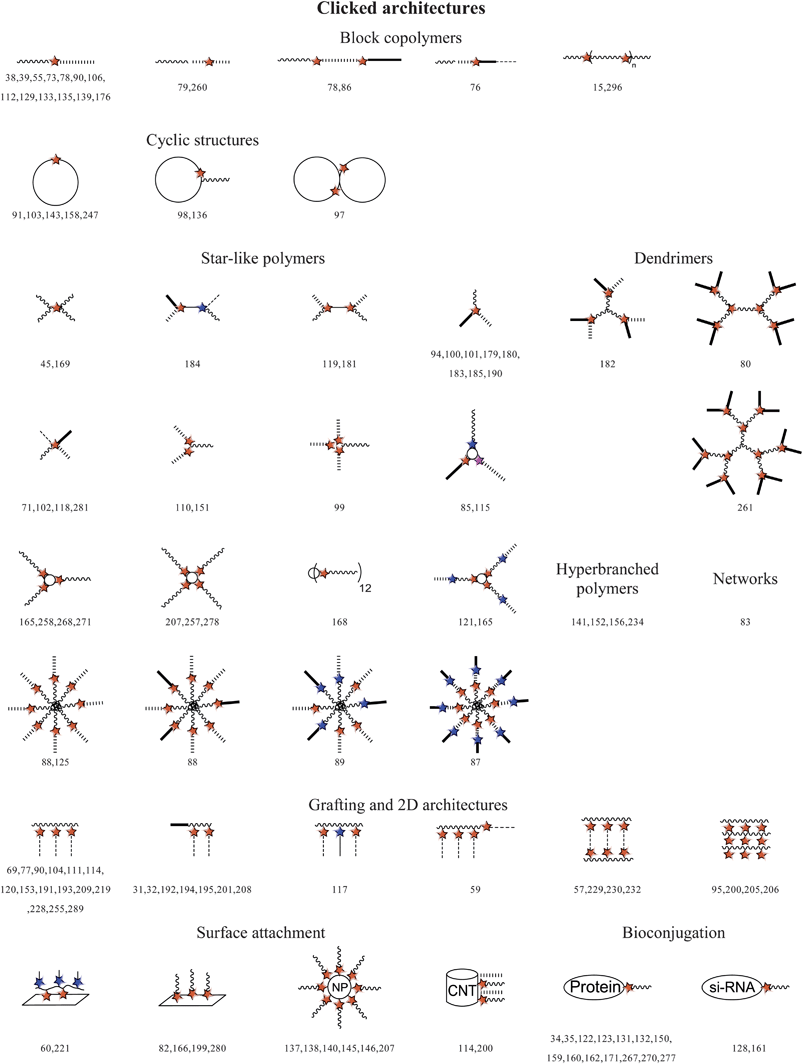 | ||
Scheme 7 Schematic representation of selected clicked architectures: ( = polymer chain; = polymer chain;  = block segments; = block segments;  = crosslinked polymer, = crosslinked polymer,  , ,  , ,  = different clicked functions, = different clicked functions,  = multifunctional core. = multifunctional core. | ||
Utilizing the different approaches (clickable initiator, clickable monomer and postmodification) different types of highly functional polymers are accessible and can act as building blocks for the preparation of more complex structures. Several clickable polymers can be prepared: (i) End functional polymers on one or on both sites, (ii) mid-chain functional polymers, (iii) side-chain functional polymers and (iv) combinations of them (Scheme 6). These clickable polymers act as basic modules for further functionalization reactions to engineer more complex architectures (Scheme 7). Hereon, different linear block copolymers such as di-, ter- or quarterpolymers were prepared. The synthesis of different cyclic polymers such as eight-shaped or tadpole polymers, which are limited for other non-click methods, has been realized using the click concept. These building blocks were also often used for the preparation of star-shaped polymers with 3 up to 12 arms as well as H-shaped or miktoarm star polymers. In this respect, two strategies arise for the synthesis of star-like polymers by clicking: (i) Using mid-chain functional polymers or (ii) end functional polymers onto a multifunctional core. Furthermore, several dendrimers and hyperbranched architectures were also realized by combining CRP and click chemistry. An elegant example represents the thiol-yne reaction for the preparation of hyperbranched polymers.
Another focus of polymer research is the grafting approach of side-chain functional polymers, which can be prepared by using the clickable monomer approach or the postmodification approach of pendant groups. The main advantage for this strategy is the facile tuning of polymer properties for a specific application. One clickable polymer backbone can act as building block for a variety of functionalization reactions (Scheme 6). Moreover, surface patterning is also of interest in material science. Polymers were clicked via CuAAC to different surfaces such as carbon nanotubes or nanoparticles (Si, Au). Also the construction of multilayer systems (layer-by-layer approach) are possible by using the 1,3-dipolar cycloaddition and CRP.
The interest in polymer-functionalized biomaterials strongly increased in the last years due to the facile access by using click chemistry for efficient conjugation and CRPs for tailoring the polymeric architectures. In this vein, different functional polymers were coupled with several proteins and siRNA.
8. Conclusion and outlook
In summary, it is clearly demonstrated in recent years that the combination of controlled/“living” radical polymerization (CRP) techniques and click reactions has become an inevitable route for preparing highly functional tailor-made macromolecules. This combination has been tremendously advanced since the introduction of the concept of click chemistry in 2001 by the cumulative efforts of a large number of research groups all over the globe. These developments on the preparation of new well-defined clickable polymers enabled straightforward access to demanding polymer structures such as cyclic and miktoarm star polymers.Two combinations of CRP with click chemistry seem to be the perfect match: ATRP in combination with azide cycloaddition and RAFT with thio-click chemistry. Halogen-terminated polymers are directly obtained by ATRP and can be transformed to azide-terminated polymers using a simple azidation procedure. As a limitation, this combination may not be suitable for the preparation of clickable polymers of high molar masses, since a high end-group fidelity of the halogen-terminated polymers is only assured for low molar masses and low conversions. Even the most efficient postmodification reaction will not overcome this inherent problem. Nevertheless, ATRP and azide-alkyne click reactions are one of the most prominent combinations to prepare functional materials. Alternatively, thiol-terminated polymers are easily accessible from polymers prepared using the RAFT technique by reduction of the CTA end group. It has been demonstrated that these thiol-terminated polymers can be clicked not only to unsaturated double bonds but also to alkynes, bromo, and para-fluoro groups making the combination of RAFT polymerizations with thio-click chemistry a powerful method. It should be noted that certain kinds of radical thiol-ene reactions do not fulfill the click criterion of a high reaction efficiency if they are applied for polymer-polymer conjugation.
We are confident that the combination of CRP and click chemistry methods will continue flourishing in the near future leading to new functional polymeric materials. Eventually, this will lead to a further establishment of CRP in combination with click chemistry as a scientific tool rather than being a separate research topic. Furthermore, it is evident that new trends in click chemistry, e.g. the development of advanced metal-free click reactions employing more reactive clickable units such as cyclooctynes as click counterparts to nitrones or nitrile oxides, will be combined with CRP methods in the near future, which will further expand the potential field of applications.
Abbreviations
| AcOSt | 4-acetoxystyrene |
| AA | acrylic acid |
| AIBN | azoisobutyronitrile |
| AM | acrylamide |
| AN | acrylonitrile |
| APBIB | 3-azidopropyl 2-bromoisobutyrate |
| ATRP | atom transfer radical polymerization |
| ATREP | atom transfer radical emulsion polymerization |
| BBiBE | 1,2-bis(bromoisobutyryloxy)ethane |
| BDAT | S,S′-bis(α,α′-dimethyl-α′′-acetic acid) trithiocarbonate |
| BDB | benzyl dithiobenzoate |
| BETP | benzyl 2-(ethylthiocarbonothioylthio) propanoate |
| BICDT | benzyl 1H-imidazole-1-carbodithioate |
| bpy | 2,2′-bipyridine |
| BlocBuilder® | N-(2-methylpropyl)-N-(1-diethylphosphono-2,2-dimethylpropyl)-O-(2-carboxylprop-2-yl) hydroxylamine |
| BMP | 2-bromo-2-methyl-propionate |
| BMPA | 2-bromo-2-methyl-propionamide |
| BMPABE | 2-bromo-2-methyl-propionic acid benzyl ester |
| BPIT | butyl phthalimidomethyl trithiocarbonate |
| BPN | 2-brompropionnitrile |
| BPO | dibenzoylperoxide |
| BSPA | 3-benzylsulfanyl-thiocarbonylsulfanyl propionic acid |
| CAN | ceric ammonium nitrate |
| CBDB | 2-cyano-2-butyl dithiobenzoate |
| CBDN | α-cyanobenzyl dithionaphthalate |
| CDB | 2-phenylpropan-2-yl dithiobenzoate (cumyl dithiobenzoate) |
| CNT | carbon nanotube |
| ε–CL | ε–caprolactone |
| CPADB | (4-cyanopentanoic acid) dithiobenzoate |
| CPDB | (2-(2-cyano-propyl)) dithiobenzoate |
| CTA | chain transfer agent |
| CRP | controlled radical polymerization |
| DDAT | S-1-dodecyl-S′-(α,α′-dimethyl-α′′-acetic acid) trithiocarbonate |
| DDET | S-1-dodecyl-S′-(α,α′-dimethyl-α′′-ethyl acetate) trithiocarbonate |
| DEGMA | di(ethylene glycol) methylether methacrylate |
| DIPEA | S-dodecyl-S′-(α,α-dimethylpentafluorophenyl acetate) trithiocarbonate |
| DMAM | N,N-dimethylacrylamide |
| DEAM | N,N-diethylacrylamide |
| DEAEMA | 2-(diethylamino) ethyl methacrylate |
| DMAEMA | 2-(dimethylamino) ethyl methacrylate |
| EBiB | ethyl 2-bromoisobutyrate |
| EA | ethyl acrylate |
| EEA | 1-ethoxyethyl acrylate |
| EHA | 2-ethylhexyl acrylate |
| EtSO2N3 | ethanesulfonyl azide |
| FMA | furfuryl methacrylate |
| GMA | glycidyl methacrylate |
| HEA | 2-hydroxyethyl acrylate |
| HEMA | 2-hydroxyethyl methacrylate |
| HMA | hostasol methacrylate |
| HMTETA | 1,1,4,7,10,10-hexamethyl triethylenetetramine |
| HPMA | 2-hydroxypropyl methacrylate |
| HPMAM | 2-hydroxypropyl methacrylamide |
| iBoA | i-bornyl acrylate |
| pHSt | 4-hydroxystyrene |
| KSPMA | potassium 3-sulfopropyl methacrylate |
| LMA | lauryl methacrylate |
| MA | methyl acrylate |
| MAA | methacrylic acid |
| MCPMDB | (S)-methoxycarbonylphenylmethyl dithiobenzoate |
| Me6TREN | tris(2-(dimethylamino)-ethyl) amine |
| MMA | methyl methacrylate |
| mPEG | linear methoxy poly(ethylene glycol) |
| MPPCTTA | methyl 2-phenyl-2-(phenylcarbonothioylthio) acetate |
| NaCp | cyclopentadienide |
| nBA | n-butyl acrylate |
| nBMA | n-butyl methacrylate |
| NIPAM | N-isopropylacrylamide |
| NiCp2 | nickelocene |
| NMP | nitroxide-mediated radical polymerization |
| NP | nanoparticle |
| NVP | N-vinylpyrrolidone |
| OEGA | oligo(ethylene glycol) methylether acrylate |
| OEGMA | oligo(ethylene glycol) methylether methacrylate |
| OPA | (2-oxopropyl)acrylate |
| PBP | propargyl 2-bromopropionate |
| PDB | 1-phenylethyl dithiobenzoate |
| PEB | 1-phenylethylbromide |
| PEDT | S-1-phenylethyl-S′-dodecyl-trithiocarbonate |
| PFPPCV | pentafluorophenyl-(4-phenylthiocarbonylthio-4-cyanovalerate) |
| PFS | pentafluorostyrene |
| PMDETA | N,N,N′,N′′,N′′-pentamethyldiethylene-triamine |
| poC | postclick |
| prC | preclick |
| RAFT | reversible addition-fragmentation chain transfer polymerization |
| ROP | ring-opening polymerization |
| SG1 | N-(2-methylpropyl)-N-(1-diethylphosphono-2,2-dimethylpropyl)-N-oxyl |
| SMA | solketal methacrylate |
| St | styrene |
| tBA | t-butyl acrylate |
| tBMA | t-butyl methacrylate |
| TBAF | tetrabutylammonium fluoride |
| tBOSt | 4-t-butyloxystyrene |
| TBDMS | t-butyldimethyl-silyl |
| TEMPO | 2,2,6,6-tetramethylpiperidinylnitroxide |
| THPA | tetrahydropyran acrylate |
| TIPNO | 2,2,5-trimethyl-4-phenyl-3-azahexane-3-nitroxide |
| TIPS | triisopropylsilyl |
| TMS | trimethylsilyl |
| TosCl | p-toluenesulfonyl chloride |
| VAc | vinylacetate |
| VBA | vinylbenzyl azide |
| 4VP | 4-vinylpyridine. |
Acknowledgements
Financial support of the Dutch Polymer Institute (DPI) and of the Thuringian Ministry for Education, Science and Culture (grant #B514-09051, NanoConSens) is gratefully acknowledged.Notes and references
- M. Szwarc, Nature, 1956, 178, 1168–1169 CrossRef CAS.
- M. Szwarc, M. Levy and R. Milkovich, J. Am. Chem. Soc., 1956, 78, 2656–2657 CrossRef CAS.
- V. Ladmiral, G. Mantovani, G. J. Clarkson, S. Cauet, J. L. Irwin and D. M. Haddleton, J. Am. Chem. Soc., 2006, 128, 4823–4830 CrossRef CAS.
- E. R. Gillies and J. M. J. Fréchet, Drug Discovery Today, 2005, 10, 35–43 CrossRef CAS.
- K. Matyjaszewski, Prog. Polym. Sci., 2005, 30, 858–875 CrossRef CAS.
- M. Ouchi, T. Terashima and M. Sawamoto, Chem. Rev., 2009, 109, 4963–5050 CrossRef CAS.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS.
- M. Kamigaito, T. Ando and M. Sawamoto, Chem. Rev., 2001, 101, 3689–3746 CrossRef CAS.
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661–3688 CrossRef CAS.
- R. K. Iha, K. L. Wooley, A. M. Nyström, D. J. Burke, M. J. Kade and C. J. Hawker, Chem. Rev., 2009, 109, 5620–5686 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2006, 59, 669–692 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2009, 62, 1402–1472 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- D. Fournier, R. Hoogenboom and U. S. Schubert, Chem. Soc. Rev., 2007, 36, 1369–1380 RSC.
- W. H. Binder and R. Sachsenhofer, Macromol. Rapid Commun., 2007, 28, 15–54 CrossRef CAS.
- Wolfgang H. Binder and Robert Sachsenhofer, Macromol. Rapid Commun., 2008, 29, 952–981 CrossRef CAS.
- C. R. Becer, R. Hoogenboom and U. S. Schubert, Angew. Chem., Int. Ed., 2009, 48, 4900–4908 CrossRef CAS.
- J.-F. Lutz, Angew. Chem., Int. Ed., 2007, 46, 1018–1025 CrossRef CAS.
- A. B. Lowe, Polym. Chem., 2010, 1, 17–36 RSC.
- P. L. Golas and K. Matyjaszewski, Chem. Soc. Rev., 2010, 39, 1338–1354 RSC.
- C. Barner-Kowollik and A. J. Inglis, Macromol. Chem. Phys., 2009, 210, 987–992 CrossRef CAS.
- D. D. Diaz, S. Punna, P. Holzer, A. K. McPherson, K. B. Sharpless, V. V. Fokin and M. G. Finn, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 4392–4403 CrossRef CAS.
- P. Wu, A. K. Feldman, A. K. Nugent, C. J. Hawker, A. Scheel, B. Voit, J. Pyun, J. M. J. Frechet, K. B. Sharpless and V. V. Fokin, Angew. Chem., Int. Ed., 2004, 43, 3928–3932 CrossRef CAS.
- W. D. Sharpless, P. Wu, T. V. Hansen and J. G. Lindberg, J. Chem. Educ., 2005, 82, 1833–1836 CrossRef CAS.
- J.-F. Lutz, Angew. Chem., Int. Ed., 2008, 47, 2182–2184 CrossRef CAS.
- A. J. Inglis and C. Barner-Kowollik, Macromol. Rapid Commun., 2010, 31, 1247–1266 CrossRef CAS.
- C. E. Hoyle, T. Y. Lee and T. Roper, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5301–5338 CrossRef CAS.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS.
- A. B. Lowe, C. E. Hoyle and C. N. Bowman, J. Mater. Chem., 2010, 20, 4745–4750 RSC.
- R. Hoogenboom, Angew. Chem., Int. Ed., 2010, 49, 3415–3417 CAS.
- C. Ott, R. Hoogenboom and U. S. Schubert, Chem. Commun., 2008, 3516–3518 RSC.
- C. R. Becer, K. Babiuch, D. Pilz, S. Hornig, T. Heinze, M. Gottschaldt and U. S. Schubert, Macromolecules, 2009, 42, 2387–2394 CrossRef CAS.
- I. Singh, Z. Zarafshani, J.-F. Lutz and F. Heaney, Macromolecules, 2009, 42, 5411–5413 CrossRef CAS.
- D. Bontempo, K. L. Heredia, B. A. Fish and H. D. Maynard, J. Am. Chem. Soc., 2004, 126, 15372–15373 CrossRef CAS.
- K. L. Heredia, D. Bontempo, T. Ly, J. T. Byers, S. Halstenberg and H. D. Maynard, J. Am. Chem. Soc., 2005, 127, 16955–16960 CrossRef CAS.
- J. Liu, V. Bulmus, C. Barner-Kowollik, M. H. Stenzel and T. P. Davis, Macromol. Rapid Commun., 2007, 28, 305–314 CrossRef CAS.
- H. Durmaz, A. Dag, O. Altintas, T. Erdogan, G. Hizal and U. Tunca, Macromolecules, 2007, 40, 191–198 CrossRef CAS.
- S. Sinnwell, A. J. Inglis, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, Chem. Commun., 2008, 2052–2054 RSC.
- A. J. Inglis, S. Sinnwell, M. H. Stenzel and C. Barner-Kowollik, Angew. Chem. Int. Ed., 2009, 48, 2411–2414 CAS.
- R. Huisgen, Angew. Chem., Int. Ed. Engl., 1963, 2, 565–598 CrossRef.
- P. Cintas, A. Barge, S. Tagliapietra, L. Boffa and G. Cravotto, Nat. Protoc., 2010, 5, 607–616 Search PubMed.
- N. J. Agard, J. A. Prescher and C. R. Bertozzi, J. Am. Chem. Soc., 2004, 126, 15046–15047 CrossRef CAS.
- S. S.v. Berkel, A. J. Dirks, S. A. Meeuwissen, D. L. L. Pingen, O. C. Boerman, P. Laverman, F. L.v. Delft, J. J. L. M. Cornelissen and F. P. J. T. Rutjes, ChemBioChem, 2008, 9, 1805–1815 CrossRef CAS.
- C. J. Duxbury, D. Cummins and A. Heise, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3795–3802 CrossRef CAS.
- E. Gungor, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6703–6711 CrossRef CAS.
- F. Himo, T. Lovell, R. Hilgraf, V. V. Rostovtsev, L. Noodleman, K. B. Sharpless and V. V. Fokin, J. Am. Chem. Soc., 2005, 127, 210–216 CrossRef CAS.
- Z. P. Demko and K. B. Sharpless, J. Org. Chem., 2001, 66, 7945–7950 CrossRef CAS.
- N. V. Tsarevsky, K. V. Bernaerts, B. Dufour, F. E. Du Prez and K. Matyjaszewski, Macromolecules, 2004, 37, 9308–9313 CrossRef CAS.
- S. Bräse, C. Gil, K. Knepper and V. Zimmermann, Angew. Chem., Int. Ed., 2005, 44, 5188–5240 CrossRef.
- B. Yu, J. W. Chan, C. E. Hoyle and A. B. Lowe, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3544–3557 CrossRef CAS.
- C. E. Hoyle, A. B. Lowe and C. N. Bowman, Chem. Soc. Rev., 2010, 39, 1355–1387 RSC.
- K. L. Killops, L. M. Campos and C. J. Hawker, J. Am. Chem. Soc., 2008, 130, 5062–5064 CrossRef CAS.
- S. P. S. Koo, M. M. Stamenovic, R. A. Prasath, A. J. Inglis, F. E. Du Prez, C. Barner-Kowollik, W. V. Camp and T. Junkers, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1699–1713 CrossRef CAS.
- H. Li, B. Yu, H. Matsushima, C. E. Hoyle and A. B. Lowe, Macromolecules, 2009, 42, 6537–6542 CrossRef CAS.
- A. J. Inglis, M. H. Stenzel and C. Barner-Kowollik, Macromol. Rapid Commun., 2009, 30, 1792–1798 CrossRef CAS.
- A. Dag, H. Durmaz, V. Kirmizi, G. Hizal and U. Tunca, Polym. Chem., 2010, 1, 621–623 RSC.
- A. A. Kavitha and N. K. Singha, ACS Appl. Mater. Interfaces, 2009, 1, 1427–1436 Search PubMed.
- K. L. Heredia and H. D. Maynard, Org. Biomol. Chem., 2007, 5, 45–53 RSC.
- N. V. Tsarevsky, S. A. Bencherif and K. Matyjaszewski, Macromolecules, 2007, 40, 4439–4445 CrossRef CAS.
- S. Fleischmann, K. Hinrichs, U. Oertel, S. Reichelt, K.-J. Eichhorn and B. Voit, Macromol. Rapid Commun., 2008, 29, 1177–1185 CrossRef CAS.
- H. Kwart and K. King, Chem. Rev., 1968, 68, 415–447 CrossRef CAS.
- J. Sauer and R. Sustmann, Angew. Chem., Int. Ed. Engl., 1980, 19, 779–807 CrossRef.
- T. Dispinar, R. Sanyal and A. Sanyal, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 4545–4551 CrossRef CAS.
- J. A. Syrett, G. Mantovani, W. R. S. Barton, D. Price and D. M. Haddleton, Polym. Chem., 2010, 1, 102–106 RSC.
- A. J. Inglis, L. Nebhani, O. Altintas, F. G. Schmidt and C. Barner-Kowollik, Macromolecules, 2010, 43, 5515–5520 CrossRef CAS.
- J. Geng, G. Mantovani, L. Tao, J. Nicolas, G. Chen, R. Wallis, D. A. Mitchell, B. R. G. Johnson, S. D. Evans and D. M. Haddleton, J. Am. Chem. Soc., 2007, 129, 15156–15163 CrossRef CAS.
- R. J. Thibault, K. Takizawa, P. Lowenheilm, B. Helms, J. L. Mynar, J. M. J. Frechet and C. J. Hawker, J. Am. Chem. Soc., 2006, 128, 12084–12085 CrossRef CAS.
- V. L. G. Mantovani, L. Tao and D. M. Haddleton, Chem. Commun., 2005, 2089–2091 RSC.
- J. Geng, J. Lindqvist, G. Mantovani and D. M. Haddleton, Angew. Chem., Int. Ed., 2008, 47, 4180–4183 CrossRef CAS.
- L. Mespouille, M. Vachaudez, F. Suriano, P. Gerbaux, O. Coulembier, P. Degee, R. Flammang and P. Dubois, Macromol. Rapid Commun., 2007, 28, 2151–2158 CrossRef CAS.
- O. Altintas, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 1218–1228 CrossRef CAS.
- E. Gungor, H. Durmaz, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 4459–4468 CrossRef CAS.
- J. A. Opsteen and J. C. M. van Hest, Chem. Commun., 2005, 57–59 RSC.
- M. A. Bennett, Chem. Rev., 1962, 62, 611–652 CrossRef CAS.
- R. Nast, Coord. Chem. Rev., 1982, 47, 89–124 CrossRef CAS.
- A. Hasneen, H. S. Han and H.-J. Paik, React. Funct. Polym., 2009, 69, 681–687 CrossRef CAS.
- B. S. Sumerlin, N. V. Tsarevsky, G. Louche, R. Y. Lee and K. Matyjaszewski, Macromolecules, 2005, 38, 7540–7545 CrossRef CAS.
- J. A. Opsteen and J. C. M. Van Hest, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 2913–2924 CrossRef CAS.
- Z. Zarafshani, O. Akdemir and J.-F. Lutz, Macromol. Rapid Commun., 2008, 29, 1161–1166 CrossRef CAS.
- C. N. Urbani, C. A. Bell, D. Lonsdale, M. R. Whittaker and M. J. Monteiro, Macromolecules, 2008, 41, 76–86 CrossRef CAS.
- G. D. Fu, L. Q. Xu, F. Yao, K. Zhang, X. F. Wang, M. F. Zhu and S. Z. Nie, ACS Appl. Mater. Interfaces, 2009, 1, 239–243 Search PubMed.
- G. J. Chen, L. Tao, G. Mantovani, V. Ladmiral, D. P. Burt, J. V. Macpherson and D. M. Haddleton, Soft Matter, 2007, 3, 732–739 RSC.
- L. Q. Xu, F. Yao, G. D. Fu and L. Shen, Macromolecules, 2009, 42, 6385–6392 CrossRef CAS.
- N. V. Tsarevsky, B. S. Sumerlin and K. Matyjaszewski, Macromolecules, 2005, 38, 3558–3561 CrossRef CAS.
- C. H. Li, Z. S. Ge, H. W. Liu and S. Y. Liu, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 4001–4013 CrossRef CAS.
- W. Lin, Q. Fu, Y. Zhang and J. Huang, Macromolecules, 2008, 41, 4127–4135 CrossRef CAS.
- H. Durmaz, A. Dag, D. Gursoy, A. L. Demirel, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1557–1564 CrossRef CAS.
- H. Durmaz, A. Dag, E. Erdogan, A. L. Demirel, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 99–108 CrossRef CAS.
- A. Dag, H. Durmaz, V. Kirmizi, G. Hizal and U. Tunca, Polym. Chem., 2010, 1, 621–623 RSC.
- W. Van Camp, V. Germonpre, L. Mespouille, P. Dubois, E. J. Goethals and F. E. Du Prez, React. Funct. Polym., 2007, 67, 1168–1180 CrossRef CAS.
- J. Xu, J. Ye and S. Y. Liu, Macromolecules, 2007, 40, 9103–9110 CrossRef CAS.
- L. Mespouille, M. Vachaudez, F. Suriano, P. Gerbaux, W. Van Camp, O. Coulembier, P. Degee, R. Flammang, F. Du Prez and P. Dubois, React. Funct. Polym., 2008, 68, 990–1003 CrossRef CAS.
- W. A. Braunecker and K. Matyjaszewski, Prog. Polym. Sci., 2007, 32, 93–146 CrossRef CAS.
- Y. F. Zhang, L. Hao, J. M. Hu, C. H. Li and S. Y. Liu, Macromol. Rapid Commun., 2009, 30, 941–947 CrossRef CAS.
- C. J. Huang and F. C. Chang, Macromolecules, 2009, 42, 5155–5166 CrossRef CAS.
- J.-M. Schumers, J.-F. Gohy and C.-A. Fustin, Polym. Chem., 2010, 1, 161–163 RSC.
- G. Y. Shi and C. Y. Pan, Macromol. Rapid Commun., 2008, 29, 1672–1678 CrossRef CAS.
- Y. Q. Dong, Y. Y. Tong, B. T. Dong, F. S. Du and Z. C. Li, Macromolecules, 2009, 42, 2940–2948 CrossRef CAS.
- J. Zhu, X. L. Zhu, E. T. Kang and K. G. Neoh, Polymer, 2007, 48, 6992–6999 CrossRef CAS.
- Y. F. Zhang, C. H. Li and S. Y. Liu, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3066–3077 CrossRef CAS.
- G. H. Deng, D. Y. Ma and Z. Z. Xu, Eur. Polym. J., 2007, 43, 1179–1187 CrossRef CAS.
- G. W. Wang, X. L. Luo, C. Liu and J. L. Huang, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 2154–2166 CrossRef CAS.
- B. A. Laurent and S. M. Grayson, J. Am. Chem. Soc., 2006, 128, 4238–4239 CrossRef CAS.
- C. H. Li, Z. S. Ge, J. Fang and S. Y. Liu, Macromolecules, 2009, 42, 2916–2924 CrossRef CAS.
- V. Ladmiral, T. M. Legge, Y. L. Zhao and S. Perrier, Macromolecules, 2008, 41, 6728–6732 CrossRef CAS.
- W. Agut, D. Taton and S. Lecommandoux, Macromolecules, 2007, 40, 5653–5661 CrossRef CAS.
- S. Pfeifer, Z. Zarafshani, N. Badi and J.-F. Lutz, J. Am. Chem. Soc., 2009, 131, 9195–9197 CrossRef CAS.
- Y. Li, J. W. Yang and B. C. Benicewicz, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 4300–4308 CrossRef CAS.
- M. Urien, H. Erothu, E. Cloutet, R. C. Hiorns, L. Vignau and H. Cramail, Macromolecules, 2008, 41, 7033–7040 CrossRef CAS.
- C. H. Li, J. M. Hu, J. Yin and S. Y. Liu, Macromolecules, 2009, 42, 5007–5016 CrossRef CAS.
- X. Jiang, M. C. Lok and W. E. Hennink, Bioconjugate Chem., 2007, 18, 2077–2084 CrossRef CAS.
- P. D. Topham, N. Sandon, E. S. Read, J. Madsen, A. J. Ryan and S. P. Armes, Macromolecules, 2008, 41, 9542–9547 CrossRef CAS.
- A. Narumi, K. Fuchise, R. Kakuchi, A. Toda, T. Satoh, S. Kawaguchi, K. Sugiyama, A. Hirao and T. Kakuchi, Macromol. Rapid Commun., 2008, 29, 1126–1133 CrossRef CAS.
- Y. Zhang, H. K. He and C. Gao, Macromolecules, 2008, 41, 9581–9594 CrossRef CAS.
- A. Dag, H. Durmaz, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 302–313 CrossRef CAS.
- M. Li, P. De, S. R. Gondi and B. S. Sumerlin, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5093–5100 CrossRef CAS.
- A. Dag, H. Durmaz, E. Demir, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6969–6977 CrossRef CAS.
- O. Altintas, G. Hizal and U. Tunca, Des. Monomers Polym., 2009, 12, 83–98 Search PubMed.
- H. Durmaz, F. Karatas, U. Tunca and G. Hizal, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 3947–3957 CrossRef CAS.
- B. Gacal, H. Durmaz, M. A. Tasdelen, G. Hizal, U. Tunca, Y. Yagci and A. L. Demirel, Macromolecules, 2006, 39, 5330–5336 CrossRef CAS.
- H. Durmaz, A. Dag, A. Hizal, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7091–7100 CrossRef CAS.
- G. Mantovani, F. Lecolley, L. Tao, D. M. Haddleton, J. Clerx, J. J. L. M. Cornelissen and K. Velonia, J. Am. Chem. Soc., 2005, 127, 2966–2973 CrossRef CAS.
- B. Le Droumaguet, G. Mantovani, D. M. Haddleton and K. Velonia, J. Mater. Chem., 2007, 17, 1916–1922 RSC.
- M. Erdogan, G. Hizal, Ü. Tunca, D. Hayrabetyan and Ö. Pekcan, Polymer, 2002, 43, 1925–1931 CrossRef CAS.
- A. Dag, H. Durmaz, U. Tunca and G. Hizal, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 178–187 CrossRef CAS.
- W. Zhang, W. Zhang, Z. Zhang, J. Zhu, Q. Pan and X. Zhu, Polym. Bull., 2009, 63, 467–483 CrossRef CAS.
- L. M. Campos, K. L. Killops, R. Sakai, J. M. J. Paulusse, D. Damiron, E. Drockenmuller, B. W. Messmore and C. J. Hawker, Macromolecules, 2008, 41, 7063–7070 CrossRef CAS.
- V. Vázquez-Dorbatt, Z. P. Tolstyka, C.-W. Chang and H. D. Maynard, Biomacromolecules, 2009, 10, 2207–2212 CrossRef CAS.
- A. Klaikherd, S. Ghosh and S. Thayumanavan, Macromolecules, 2007, 40, 8518–8520 CrossRef CAS.
- N. V. Tsarevsky and K. Matyjaszewski, Macromolecules, 2002, 35, 9009–9014 CrossRef CAS.
- H. D. Maynard, K. L. Heredia, R. C. Li, D. P. Parra and V. Vázquez-Dorbatt, J. Mater. Chem., 2007, 17, 4015–4017 RSC.
- K. L. Heredia, Z. P. Tolstyka and H. D. Maynard, Macromolecules, 2007, 40, 4772–4779 CrossRef CAS.
- D. Quemener, T. P. Davis, C. Barner-Kowollik and M. H. Stenzel, Chem. Commun., 2006, 5051–5053 RSC.
- R. K. O'Reilly, M. J. Joralemon, C. J. Hawker and K. L. Wooley, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 5203–5217 CrossRef CAS.
- S. R. S. Ting, A. M. Granville, D. Quemener, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, Aust. J. Chem., 2007, 60, 405–409 CrossRef CAS.
- G. Y. Shi, X. Z. Tang and C. Y. Pan, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 2390–2401 CrossRef CAS.
- R. Ranjan and W. J. Brittain, Macromol. Rapid Commun., 2008, 29, 1104–1110 CrossRef CAS.
- R. Ranjan and W. J. Brittain, Macromol. Rapid Commun., 2007, 28, 2084–2089 CrossRef CAS.
- A. J. D. Magenau, N. Martinez-Castro, D. A. Savin and R. F. Storey, Macromolecules, 2009, 42, 8044–8051 CrossRef CAS.
- T. Zhang, Y. P. Wu, X. M. Pan, Z. H. Zheng, X. B. Ding and Y. X. Peng, Eur. Polym. J., 2009, 45, 1625–1633 CrossRef CAS.
- D. Konkolewicz, A. Gray-Weale and S. Perrier, J. Am. Chem. Soc., 2009, 131, 18075–18077 CrossRef CAS.
- N. Akeroyd, R. Pfukwa and B. Klumperman, Macromolecules, 2009, 42, 3014–3018 CrossRef CAS.
- A. S. Goldmann, D. Quémener, P. E. Millard, T. P. Davis, M. H. Stenzel, C. Barner-Kowollik and A. H. E. Mueller, Polymer, 2008, 49, 2274–2281 CrossRef CAS.
- L. Barner, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, Macromol. Rapid Commun., 2007, 28, 539–559 CrossRef CAS.
- R. Ranjan and W. J. Brittain, Macromolecules, 2007, 40, 6217–6223 CrossRef CAS.
- T. Zhang, Z. H. Zheng, X. B. Ding and Y. X. Peng, Macromol. Rapid Commun., 2008, 29, 1716–1720 CrossRef CAS.
- S. Puttick, D. J. Irvine, P. Licence and K. J. Thurecht, J. Mater. Chem., 2009, 19, 2679–2682 RSC.
- S. R. Gondi, A. P. Vogt and B. S. Sumerlin, Macromolecules, 2007, 40, 474–481 CrossRef CAS.
- P. De, S. R. Gondi and B. S. Sumerlin, Biomacromolecules, 2008, 9, 1064–1070 CrossRef CAS.
- M. Li, P. De, S. R. Gondi and B. S. Sumerlin, Macromol. Rapid Commun., 2008, 29, 1172–1176 CrossRef CAS.
- A. Vora, K. Singh and D. C. Webster, Polymer, 2009, 50, 2768–2774 CrossRef CAS.
- A. P. Vogt, S. R. Gondi and B. S. Sumerlin, Aust. J. Chem., 2007, 60, 396–399 CrossRef CAS.
- D. Quemener, M. Le Hellaye, C. Bissett, T. P. Davis, C. Barner-Kowollik and M. H. Stenzel, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 155–173 CrossRef CAS.
- J. Bernard, M. Save, B. Arathoon and B. Charleux, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 2845–2857 CrossRef CAS.
- F. Chen, Z. P. Cheng, J. Zhu, W. Zhang and X. L. Zhu, Eur. Polym. J., 2008, 44, 1789–1795 CrossRef CAS.
- A. P. Vogt and B. S. Sumerlin, Macromolecules, 2008, 41, 7368–7373 CrossRef CAS.
- Z. S. An, W. Tang, M. H. Wu, Z. Jiao and G. D. Stucky, Chem. Commun., 2008, 6501–6503 RSC.
- X. P. Qiu, F. Tanaka and F. M. Winnik, Macromolecules, 2007, 40, 7069–7071 CrossRef CAS.
- C. Boyer, J. Liu, V. Bulmus, T. P. Davis, C. Barner-Kowollik and M. H. Stenzel, Macromolecules, 2008, 41, 5641–5650 CrossRef CAS.
- J. Liu, V. Bulmus, D. L. Herlambang, C. Barner-Kowollik, M. H. Stenzel and T. P. Davis, Angew. Chem., Int. Ed., 2007, 46, 3099–3103 CrossRef CAS.
- K. L. Heredia, T. H. Nguyen, C.-W. Chang, V. Bulmus, T. P. Davis and H. D. Maynard, Chem. Commun., 2008, 3245–3247 RSC.
- C. Boyer, V. Bulmus, J. Liu, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, J. Am. Chem. Soc., 2007, 129, 7145–7154 CrossRef CAS.
- C. Boyer, J. Liu, L. Wong, M. Tippett, V. Bulmus and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7207–7224 CrossRef.
- J. Liu, H. Liu, C. Boyer, V. Bulmus and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 899–912 CrossRef CAS.
- S. Sinnwell, A. J. Inglis, M. H. Stenzel and C. Barner-Kowollik, Macromol. Rapid Commun., 2008, 29, 1090–1096 CrossRef CAS.
- L. Nebhani, P. Gerstel, P. Atanasova, M. Bruns and C. Barner-Kowollik, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 7090–7095 CrossRef.
- L. Nebhani, S. Sinnwell, C. Y. Lin, M. L. Coote, M. H. Stenzel and C. Barner-Kowollik, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 6053–6071 CrossRef CAS.
- S. Sinnwell, M. Lammens, M. H. Stenzel, F. E. Du Prez and C. Barner-Kowollik, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 2207–2213 CrossRef CAS.
- A. J. Inglis, S. Sinnwell, T. P. Davis, C. Barner-Kowollik and M. H. Stenzel, Macromolecules, 2008, 41, 4120–4126 CrossRef CAS.
- S. Sinnwell, C. V. Synatschke, T. Junkers, M. H. Stenzel and C. Barner-Kowollik, Macromolecules, 2008, 41, 7904–7912 CrossRef CAS.
- P. De, M. Li, S. R. Gondi and B. S. Sumerlin, J. Am. Chem. Soc., 2008, 130, 11288–11289 CrossRef CAS.
- S. Marque, H. Fischer, E. Baier and A. Studer, J. Org. Chem., 2001, 66, 1146–1156 CrossRef CAS.
- M. Rodlert, E. Harth, I. Rees and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 4749–4763 CrossRef CAS.
- Nicole L. Hill and Rebecca Braslau, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 2341–2349 CrossRef.
- J. Dao, D. Benoit and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 1998, 36, 2161–2167 CrossRef CAS.
- S. Fleischmann, H. Komber, D. Appelhans and B. I. Voit, Macromol. Chem. Phys., 2007, 208, 1050–1060 CrossRef CAS.
- G. F. D'Alelio and R. C. Evers, J. Polym. Sci., Part A-1, 1967, 5, 999–1014 CrossRef CAS.
- L. B. Sessions, L. A. Miinea, K. D. Ericson, D. S. Glueck and R. B. Grubbs, Macromolecules, 2005, 38, 2116–2121 CrossRef CAS.
- O. Altintas, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 5699–5707 CrossRef CAS.
- A. Gozgen, A. Dag, H. Durmaz, O. Sirkecioglu, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 497–504 CrossRef CAS.
- E. Gungor, G. Cote, T. Erdogan, H. Durmaz, A. L. Demirel, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 1055–1065 CrossRef CAS.
- O. Altintas, A. L. Demirel, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5916–5928 CrossRef CAS.
- O. Altintas, B. Yankul, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 3588–3598 CrossRef CAS.
- E. Gungor, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3409–3418 CrossRef CAS.
- E. H. H. Wong, M. H. Stenzel, T. Junkers and C. Barner-Kowollik, Macromolecules, 2010, 43, 3785–3793 CrossRef CAS.
- T. Junkers, E. H. H. Wong, M. H. Stenzel and C. Barner-Kowollik, Macromolecules, 2009, 42, 5027–5035 CrossRef CAS.
- E. H. H. Wong, T. Junkers and C. Barner-Kowollik, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7273–7279 CrossRef CAS.
- W. H. Binder, D. Gloger, H. Weinstabl, G. Allmaier and E. Pittenauer, Macromolecules, 2007, 40, 3097–3107 CrossRef CAS.
- U. Tunca, Z. Ozyurek, T. Erdogan and G. Hizal, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 4228–4236 CrossRef CAS.
- H. Durmaz, F. Karatas, U. Tunca and G. Hizal, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 499–509 CrossRef CAS.
- A. Krieg, C. R. Becer, R. Hoogenboom and U. S. Schubert, Macromol. Symp., 2009, 275–276, 73–81 CrossRef CAS.
- J. T. Wiltshire and G. G. Qiao, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 1485–1498 CrossRef CAS.
- M. Malkoch, R. J. Thibault, E. Drockenmuller, M. Messerschmidt, B. Voit, T. P. Russell and C. J. Hawker, J. Am. Chem. Soc., 2005, 127, 14942–14949 CrossRef CAS.
- R. K. O'Reilly, M. J. Joralemon, C. J. Hawker and K. L. Wooley, Chem.–Eur. J., 2006, 12, 6776–6786 CrossRef CAS.
- S. Fleischmann, H. Komber and B. Voit, Macromolecules, 2008, 41, 5255–5264 CrossRef CAS.
- T. Ishizone, G. Uehara, A. Hirao, S. Nakahama and K. Tsuda, Macromolecules, 1998, 31, 3764–3774 CrossRef CAS.
- F. Bertini, G. Audisio, J. Kiji and M. Fujita, J. Anal. Appl. Pyrolysis, 2003, 68–69, 61–81 CrossRef CAS.
- M. M. Martin and E. B. Sanders, J. Am. Chem. Soc., 1967, 89, 3777–3782 CrossRef CAS.
- R.-V. Ostaci, D. Damiron, Y. Grohens, L. Léger and E. Drockenmuller, Langmuir, 2010, 26, 1304–1310 CrossRef CAS.
- Y. Zhang, H. He, C. Gao and J. Y. Wu, Langmuir, 2009, 25, 5814–5824 CrossRef CAS.
- J. Stadermann, S. Fleischmann, M. Messerschmidt, H. Komber and B. Voit, Macromol. Symp., 2009, 275–276, 35–42 CrossRef CAS.
- C. Ladaviere, N. Dorr and J. P. Claverie, Macromolecules, 2001, 34, 5370–5372 CrossRef CAS.
- R. K. O'Reilly, M. J. Joralemon, C. J. Hawker and K. L. Wooley, New J. Chem., 2007, 31, 718–724 RSC.
- T. Junkers and C. Barner-Kowollik, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7585–7605 CrossRef CAS.
- G. K. Such, J. F. Quinn, A. Quinn, E. Tjipto and F. Caruso, J. Am. Chem. Soc., 2006, 128, 9318–9319 CrossRef CAS.
- G. K. Such, E. Tjipto, A. Postma, A. P. R. Johnston and F. Caruso, Nano Lett., 2007, 7, 1706–1710 CrossRef CAS.
- A. R. de Luzuriaga, N. Ormategui, H. J. Grande, I. Odriozola, J. A. Pornposo and I. Loinaz, Macromol. Rapid Commun., 2008, 29, 1156–1160 CrossRef.
- X. W. Zhang, X. M. Lian, L. Liu, J. Zhang and H. Y. Zhao, Macromolecules, 2008, 41, 7863–7869 CrossRef CAS.
- D. Damiron, M. Desorme, R. V. Ostaci, S. Al Akhrass, T. Hamaide and E. Drockenmuller, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3803–3813 CrossRef CAS.
- D. Gromadzki, J. Lokaj, P. Cernoch, O. Diat, F. Nallet and P. Stepánek, Eur. Polym. J., 2008, 44, 189–199 CrossRef CAS.
- M. Aldhoun, A. Massi and A. Dondoni, J. Org. Chem., 2008, 73, 9565–9575 CrossRef CAS.
- V. Aureggi and G. Sedelmeier, Angew. Chem., Int. Ed., 2007, 46, 8440–8444 CrossRef CAS.
- J. Aimi, L. A. McCullough and K. Matyjaszewski, Macromolecules, 2008, 41, 9522–9524 CrossRef CAS.
- D. G. Li, J. Zhu, Z. P. Cheng, W. Zhang and X. L. Zhu, React. Funct. Polym., 2009, 69, 240–245 CrossRef CAS.
- Y. M. Zhou, K. Q. Jiang, Y. Q. Chen and S. Y. Liu, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6518–6531 CrossRef CAS.
- S. Pfeifer and J.-F. Lutz, Chem.–Eur. J., 2008, 14, 10949–10957 CrossRef CAS.
- G. Li, H. T. Zheng and R. K. Bai, Macromol. Rapid Commun., 2009, 30, 442–447 CrossRef CAS.
- X. Z. Jiang, J. Y. Zhang, Y. M. Zhou, J. Xu and S. Y. Liu, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 860–871 CrossRef CAS.
- Y. Li and B. C. Benicewicz, Macromolecules, 2008, 41, 7986–7992 CrossRef CAS.
- J. Y. Zhang, Y. M. Zhou, Z. Y. Zhu, Z. S. Ge and S. Y. Liu, Macromolecules, 2008, 41, 1444–1454 CrossRef CAS.
- L. A. Canalle, S. S.v. Berkel, L. T.d. Haan and J. C. M.v. Hest, Adv. Funct. Mater., 2009, 19, 3464–3470 CrossRef CAS.
- L. González, A. Rodríguez, J. L.d. Benito and A. Marcos-Fernández, J. Appl. Polym. Sci., 1997, 63, 1353–1359 CrossRef CAS.
- J. E. Leffler and H. H. Gibson, J. Am. Chem. Soc., 1968, 90, 4117–4121 CrossRef CAS.
- C. J. Ruud, J. Jia and G. L. Baker, Macromolecules, 2000, 33, 8184–8191 CrossRef CAS.
- C. K. Govindan, Org. Process Res. Dev., 2002, 6, 74–77 Search PubMed.
- G. T. Anderson, J. R. Henry and S. M. Weinreb, J. Org. Chem., 1991, 56, 6946–6948 CrossRef.
- W. Broeckx, N. Overbergh, C. Samyn, G. Smets and G. L'abbé, Tetrahedron, 1971, 27, 3527–3534 CrossRef CAS.
- A. Bousquet, C. Barner-Kowollik and M. H. Stenzel, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1773–1781 CrossRef CAS.
- A. A. Kavitha and N. K. Singha, Macromol. Chem. Phys., 2007, 208, 2569–2577 CrossRef CAS.
- A. A. Kavitha and N. K. Singha, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 4441–4449 CrossRef CAS.
- A. A. Kavitha, A. Choudhury and N. K. Singha, Macromol. Symp., 2006, 240, 232–237 CrossRef CAS.
- A. A. Kavitha and N. K. Singha, Macromolecules, 2010, 43, 3193–3205 CrossRef CAS.
- M. J. Kade, D. J. Burke and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 743–750 CrossRef CAS.
- Z. Jia, J. Liu, T. P. Davis and V. Bulmus, Polymer, 2009, 50, 5928–5932 CrossRef CAS.
- Z. Jia, L. Wong, T. P. Davis and V. Bulmus, Biomacromolecules, 2008, 9, 3106–3113 CrossRef CAS.
- L. Wong, C. Boyer, Z. Jia, H. M. Zareie, T. P. Davis and V. Bulmus, Biomacromolecules, 2008, 9, 1934–1944 CrossRef CAS.
- J. Zhu, Z. Di, X. Zhu and G. Chen, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 2558–2565 CrossRef CAS.
- K. Dayananda and R. Dhamodharan, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 902–915 CrossRef CAS.
- R. Krishnan and K. S. V. Srinivasan, Macromolecules, 2003, 36, 1769–1771 CrossRef CAS.
- J. L. De La Fuente, P. F. Canamero and M. Fernandez-Garcia, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 1807–1816 CrossRef CAS.
- P. F. Cañamero, J. L. de la Fuente, E. L. Madruga and M. Fernández-García, Macromol. Chem. Phys., 2004, 205, 2221–2228 CrossRef CAS.
- R. Krishnan and K. S. V. Srinivasan, Macromolecules, 2004, 37, 3614–3622 CrossRef CAS.
- G. Li, X. Zhu, J. Zhu, Z. Cheng and W. Zhang, Polymer, 2005, 46, 12716–12721 CrossRef CAS.
- Y.-Y. Yuan, Q. Du, Y.-C. Wang and J. Wang, Macromolecules, 2010, 43, 1739–1746 CrossRef CAS.
- K. Godula, D. Rabuka, K. T. Nam and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 4973–4976 CrossRef CAS.
- R. K. O'Reilly, C. J. Hawker and K. L. Wooley, Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.), 2004, 45, 780 CAS.
- G. O'Bryan, N. Ningnuek and R. Braslau, Polymer, 2008, 49, 5241–5248 CrossRef CAS.
- K. Matyjaszewski, Y. Nakagawa and S. G. Gaynor, Macromol. Rapid Commun., 1997, 18, 1057–1066 CrossRef.
- V. Coessens, Y. Nakagawa and K. Matyjaszewski, Polym. Bull., 1998, 40, 135–142 CrossRef CAS.
- V. Coessens and K. Matyjaszewski, J. Macromol. Sci., Part A: Pure Appl. Chem., 1999, 36, 667–679 Search PubMed.
- S. O. Kyeremateng, E. Amado, A. Blume and J. Kressler, Macromol. Rapid Commun., 2008, 29, 1140–1146 CrossRef CAS.
- A. Sinaga, P. Ravi, T. A. Hatton and K. C. Tam, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 2646–2656 CrossRef CAS.
- J.-F. Lutz, H. G. Borner and K. Weichenhan, Macromol. Rapid Commun., 2005, 26, 514–518 CrossRef CAS.
- J.-F. Lutz, H. G. Borner and K. Weichenhan, Macromolecules, 2006, 39, 6376–6383 CrossRef CAS.
- Q. C. Liu and Y. M. Chen, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 6103–6113 CrossRef CAS.
- A. P. Vogt and B. S. Sumerlin, Macromolecules, 2006, 39, 5286–5292 CrossRef CAS.
- H. F. Gao and K. Matyjaszewski, Macromolecules, 2006, 39, 4960–4965 CrossRef CAS.
- O. Altintas, B. Yankul, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 6458–6465 CrossRef CAS.
- B. S. Lee, J. K. Lee, W. J. Kim, Y. H. Jung, S. J. Sim, J. Lee and I. S. Choi, Biomacromolecules, 2007, 8, 744–749 CrossRef CAS.
- M. Degirmenci and N. Genli, Macromol. Chem. Phys., 2009, 210, 1617–1623 CrossRef CAS.
- Q. Liu, P. Zhao and Y. Chen, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 3330–3341 CrossRef CAS.
- P. L. Golas, N. V. Tsarevsky and K. Matyjaszewski, Macromol. Rapid Commun., 2008, 29, 1167–1171 CrossRef CAS.
- J. Kulis, C. A. Bell, A. S. Micallef, Z. Jia and M. J. Monteiro, Macromolecules, 2009, 42, 8218–8227 CrossRef CAS.
- A. Gheorghe, A. Matsuno and O. Reiser, Adv. Synth. Catal., 2006, 348, 1016–1020 CrossRef CAS.
- E. H. H. Wong, C. Boyer, M. H. Stenzel, C. Barner-Kowollik and T. Junkers, Chem. Commun., 2010, 46, 1959–1961 RSC.
- A. J. Inglis, T. Paulöhrl and C. Barner-Kowollik, Macromolecules, 2010, 43, 33–36 CrossRef CAS.
- C. Boyer, V. Bulmus and T. P. Davis, Macromol. Rapid Commun., 2009, 30, 493–497 CrossRef.
- J. W. Chan, B. Yu, C. E. Hoyle and A. B. Lowe, Polymer, 2009, 50, 3158–3168 CrossRef CAS.
- C. W. Chang, E. Bays, L. Tao, S. N. S. Alconcel and H. D. Maynard, Chem. Commun., 2009, 3580–3582 RSC.
- C. Boyer, A. Granville, T. P. Davis and V. Bulmus, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3773–3794 CrossRef CAS.
- J. W. Chan, B. Yu, C. E. Hoyle and A. B. Lowe, Chem. Commun., 2008, 4959–4961 RSC.
- V. Lima, X. Jiang, J. Brokken-Zijp, P. J. Schoenmakers, B. Klumperman and R. V. D. Linde, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 959–973 CrossRef CAS.
- X.-P. Qiu and F. M. Winnik, Macromol. Rapid Commun., 2006, 27, 1648–1653 CrossRef CAS.
- M. Nakayama and T. Okano, Biomacromolecules, 2005, 6, 2320–2327 CrossRef CAS.
- F. Segui, X.-P. Qiu and F. M. Winnik, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 314–326 CrossRef CAS.
- C. W. Scales, A. J. Convertine and C. L. McCormick, Biomacromolecules, 2006, 7, 1389–1392 CrossRef CAS.
- H. Kakwere and S. Perrier, J. Am. Chem. Soc., 2009, 131, 1889–1895 CrossRef CAS.
- A. S. Goldmann, A. Walther, L. Nebhani, R. Joso, D. Ernst, K. Loos, C. Barner-Kowollik, L. Barner and A. H. E. Mueller, Macromolecules, 2009, 42, 3707–3714 CrossRef CAS.
- G. L. Ellman, Arch. Biochem. Biophys., 1958, 74, 443–450 CrossRef CAS.
- P. J. Roth, D. Kessler, R. Zentel and P. Theato, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3118–3130 CrossRef CAS.
- L. P. Yang, H. X. Zhou, G. Y. Shi, Y. Wang and C. Y. Pan, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6641–6653 CrossRef CAS.
- D. H. Solomon, E. Rizzardo and P. Cacioli, US Patent, 4581 429, 1985.
- E. Harth, C. J. Hawker, W. Fan and R. M. Waymouth, Macromolecules, 2001, 34, 3856–3862 CrossRef CAS.
- G. O'Bryan and R. Braslau, Macromolecules, 2006, 39, 9010–9017 CrossRef CAS.
- Y. Guillaneuf, P.-E. Dufils, L. Autissier, M. Rollet, D. Gigmes and D. Bertin, Macromolecules, 2010, 43, 91–100 CrossRef CAS.
- C. Ollivier and P. Renaud, J. Am. Chem. Soc., 2001, 123, 4717–4727 CrossRef CAS.
- P. Panchaud and P. Renaud, J. Org. Chem., 2004, 69, 3205–3207 CrossRef CAS.
- X. Z. Jiang, G. Y. Zhang, R. Narain and S. Y. Liu, Langmuir, 2009, 25, 2046–2054 CrossRef CAS.
- H. Gao and K. Matyjaszewski, J. Am. Chem. Soc., 2007, 129, 6633–6639 CrossRef CAS.
- D. Cummins, C. J. Duxbury, P. Quaedflieg, P. Magusin, C. E. Koning and A. Heise, Soft Matter, 2009, 5, 804–811 RSC.
- B. Sieczkowska, M. Millaruelo, M. Messerschmidt and B. Voit, Macromolecules, 2007, 40, 2361–2370 CrossRef CAS.
- R. K. O'Reilly, M. J. Joralemon, K. L. Wooley and C. J. Hawker, Chem. Mater., 2005, 17, 5976–5988 CrossRef CAS.
- G. Temel, B. Aydogan, N. Arsu and Y. Yagci, Macromolecules, 2009, 42, 6098–6106 CrossRef CAS.
- W.-H. Ting, S. A. Dai, Y.-F. Shih, I.-K. Yang, W.-C. Su and R.-J. Jeng, Polymer, 2008, 49, 1497–1505 CrossRef CAS.
- M. J. Joralemon, R. K. O'Reilly, C. J. Hawker and K. L. Wooley, J. Am. Chem. Soc., 2005, 127, 16892–16899 CrossRef CAS.
- P. L. Golas, N. V. Tsarevsky, B. S. Sumerlin and K. Matyjaszewski, Macromolecules, 2006, 39, 6451–6457 CrossRef CAS.
- M.-A. Berthet, Z. Zarafshani, S. Pfeifer and J.-F. Lutz, Macromolecules, 2010, 43, 44–50 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |