Effect of solvents on polymerization of N-propargylamide monomer and secondary structure of polymer
Linyue
Tong
ab,
Xiaoqing
Liu
ab,
Jianping
Deng
*ab and
Wantai
Yang
*ab
aState Key Laboratory of Chemical Resource Engineering, Beijing University of Chemical Technology, Beijing, 100029, China. E-mail: dengjp@mail.buct.edu.cn; yangwt@mail.buct.edu.cn; Fax: +86-10-6443-5128; Tel: +86-10-6443-5128
bCollege of Materials Science and Engineering, Beijing University of Chemical Technology, Beijing, 100029, China
First published on 27th August 2010
Abstract
Polymerization of M1, a chiral N-propargylamide monomer, was carried out with (nbd)Rh+B−(C6H5)4 as the catalyst in five solvents to explore the effect of solvents on polymerization. All the polymerizations occurred smoothly and provided polymers in high yield, however the number-average molecular weights of the polymers differ largely, which is attributed to the different solubility of the polymers in solvents. The helical structure and the optical activity of the polymer prepared in THF was examined by CD and UV-Vis spectroscopy measurements in the five solvents and in solvent mixtures consisting of CH2Cl2 (a relatively good solvent) and THF (a relatively not-so-good solvent) in varied ratios. The polymer could adopt helical conformations in all the solvents, but different CD intensities and UV-Vis absorptions were observed. In CH2Cl2, the polymer exhibited lower intensity in both CD effect and UV-Vis absorption, while they were higher when the polymer was examined in THF, demonstrating that solvophobic effects made large contribution for the polymer chains to adopt helical conformations.
Introduction
Helical polymers have received a great deal of attention because of their visually attractive structures and particularly excellent electronic and chiroptical properties.1–6 Over the past decade, a large variety of helical polymers have been synthesized ranging from polysilanes,6 polyisocyanides,7 polymethacrylates8 to polyacetylenes.4,5,9 Among them, polyacetylenes and their derivatives are gathering a lot of interest as typical synthetic helical polymers. The underlying driving force for the enormous investigations of helical polyacetylenes originates in the versatile conjugated polymer main chains and the functional pendant groups. A combination of the conjugated polymer backbones able to form ordered helical structures and functional groups in side chains shall create a series of advanced polymeric materials.Some groups2,4,5,9,10 have contributed largely to acetylene-based helical polymers, affording great progress in this significant research field. We have recently reported a series of mono-substituted polyacetylenes, including poly(N-propargylamide)s,11,12 poly(N-propargylsulfamide)s,13 and poly(N-propargylurea)s.14,15 The polymers with moderate pendent groups could adopt helical structures under suited conditions. For hydrophobic monomers, they frequently underwent polymerization in organic solvents. However, interestingly, we built a facile and straightforward approach to preparing hydrophobic helical polymers in aqueous media.16 More importantly, we obtained emulsions consisting of helical polymers,17 and even core/shell nanoparticles consisting of helical substituted polyacetylenes (the core) and vinyl polymers (the shell).18 In another more recent study, we developed asymmetric polymerizations of achiral monomers in chiral micelles.19
The studies from us and in particular from the Masuda,4 Yashima,2 and Tang5 groups demonstrated that, for mono-substituted polyacetylenes to form helical conformations, some key factors should be taken into account, including internal factors (e.g., length20 and the bulkiness21 of the pendent groups) and external factors (e.g., temperature22). In the course of research, we found that solvents considerably affected both the polymerization of the monomers and the secondary structures of the resulting polymers. Nevertheless, only a few reports could be found in literature focusing on this important research topic.23,24 Thus detailed and systemic studies on this theme are still required for a complete understanding of the artificially helical polymers.
The present study is therefore devoted to a detailed exploration of the effects of solvents in two aspects: the polymerization of monomers and the secondary structures of the obtained polymers. To this end, M1 (shown in Scheme 1) was taken as a representative monomer and was polymerized in different solvents. Poly(1) (derived from M1) was selected as a model due to its relatively good solubility among the helical polyacetylenes of the same type.25 The polymer obtained was then subjected to CD and UV-Vis spectroscopic analysis. For a further deep understanding of the effects of solvents, poly(1) was investigated in solvent mixtures consisting of a relatively good solvent and a not-so-good solvent in varied ratios. The present investigations provide deep insights into the helical substituted polyacetylenes.
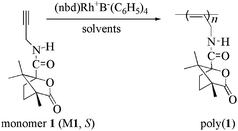 | ||
| Scheme 1 The structure of the monomer (M1) and the polymer. | ||
Experimental
Measurements
Number-average molecular weights (Mn) and molecular weight polydispersity indices (Mw/Mn) of the polymers were determined by GPC (Waters 515–2410 system) calibrated by using polystyrenes as standards and THF as an eluent. Circular dichroism (CD) and UV-Vis absorption spectra were recorded on a JASCO J-810 spectropolarimeter. Specific rotations ([α]) were measured on a JASCO P-1020 digital polarimeter with a sodium lamp as light source at room temperature.Materials
The solvents, THF (tetrahydrofuran), DMF (dimethyl formamide), CHCl3, CH2Cl2, and C7H8 (toluene) were freshly distilled under reduced pressure before use. (nbd)Rh+B−(C6H5)4 was prepared according to the literature.26 The monomer (M1) was prepared by the method reported previously.25Polymerization
A detailed description of polymerization was reported in earlier articles.11–16,27 A typical polymerization procedure is briefly stated as follows. Polymerization was performed in the presence of Rh-based catalyst in a dry solvent at 30 °C for 1 h under nitrogen. The concentration of M1 was 0.2 M, and the concentration of Rh-catalyst was 2 mM. After polymerization, the resulting solution was poured into a large amount of hexane to precipitate the polymer. Herein, it should be pointed out that when CHCl3, CH2Cl2, THF, and C7H8 were used as the solvent, precipitation of the resulting polymer could be achieved by directly pouring the polymerization solution into a large amount of hexane. However, when DMF was employed as the solvent, the polymer precipitated with much difficulty when simply pouring the polymerization solution into hexane. In this case, a small amount of THF should be added in the polymerization solution first so as to transfer the DMF system to a THF system, and then a large amount of hexane was added to the mixture to precipitate the polymer. The polymers were filtered, washed with solvents, and then dried under reduced pressure.Results and discussion
Effects of solvents on polymerization
Substituted acetylene monomers could be polymerized with various transition metal catalysts, to which Masuda et al.,28 Tabata et al.,29 and Tang and co-workers30 have made large contributions. Rh-based catalysts were found to be able to efficiently polymerize mono-substituted acetylene monomers carrying polar functional groups in protic solvents and even in water31,32 to prepare polymers with high stereoregularity, predominantly cis structures.33,34 This advantage of Rh catalysts means that they have been widely employed in the polymerization of mono-substituted acetylenes. Accordingly, Rh catalyst, (nbd)Rh+B−(C6H5)4, was selected to polymerize M1 in five solvents in the present study, including THF, DMF, CHCl3, CH2Cl2, and C7H8. It was found that this catalyst was indeed highly efficient in spite of the altering nature of solvents, as will be discussed later on. All the polymers were found to possess a high stereoregularity, i.e., a high cis content, approx. 100%, like the other polymers of the same type.11–22,24,25Five solvents were employed for the polymerization of M1. The polymerization results are listed in Table 1. It was found that the yields of poly(1) were quite high, i.e., nearly 100%, except for the poly(1) obtained from DMF, since the polymer was hardly precipitated totally when DMF was used as solvent. This situation was mentioned in Experimental section. Among the prepared polymers, poly(1)–THF (THF indicates the solvent in which M1 was polymerized to afford poly(1)–THF. The same is true with the other designations below) was a pale yellow solid, while the other four polymers were light yellow solid but felt more rubbery than poly(1)–THF. The different appearance of the poly(1)s should be closely related to their different molecular weights. Poly(1)s with moderate number-average molecular weights (Mn = 4,600–10,000) were obtained and their polydispersity indices (Mw/Mn) were in a range of 1.3–2.7. Thus Mns of the formed polymers demonstrated a clear solvent-dependence although all the polymerizations were carried out under the same conditions only with the exception of the solvent.
| poly(1)a | Yield (%)b | M n | M w/Mnc | [α]D (deg)d |
|---|---|---|---|---|
| a The solvents following poly(1) indicate the solvent from which the polymer was obtained. b Hexane-insoluble part. c Measured by GPC (THF as solvent, polystyrenes as standards). d Measured by polarimeter at room temperature, c = 0.090–0.1 g/dL in CHCl3 for polymerization with the soluble part as sample. | ||||
| poly(1)–THF | 96 | 4,600 | 2.70 | −392 |
| poly(1)–DMF | 82 | 7,100 | 1.47 | −659 |
| poly(1)–CHCl3 | 97 | 9,000 | 1.36 | −782 |
| poly(1)–CH2Cl2 | 98 | 10,000 | 1.53 | −917 |
| poly(1)–C7H8 | 99 | 10,300 | 1.30 | −544 |
When THF was used as the solvent for polymerization, the Mn of poly(1)–THF was just 4,600, much lower then those of the other poly(1)s. The low Mn of poly(1)–THF was attributable to the relatively lower solubility of poly(1) in THF. This consideration was well evidenced by the solubility examination of poly(1) in the five solvents, as demonstrated in Table 2.
Table 2 shows CH2Cl2 and C7H8 are good solvent for the five poly(1)s with known Mns, since all of them could dissolve completely in these two solvents. For the other solvents, DMF, THF, and CHCl3, only the poly(1)s with relatively lower Mns (≤9000) could entirely dissolve in them. The poly(1)s with Mns being higher than 10000 only partly dissolve in these three solvents. From these results, it is indicated that DMF, THF, and CHCl3 are not-so-good solvent for poly(1), when compared with CH2Cl2 and C7H8, both of which can be considered as good solvent for poly(1) in the present study.
In Table 1, the data on specific rotation changed drastically for the five poly(1)s. This result can be originated from two reasons. The first one is related to the solubility of poly(1)s in the solvent, which affected the secondary structures of the polymers and their optical activities. This will be discussed in more detail later. The second reason is due to the variations in Mns of poly(1)s. It has been revealed by different research groups that the molecular weight of a polymer affected its optical activity to a large degree. According to the studies from Angiolini35 and Gu,36 the optical rotation increased with an increase in molecular weight within a specific range. When the molecular weight exceeded this range, optical rotation would not increase further. Nevertheless, for polymers of different types, this molecular weight range would differ greatly. In Table 1, we also can see that the optical rotation increased with the increase in molecular weight of the polymer, with the exception of poly(1)-C7H8. The detailed reason is not clear at present.
Poly(1) from M1 was known to form helices by CD and UV-Vis spectra analysis, according to the earlier excellent work from Masuda et al.25 To elucidate the effects of solvents on the secondary structure of the polymers, we employed poly(1)–THF as the polymer model since it could dissolve with much ease in all the five solvents. Fig. 1 presents the obtained CD and UV-Vis spectra. In previous studies, it has been proved repeatedly that, for poly(N-propargylamide)s, when the polymer showed a strong UV-Vis absorption in a wavelength frequently between 350–400 nm, the polymer was considered to form helical conformations under the examined conditions. Furthermore, for the polymers able to form a preferential helicity, intense CD effects also could be observed in the CD spectra.11–22
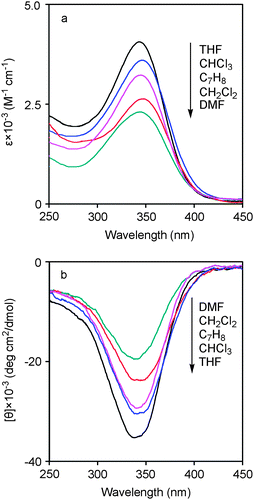 | ||
| Fig. 1 (a) UV-Vis and (b) CD spectra of poly(1)–THF, which was obtained from THF, measured in different solvents at room temperature (c = 0.1 mM). | ||
In Fig. 1a, poly(1)–THF showed intense UV-Vis absorptions around 350 nm in all the solvents examined. However, the same polymer demonstrated different absorbency in different solvents. The UV-Vis absorption decreased in the order of THF, CHCl3, C7H8, CH2Cl2, and DMF. In Fig. 1b, similar observations to Fig. 1a were demonstrated, namely, the absolute value of CD signal intensity increased in the same sequence as above. DMF is so polar that, when used as the solvent, it destroyed the hydrogen bonds formed intramolecularly. Such intramolecular hydrogen bonds play vital roles for the polymer main chains to adopt stable helices.15,20
On the basis of the discussion above, namely, C7H8 and CH2Cl2 are good solvents while THF and CHCl3 are not-so-good solvents for poly(1), we considered that in the good solvents, poly(1) tended to form disordered coils due to the relatively free mobility of the polymer chains, while in the not-so-good solvents, the polymer chains are ready to adopt ordered helical conformations due to their relatively lower mobility. This solvophobic effect has been well investigated and was found to be favorable for the formation of helical structures.37,38 For instance, the Moore group has reported solvophobically driven helices observed in phenylene ethynylene macrocycles and oligomers.39,40 Adisa and colleagues also explored the solvophobic effect on polymeric folding.41 More recently, Mikami and coworkers reported the helical folding prompted by solvophobic effect in poly(naphthalenecarboxamide).42 Similar effects were also observed in conjugated oligo(o-phenyleneethynylene-alt-p-phenyleneethynylene) systems.43
The above consideration was further verified by examination of the secondary structures of poly(1)–THF in solvent mixtures consisting of a good solvent, CH2Cl2, and a not-so-good solvent, THF, in varied ratios. The obtained results are presented in Fig. 2. Fig. 2a shows that with the increase in CH2Cl2 in the mixed solvent, UV-Vis absorption decreased gradually, while Fig. 2b demonstrates the similar tendency in CD effects to UV-Vis absorption observed in Fig. 2a. Hence Fig. 2 clearly shows the effects of solvents on the secondary structures of poly(1)–THF. In order to provide a more visually clear description, both UV-Vis absorption and CD signal intensity are plotted as a function of THF content in the mixed solvent, as illustrated in Fig. 3. The solvent-dependence of the helical structures, which are reflected by ε and [θ], can be clearly observed.
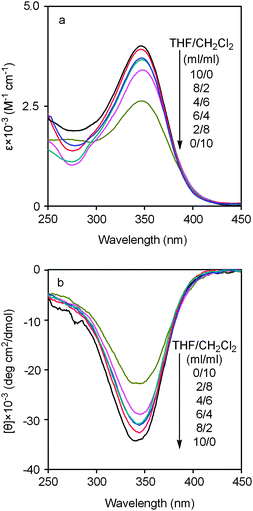 | ||
| Fig. 2 (a) UV-Vis and (b) CD spectra of poly(1)–THF, which was obtained from THF, measured at room temperature in mixed solvents consisting of THF–CH2Cl2 = 0/10, 2/8, 4/6, 6/4, 8/2, 10/0 in ml/ml (c = 0.1 mM). | ||
![The solvent-dependence of ε and [θ] of poly(1)–THF, according to Fig. 2.](/image/article/2010/PY/c0py00116c/c0py00116c-f3.gif)
According to Fig. 1–3, we proposed a schematic representation of the secondary structures of poly(1)–THF in a good solvent and in a not-so-good solvent, as displayed in Scheme 2. In a good solvent, the polymer chains moved freely, and hence there are more disordered chain segments and turning points between helices. Moreover, conformational transitions occurred more readily between helices and random coils. In the contrast, in a not-so-good solvent, the mobility of the polymer chains was hindered to a large degree due to the not-so-good solubility of the polymer, from which the polymer chains are ready to adopt order helical conformations. The investigations in the present study will be helpful for us to understand more well of the formation of helical structures in artificial polymers.
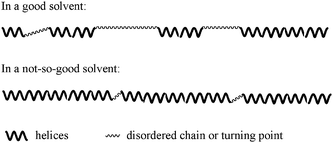 | ||
| Scheme 2 A schematic representation of a polymer chain in a good solvent and in a not-so-good solvent. | ||
Conclusions
The polymerization of M1 was carried out in five solvents. The secondary structures of the polymers were examined by polarimeter, CD and UV-Vis spectroscopic methods. It was found that the solubility of the polymer affected largely the Mns of the monomer and the secondary structures of the polymers. In a good solvent, the polymer chains tend to adopt disordered conformations, while in a not-so-good solvent, the polymer chains tend to form helical conformations. Solvophobic effects were observed in the polymers in not-so-good solvents.Acknowledgements
The project was supported by the “Program for New Century Excellent Talents in University” (NCET-06-0096), the “National Science Foundation of China” (20974007), and the “Program for Changjiang Scholars and Innovative Research Team in University” (PCSIRT, IRT0706).Notes and references
- J. H. K. Ky Hirschberg, L. Brunsveld, A. Ramzi, A. J. M. Jef Vekemans, R. P. Sijbesma and E. W. Meijer, Nature, 2000, 407, 167 CrossRef CAS.
- E. Yashima, K. Maeda, H. Iika, Y. Furusho and K. Nagai, Chem. Rev., 2009, 109, 6102 CrossRef CAS.
- M. M. Green, J. W. Park, T. Sato, A. Teramoto, S. Lifson, R. L. B. Selinger and J. V. Selinger, Angew. Chem., Int. Ed., 1999, 38, 3138 CrossRef.
- T. Masuda, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 165 CrossRef CAS.
- J. E. Y. Lam and B. Z. Tang, Acc. Chem. Res., 2005, 38, 745 CrossRef CAS.
- M. Fujiki, Top. Curr. Chem., 2008, 284, 119 CAS.
- T. Kajitani, H. Z. Lin, K. Nagai, K. Okoshi, H. Onouchi and E. Yashima, Macromolecules, 2009, 42, 560 CrossRef CAS.
- S. Habaue and Y. Okamoto, Chem. Rec., 2001, 1, 46 CrossRef CAS.
- J. G. Rudick and V. Percec, Acc. Chem. Res., 2008, 41, 1641 CrossRef CAS.
- T. Aoki, T. Kaneko, N. Maruyama, A. Sumi, M. Takahashi, T. Sato and M. Teraguchi, J. Am. Chem. Soc., 2003, 125, 6346 CrossRef CAS.
- J. P. Deng, W. G. Zhao, J. M. Wang, Z. G. Zhang and W. T. Yang, Macromol. Chem. Phys., 2007, 208, 218 CrossRef CAS.
- J. P. Deng, J. Tabei, M. Shiotsuki, F. Sanda and T. Masuda, Macromolecules, 2004, 37, 7156 CrossRef CAS.
- Z. G. Zhang, J. P. Deng and W. T. Yang, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 500 CrossRef CAS.
- J. P. Deng, X. F. Luo, W. G. Zhao and W. T. Yang, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 4112 CrossRef CAS.
- X. F. Luo, J. Chang, J. P. Deng and W. T. Yang, React. Funct. Polym., 2010, 70, 116 CrossRef CAS.
- L. Ding, X. F. Jiao, J. P. Deng, W. G. Zhao and W. T. Yang, Macromol. Rapid Commun., 2009, 30, 120 CrossRef CAS.
- J. P. Deng, B. Chen, X. F. Luo and W. T. Yang, Macromolecules, 2009, 42, 933 CrossRef CAS.
- B. Chen, J. P. Deng, X. Q. Liu and W. T. Yang, Macromolecules, 2010, 43, 3177 CrossRef CAS.
- X. F. Luo, L. Li, J. P. Deng, T. T. Guo and W. T. Yang, Chem. Commun., 2010, 46, 2745 RSC.
- J. P. Deng, J. Tabei, M. Shiotsuki, F. Sanda and T. Masuda, Macromolecules, 2004, 37, 1891 CrossRef CAS.
- J. P. Deng, J. Tabei, M. Shiotsuki, F. Sanda and T. Masuda, Macromolecules, 2004, 37, 5149 CrossRef CAS.
- J. P. Deng, J. Tabei, M. Shiotsuki, F. Sanda and T. Masuda, Macromol. Chem. Phys., 2004, 205, 1103 CrossRef CAS.
- H. Goto, E. Yashima and Y. Okamoto, Chirality, 2000, 12, 396 CrossRef CAS.
- J. Tabei, R. Nomura, S. Shiotsuki, F. Sanda and T. Masuda, Macromol. Chem. Phys., 2005, 206, 323 CrossRef CAS.
- J. Tabei, R. Nomura and T. Masuda, Macromolecules, 2002, 35, 5405 CrossRef CAS.
- R. R. Schrock and J. A. Osborn, Inorg. Chem., 1970, 9, 2339 CrossRef CAS.
- J. P. Deng, J. Tabei, M. Shiotsuki, F. Sanda and T. Masuda, Macromolecules, 2004, 37, 9715 CrossRef CAS.
- T. Masuda, E. Isobe, T. Higashimura and K. Takada, J. Am. Chem. Soc., 1983, 105, 7473 CrossRef CAS.
- M. Tabata, T. Sone and Y. Sadahiro, Macromol. Chem. Phys., 1999, 200, 265 CrossRef CAS.
- K. Xu, H. Peng, J. W. Y. Lam, T. W. H. Poon, Y. P. Dong, H. Y. Xu, Q. Sun, K. K. L. Cheuk, F. Salhi, P. P. S. Lee and B. Z. Tang, Macromolecules, 2000, 33, 6918 CrossRef CAS.
- K. Maeda, H. Goto and E. Yashima, Macromolecules, 2001, 34, 1160 CrossRef CAS.
- B. Z. Tang, W. H. Poon, S. M. Leung, W. H. Leung and H. Peng, Macromolecules, 1997, 30, 2209 CrossRef CAS.
- M. Tabata, Y. Tanaka, Y. Sadahiro, T. Sone, K. Yokota and I. Miura, Macromolecules, 1997, 30, 5200 CrossRef CAS.
- M. V. Russo, A. Furlani and R. D'Amato, J. Polym. Sci., Part A: Polym. Chem., 1998, 36, 93 CrossRef CAS.
- L. Angiolini, T. Benelli, L. Giorgini and E. Salatelli, Polymer, 2006, 47, 1875 CrossRef CAS.
- H. Gu, Y. Nakamura and T. Sato, Macromolecules, 1995, 28, 1016 CrossRef CAS.
- T. Giorgi, S. Lena, P. Mariani, M. A. Cremonini, S. Masiero, S. Pieraccini, J. P. Rabe, P. Samori, G. P. Spada and G. Gottarelli, J. Am. Chem. Soc., 2003, 125, 14741 CrossRef CAS.
- J.-L. Hou, M.-X. Jia, X.-K. Jiang, Z.-T. Li and G.-J. Chen, J. Org. Chem., 2004, 69, 6228 CrossRef CAS.
- S. Lahiri, U. L. Thompson and J. S. Moore, J. Am. Chem. Soc., 2000, 122, 11315 CrossRef CAS.
- R. B. Prince, T. Okada and J. S. Moore, Angew. Chem., Int. Ed., 1999, 38, 233 CrossRef CAS.
- B. Adisa and D. A. Bruce, J. Phys. Chem. B, 2005, 109, 19952 CrossRef CAS.
- K. Mikami, A. Tanatani, A. Yokoyama and T. Yokozawa, Macromolecules, 2009, 42, 3849 CrossRef CAS.
- N. Zhu, W. Hu, S. Han, Q. Wang and D. Zhao, Org. Lett., 2008, 10, 4283 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |