‘Isothermal’ phase transitions and supramolecular architecture changes in thermoresponsive polymers via acid-labile side-chains†
Felicity
Heath
a,
Aram Omer
Saeed
a,
Sivanand S.
Pennadam
a,
Kristofer J.
Thurecht
b and
Cameron
Alexander
*a
aThe School of Pharmacy, Boots Science Building, University of Nottingham, University Park, Nottingham, NG7 2RD, UK. E-mail: cameron.alexander@nottingham.ac.uk; Fax: +44 (0)115 951 5102; Tel: +44 (0)846 7678
bAustralian Institute for Bioengineering and Nanotechnology (AIBN), Corner College and Cooper Roads (Building 75), The University of Queensland, Brisbane, Queensland 4072, Australia
First published on 5th July 2010
Abstract
Polymers designed to change their conformation via a phase transition triggered by acidic cleavage of a hydrophobic side-chain have been synthesized and characterised. The new materials were prepared by co-polymerising N-isopropylacrylamide with an acetal-containing pH-sensitive monomer N-(2-(2,4,6-trimethoxyphenyl)-1,3-dioxan-5-yl)acrylamide (TMPDA) and then grafting the resultant linear co-polymers to branched poly(ethyleneimine). The final three-component polycations exhibited Lower Critical Solution Temperature (LCST) behaviour. The structures of these polymers, their solution behaviour and their self-association were characterized by DLS and TEM in water and buffer solutions. The acid-triggered hydrolysis of trimethoxybenzeneacetal side-chains on the poly(N-isopropylacrylamide-co-TMPDA) grafts resulted in changes in lower critical solution temperatures and in solution self-assembly; thus in effect creating an ‘isothermal’ phase transition. The changes in polymer conformation, at acidity levels corresponding to those in cell endosomes, offer promise for these polymers to act as controlled release materials.
Introduction
Materials that respond to external stimuli have been a focus of research for many applications, including sensing, diagnostics, computation, actuation and controlled release.1–8 Within the biomedical sector, responsive polymers have been considered as ‘smart’ therapeutic agents,9 as carriers for drugs10 and as supports for tissue engineering.11–13 For optimal use in the biological environment, responsive materials must change their properties, for example to protect a delicate biopolymer or cell cargo then release it at the target site, under as mild a stimulus as possible. Many groups have reported the use of thermoresponsive materials, based on co-polymers of N-isopropylacrylamide for drug delivery applications, wherein the coil-to-globule phase transition of the polymer at its Lower Critical Solution Temperature (LCST) alters its solution properties and ability to encapsulate a drug.14–17 For biopolymer drugs such as nucleic acids, enhanced transgene expression has been reported when cells incubated with cationic poly(N-isopropylacrylamide) (PNIPAm) co-polymer complexes with nucleic acids experienced a temperature below that of the polymer LCST.18–24 Although the detailed mechanisms by which PNIPAm co-polymers change transfection efficiency through a temperature change are not fully understood, the studies suggest that a change in hydrophilicity and polymer architecture is an important contributory factor. However, the use of temperature stimuli to invoke changes in polymer behaviour is sub-optimal for many applications and is not easily executed in vivo. Therefore, we have been developing strategies to cause phase transitions and supramolecular architecture modifications in polymers without a temperature change. In this paper we report the synthesis and characterization of PNIPAm-based cationic co-polymers that associate into micellar-like assemblies under normal physiological conditions but undergo a change in LCST triggered by side-chain hydrolysis that perturbs the overall supramolecular architecture—in effect an ‘isothermal’ temperature response. The overall effect is a loss of self-association behaviour and a disruption of micellar assembly at pH values around those occurring in the endosomal compartments of cells and in certain areas of solid tumours. The ability to change polymer properties through external or biologically relevant stimuli25,26 is potentially advantageous for many materials applications including logic operations, sensing, actuation as well as controlled release.27–30Results and discussion
We based our strategy for changing the phase-transition temperature of a polymer on the well-known properties of PNIPAm co-polymers, whereby incorporating a hydrophilic co-monomer raises the LCST while co-polymerisation with a hydrophobic co-monomer reduces the LCST. We further refined the approach to utilise a biologically relevant trigger for the phase change, in this case the drop in pH from 7.4 in the cytosol to pH 5–6 in the late endosome/lysosome.31 The intention was to cleave a hydrophobic group from a PNIPAm co-polymer side-chain, leaving a hydrophilic moiety, which in turn would increase the LCST and change the self-association of the polymer without requiring a temperature shock. We also intended to attach the PNIPAm chains to a second component, that could bind biopolymers via charge–charge interactions, and chose for this section poly(ethyleneimine) (PEI), which is widely used for in vitro and in vivo gene delivery experiments.32,33 Overall, we intended that the resultant terpolymer should assemble into micellar or vesicular structures above an initial LCST in order to present a positively charged outer surface, suitable for binding negatively charged biopolymers. However, at pH values ∼ 5 to 6, as postulated to occur in endosomal compartments, the LCST should be changed through side-chain cleavage, such that the micelles disassemble to smaller fragments with no supramolecular structure. The schematic of the polymer design, utilising a cationic PEI block and acid-labile PNIPAm segments grafted onto it, is shown in Fig. 1. | ||
| Fig. 1 Schematic of phase transition temperature changes with pH-cleavable side-chain functionality. Association of hydrophobic side-chains above LCST 1 drives self-assembly: loss of hydrophobic groups at pH 5.6 changes polymer association behaviour such that LCST of the hydrolysed polymer is greater than the LCST of the non-hydrolysed polymers (LCST 2 > LCST 1). | ||
The concept of a change in the properties of a co-polymer side-chain leading to an increase in LCST was first reported by the Hennink group34,35 but had not previously been used to change association behaviour of terpolymers. We accordingly designed a new monomer that could be co-polymerised with NIPAm and which should lose a hydrophobic aromatic moiety and become water-soluble at pH 5–6. Acetal chemistry offers many advantages for controlled release and drug delivery applications36–39 as the stability of this link is considerably lower at lower pH values e.g. 5–6 compared to normal physiological pH. The synthesis of the pH-sensitive acetal-containing monomer, N-(2-(2,4,6-trimethoxyphenyl)-1,3-dioxan-5-yl)acrylamide (I), was based on the methodology developed by Gillies and Frechet40 for degradable main-chain polymers, and is shown in Fig. 2.
 | ||
| Fig. 2 Synthesis of monomer and polymers: (a) pH-sensitive monomer N-(2-(2,4,6-trimethoxyphenyl)-1,3-dioxan-5-yl)acrylamide (I) (TMPDA) and (b and c) polymer preparation routes. | ||
Polymerisation of monomer TMPDA (I) with NIPAm in different co-monomer ratios yielded polymers P2, P3 and P5. The homopolymer, PNIPAm, was synthesised as a control thermo-sensitive polymer (P1) and polymer P4 was synthesized through the co-polymerisation of (I) with NIPAm and N-(1,3-dihydroxypropan-2-yl)methacrylamide (DHPMA) (Table 1). In all cases 2-aminoethanethiol was used as a chain transfer agent to control molecular mass and to provide an amine-terminus for further grafting.
| Polymer | M n (GPC)a/kDa | PDIa | M n (NMR)/kDab | Cloud point at pH 7.4c/°C | |
|---|---|---|---|---|---|
| a Eluent: DMAc and 0.1% LiBr. b End-group analysis in DMSO-d6. c Cloud points determined at 5 mg mL−1, except for P5 (2 mg mL−1) due to solubility constraints. | |||||
| P1 | PNIPAm | 13.7 | 3.2 | 13.8 | 32 |
| P2 | PNIPAm-co-TMPDA (99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) :1) |
6.1 | 2.7 | 9.5 | 33 |
| P3 | PNIPAm-co-TMPDA (95:5) |
6.0 | 2.6 | 8.9 | 23 |
| P4 | PNIPAm-co-TMPDA-co-DHPMA (88:6:6) |
34.0 | 2.8 | 7.1 | 21 |
| P5 | PNIPAm-co-TMPDA (91:9) |
11.1 | 2.9 | 12.2 | 16 |
Primary characterisation of the molar mass of the polymers was carried out by 1H NMR in DMSO-d6 using end-group integral analysis comparing the isopropylamide methyl signals at δ = 1.18–0.88 ppm to trimethoxyphenyl protons at δ = 6.2 ppm and aminoethanethioether protons centred at 2.95 and 2.7 ppm. Gel Permeation Chromatography (GPC) was used as a secondary method of molar mass analysis: chromatography was carried out in organic solvents on account of the difficulties in obtaining accurate molar masses for PNIPAm materials in water.41 Agreement in molar masses via the two different methods was good for the homopolymer P1, with a variation of less than 5% but less so for the co-polymers most likely due to the poorer correlation in GPC compared to PMMA standards.
The primary phase transition responses for these materials were evaluated through cloud-point determinations at pH 7.4, i.e. normal physiological pH. As expected, substitution of the hydrophobic TMPDA co-monomer lowered the LCST (as determined by cloud point at constant concentration) compared to PNIPAm homopolymer P1. The phase transitions were then assessed as the pH was changed from 7.4 to 5.6, to reflect the expected pH values for plasma and endosomal compartments, respectively. In all cases where the acid-labile TMPDA co-monomer was included, the cloud point increased over time at pH 5.6. Representative temperature–turbidity graphs for TMPDA-substituted co-polymers P3 and P5 are shown in Fig. 3.
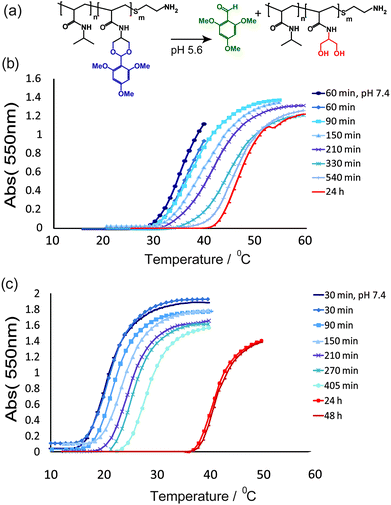 | ||
| Fig. 3 (a) Scheme showing the hydrolysis of acetal polymer side-chains at pH 5.6. Change in UV absorption at 550 nm with temperature for linear polymers P3 (b) and P5 (c) over increasing times at pH 5.6. In (b) and (c) multiple aliquots of each polymer (1 mg mL−1) in PBS (10 mM, pH 5.6) were incubated at 37 °C. An aliquot of each polymer was taken for analysis at each time point and quenched with sodium hydroxide (20 µL, 1 M) to stop further hydrolysis before UV spectrophotometric analysis. | ||
The extent of cloud point change through TMPDA cleavage in acidic media was, as expected, dependent on co-monomer ratio as measured by NMR. For P3, with 5 mol% TMPDA, the cloud point changed from 27 °C at pH 7.4 to 42 °C at pH 5.6, whereas for P5 with 9 mol% TMPDA the corresponding change was from 16–37 °C.42,43
In order to evaluate materials with a change in LCST in and around normal physiological temperature (37 °C) we selected polymers P1 (non-acid-labile), and P4 and P5 (both acid-labile and with the greatest change in LCST over the pH range). These were used to graft to branched 25 kDa poly(ethyleneimine) (PEI) as a representative polycation widely used in nucleic acid delivery systems. We adapted our prior method19 of end-grafting via the heterobifunctional linker EMCS, giving P1-graft PEI, P4-graft PEI and P5-graft PEI in 52%, 35% and 21% yields, respectively, after extensive dialysis to remove excess free PEI (Fig. 4).
 | ||
| Fig. 4 Synthetic route to responsive graft terpolymers from linear polymers P1, P4, and P5 via 2-stage heterobifunctional linker coupling. | ||
The resultant polymers were characterised by NMR as before. Graft contents were calculated from NMR integrals and molar masses obtained by GPC with triple detection. As a third method of determining molar mass and to confirm grafting densities, titration of amine end-groups using 2,4,6-trinitrobenzenesulfonic acid was performed.44
While PNIPAm-g-PEI (P1-g-PEI) showed essentially invariant cloud points at pH 7.4 and 5.6, polymers P4-g-PEI and P5-g-PEI exhibited different cloud points at the different pH values: these and other key properties of the polymers are given in Table 2.
| Polymer | M w/kDa | M n/kDa | Cloud point at pH 7.4/°C | Cloud point at pH 5.6/°C |
|---|---|---|---|---|
| P1-g-PEI | 69 | 24 | 31 | 31 |
| P4-g-PEI | 174 | 44 | 22 | 36 |
| P5-g-PEI | 57 | 21 | 22 | 40 |
The observation that the cloud point of P1-g-PEI was very similar to the parent PNIPAm P1 suggested the PNIPAm and PEI blocks within the same co-polymer chains were not strongly interacting with each other. However, for the acetal-containing polymers P4-g-PEI and P5-g-PEI the cloud points at pH 7.4 were not the same as for linear polymers P4 and P5, indicating a change in supramolecular order through introduction of the aromatic 2,4,6-(trimethoxyphenyl)-1,3-dioxan-5-yl side-chains. At pH 5.6, P4-g-PEI exhibited a cloud point just below normal physiological temperature, whereas P5-g-PEI displayed a cloud point of 40 °C.
As the pH-mediated variations in cloud point were most manifest for P5-g-PEI, experiments were carried out to monitor the rate of change of phase transition for this polymer.
The shapes of the time–temperature curves at pH 7.4 and 5.6 implied a difference in intermolecular association over this range for P5-g-PEI, whereas P1-g-PEI did not change cloud point under the same conditions and this latter polymer exhibited the same sharp time–temperature curve over both pH values (Fig. 5).
 | ||
| Fig. 5 (a) Chemical structure of P5-g-PEI both before (top structure) and after hydrolysis (lower structure). Temperature–turbidity profiles with time at pH 5.6 for (b) non-hydrolysable side-chain polymer P1-g-PEI (1 mg mL−1) and (c) hydrolysable side-chain polymer P5-g-PEI (1 mg mL−1). | ||
Based on the temperature–turbidity profiles, we estimate the hydrolysis of the acetal side-chains in the terpolymer to have been essentially complete within 210 minutes, which is faster than that observed for 5-membered acetals of similar chemistries45 but slower than the 6-membered acetals originally reported by Gillies and Frechet.40 Interestingly, the hydrolysis of side-chains appeared to take place faster for the block co-polymers compared to the linear co-polymers from which they were derived, suggesting perhaps a role for the PEI, as a highly soluble component, in accelerating the acetal degradation.
The changes in temperature–turbidity curves with pH for P5-g-PEI but not P1-g-PEI, combined with our prior observation of supramolecular association in PNIPAm-g-PEI systems,22 implied the formation of micellar structures for the non-hydrolysable polymers at pH 7.4 and 5.6, but less order for the hydrolysed TMPDA-containing graft-PEI polymer at pH 5.6.
Dynamic light scattering measurements of aqueous grafted copolymer solutions, at body temperature and pH 7.4, confirmed the formation of supramolecular assemblies for P1-g-PEI, P4-g-PEI and P5-g-PEI. Importantly, there were different DLS profiles with temperature following a pH change for 8 hours across the different co-polymers (Table 3).
| Polymer | R H at 25 °C, pH 7.4/nm | R H at 37 °C, pH 7.4/nm | R H at 37 °C, pH 5.6/nm |
|---|---|---|---|
| a Figures in parentheses are the percentage of the overall population for the particle size quoted. Data are number distributions (calculated from recorded intensity distributions) obtained from CONTIN analysis. | |||
| P1-g-PEI | 7 ± 0.8 (59) | 132 ± 20 (100) | 73 ± 16 (100) |
| P4-g-PEI | 13 ± 0.7 (100) | 73 ± 12 (100) | 52 ± 17 (100) |
| P5-g-PEI | 18 ± 1.3 (100) | 69 ± 14 (100) | 5 ± 0.5 (98) |
Experiments carried out over room temperature and physiological temperature ranges and the two pH values indicated that for P1-g-PEI polymers at room temperature and pH 7.4 the predominant fractions (59%) ranged between RH = 2–7 nm. These were likely to have been loosely associated polymer chains rather than well-structured micelles.22 The remaining fractions were made up of species 2 nm or less in size (36% by mass)46 with the residual 4% of the mass being particles of RH = 97 nm. Increasing the temperature to 37 °C (i.e. above the LCST of P1-g-PEI) resulted in complete conversion to higher-order structures of RH ≈ 132 nm in hydrodynamic radius. Changing the pH of the solution to pH 5.6 reduced the apparent particle size (RH = 73 nm), but these species were again likely to have been micellar or vesicular in nature. The reduction in volume was probably caused by increased curvature of the PEI domains at the exterior as they were increasingly protonated and a compaction of the PNIPAm core. P4-g-PEI formed slightly larger particles than P1-g-PEI at 25 °C (13 nm) but smaller supramolecular assemblies at 37 °C at both pH 7.4 and 5.6 (radius RH ≈ 73 nm and 52 nm, respectively) compared to P1-g-PEI. This suggested some higher order structures were present for this polymer across the whole pH and temperature range, albeit with much larger particle sizes at 37 °C.
By contrast, while P5-g-PEI formed particles with similar radii (18 nm) to P4-g-PEI at 25 °C and 37 °C (RH ≈ 69 nm), a marked change in DLS profile was observed at pH 5.6 and 37 °C. After the same length of time at pH 5.6 as the other polymers, P5-g-PEI polymers were present as much smaller species, with a very low proportion (2.1% by number) of >50 nm particles, and most of the sample (98% by number) centred around 5 nm. This indicated that supramolecular assemblies in P5-g-PEI formed above an initial LCST at pH 7.4, but that loss of the hydrophobic side-chains through hydrolysis at pH 5.6 resulted in an increase of LCST to above the assay temperature of 37 °C. In turn, the increase of polymer LCST resulted in loss of associative order, and micellar/vesicular disassembly as the PNIPAm-co-TMPDA chains expanded from collapsed globules to extended hydrophilic chains.
We selected polymer P5-g-PEI for further studies and carried out proton NMR experiments across temperature and pH ranges (Fig. 6). The 1H NMR spectrum of P5-g-PEI at 37 °C showed diminished signals (see arrows, Fig. 6a) for protons (a, d, e, f, j, k, and l) from the thermo-sensitive part of the polymer (P5) when raised from 25 °C to 37 °C, which we attribute to a chain collapsed conformation at this temperature.
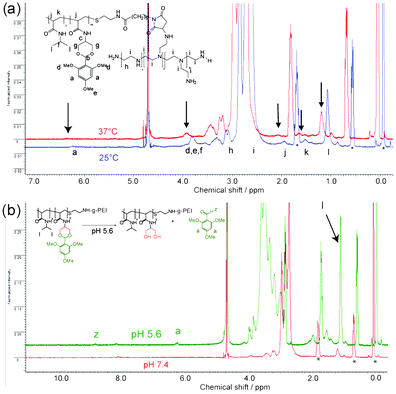 | ||
| Fig. 6 NMR spectra for polymer P5-g-PEI. In (a) are overlaid 1H NMR of P5-g-PEI in D2O at different temperatures: 25 °C (blue), a temperature before a complete phase transition has occurred and at 37 °C (red), a temperature above the polymer LCST. In (b) the polymer solutions (P5-g-PEI in D2O) are shown before and after acidification with DCl (75 µL, 2 M) and incubated at 37 °C. Spectra were recorded at 37 °C before hydrolysis (lower spectrum in red) and after hydrolysis (top spectrum in green). * indicates peaks from DSS standard. | ||
There was also peak broadening, which, combined with the reduced signal intensity, was indicative of the sequestration of this portion of the polymer into a hydrophobic micellar core surrounded by more hydrophilic PEI. After hydrolysis of P5-g-PEI under acidic conditions (Fig. 6b), sharp signals appeared in the spectrum due to the presence of free 2,4,6-trimethoxybenzaldehyde (a and z) released from the polymer. The methyl protons from the thermo-sensitive part of the polymer (peak “l”, Fig. 6b) were much increased in intensity after hydrolysis and were also less broad. After complete hydrolysis, the LCST of P5-g-PEI was above 37 °C (see Fig. 5c), thus the polymer would have adopted a chain extended conformation. Accordingly, hydrolysed P5-g-PEI was more hydrophilic in nature and an increase in signal intensity for this polymer indicated enhanced dissolution and possibly disassembly of any micellar structures that were present before hydrolysis at 37 °C.
In order to confirm that the associative properties of this polymer, once hydrolysed, were still a function of the side-chain LCST we carried out a time course experiment at pH 5.6, at temperatures below and above the LCST of the non-hydrolysed and the hydrolysed polymer. As apparent from Fig. 7(vi), higher-order structures could still be formed from the hydrolysed polymer, but only at temperatures above the LCST of the hydrolysed side-chain polymer graft, which was significantly above the LCST of the non-hydrolysed graft.
 | ||
| Fig. 7 Change in cloud point of P5-g-PEI (5 mg mL−1) with time at pH ≈ 5.6 at (a) early time points and (b) after complete hydrolysis; (i–vi) DLS of P5-g-PEI solutions at different stages of hydrolysis to show the sizes of species present after 100 minutes at pH 5.6, at temperatures (i) 15 °C, (ii) 25 °C, (iii) 37 °C, and after complete hydrolysis at temperatures (iv) 25 °C, (v) 37 °C, and (vi) 50 °C. | ||
Further indications of polymer behaviour with changes in temperature and pH were obtained by Transmission Electron Microscopy (TEM) (Fig. 8). While no evidence for >30 nm species was obtained for P1-g-PEI and P5-g-PEI when samples were prepared below LCST (data not shown), supramolecular aggregates were apparent in TEM micrographs of P1-g-PEI and P5-g-PEI prepared and rapidly dehydrated above LCST. Micelle-like aggregates of P1-g-PEI were visible (Fig. 8(i) and (ii)) at both pH 7.4 and pH 5.6.
 | ||
| Fig. 8 Transmission electron micrographs of P1-g-PEI and P5-g-PEI from solutions originally at 37 °C and rapidly dehydrated at 37 °C. (i) and (ii) show P1-g-PEI at pH 7.4 and pH 5.6 respectively. (iii) and (iv) show P5-g-PEI at pH 7.4 and pH 5.6 respectively. Images (v) and (vi) are also of P5-g-PEI at pH 7.4 and pH 5.6, respectively, at 37 °C at higher magnification. Micellar-like structures present in images (i–iii and v) are shown in cartoons to depict postulated species present from TEM and DLS analysis at the pH ranges. Note—aggregates and crystals of buffer salts present in (iv and vi) lacking similar order to structures in (i–iii and v). | ||
Fig. 8 (plates (iii) and (v)) shows vesicle-like particles of P5-g-PEI prepared above LCST, at pH 7.4, clearly indicating polymer self-assembly. Comparison of P5-g-PEI samples prepared under neutral pH conditions (Fig. 8(iii) and (v)) and under acidic conditions (Fig. 8(iv) and (vi)) confirmed the loss of association as suggested by DLS analysis, with very few micellar-like structures visible in Fig. 8(iv) and (vi) following the pH change.
Higher magnification TEM images of P5-g-PEI species (Fig. 8(v)) showed some variation in species present at pH 7.4 and 37 °C which may have been a function of the polydispersity of the graft co-polymer. In addition, lower diameters of particles were apparent in TEM compared to DLS. This was likely a consequence of dehydration under TEM sample preparation conditions as has been reported before.47 In addition, the non-spherical nature of aggregate species would have exaggerated particle size in DLS but not in TEM: inspection of Fig. 8(v) indicates that many particles were not spherical, implying variations in the packing parameters during self-assembly.
Taken together, the data from cloud-point determinations, light scattering, NMR and TEM were strongly indicative of changes in supramolecular architecture and self-assembly driven by a pH-mediated loss of hydrophobic side-chains and consequent polymer phase transition. This process occurred at a constant temperature, thus transforming the phase transition from what is conventionally a temperature-driven process to an ‘isothermal’ one. This suggests that the materials might act as switchable release systems wherein functional components such as drugs or signalling molecules could be encapsulated under one set of conditions and released under another without a potentially difficult thermal transition.
Conclusions
In this paper we have demonstrated the concept of utilising a pro-drug type strategy to invoke changes in polymer LCST, using a new pH-sensitive monomer in combination with N-isopropylacrylamide. Through the grafting of the pH-sensitive component to a pre-formed polycation, a block terpolymer was generated which exhibited changes in supramolecular order dependent on the phase transition state of the grafted polymeric block. Our primary goal for this work was to establish proof-of-principle rather than work with pharmaceutical grade materials, but PEI-based polymers and PNIPAm systems have shown promise for in vitro biomedical assays48–51 and are being evaluated for in vivo use.52 It should also be noted that the behaviour of synthetic polymers in vivo can be rather different to the behaviour of the same materials in vitro, due to the effects of complexation with biopolymers and other cellular components. However, the finding that these polymers were able to change their association from supramolecular aggregates with physiologically relevant pH stimuli is promising for biomedical applications. We are currently investigating the effects of phase changes in these pH- and ‘isothermal’ responsive terpolymers for endosomally triggered nucleic acid release and will report the biological data in a future manuscript.Experimental
Chemicals
Reagents, monomers and solvents for synthesis were obtained in the highest available purity and were used as received. Serinol (2-amino-1,3-propanediol) and dimethylsulfoxide (DMSO) were obtained from Alfa Aesar; ethyl trifluoroacetate, methanol, N-isopropylacrylamide (NIPAm), 2,4,6-trimethoxybenzaldehyde, dry tetrahydrofuran (THF), acryloyl chloride, 2-aminoethanethiol hydrochloride and poly(ethyleneimine) (PEI) from Aldrich; triethylamine and p-toluene sulfonic acid (pTSA) from Fluka Biochemika; and azo-iso butyronitrile (AIBN) and phosphate buffered saline (PBS) were from Fisher Scientific. N-(ε-Maleimidocaproyloxy)succinimide ester (EMCS) was obtained from Pierce. PEI (FW ∼25 kDa, Aldrich) was dialysed (3 kDa cut-off) against deionised water (5 × 1000 mL) and lyophilized prior to use. Inhibitors were removed from N-isopropylacrylamide (NIPAm) by recrystallisation from hexane.Synthesis of monomers
:acetone:triethylamine, 70:30:2. An off white solid was obtained 0.563 g (40%).
Melting point: 102.4–119 °C.
IR νmax (KBr)/cm−1: 3356 (NH), 3002 (CH), 2975 (CH3), 2925 (CH2), 2877 (OCH3), 2846 (CH2), 1660 (aromatic), 1629 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), 1606 (CO), 1591 (aromatic CC), 1533 (NH), 1124 (ether CO).
C), 1606 (CO), 1591 (aromatic CC), 1533 (NH), 1124 (ether CO).
1H NMR δH (399.8 MHz, CDCl3 7.28) 7.11 (1H, br d, NH), 6.37 (1H, dd, 3J 17, 2J 1.5, CHH), 6.22 (1H, dd, 3J 10.2, 2J 17, CH), 6.14 (2H, s, aromatic CH), 6.10 (1H, s, OCH), 5.71 (1H dd, 3J 10.2, 2J 1.5, CHH), 4.13 (1H, dq, NHCH(CH2)2), 4.07 (2H, dd, 3J 1.7, 2J 11.3, CHHO), 4.04 (2H, dd, 3J 1.7, 2J 11.3, CHHO), 3.86 (6H, s, 2(OCH3)), 3.81 (3H, s, OCH3).
13C NMR δC (100.5 MHz, CDCl3 77.07) 164.8 (CHCONH), 162.0 (aromatic C), 159.5 (aromatic C), 131.1 (CH), 126.5 (CH2), 107.2 (aromatic C), 96.6 (aromatic C), 91.3, 70.6 (CH(CH2)2(O)2), 56.2 (OCH3), 55.4 (OCH3), 43.9 (NHCH(CH2)2). 13C DEPT NMR (CDCl3 77.07) 131.2↓ (CH2CH), 126.5↑ (CH2CH), 96.6↓ (aromatic C), 91.3↓, 70.6↑ (CH(CH2)2(O2)), 56.2↓ (OCH3), 55.4↓ (OCH3), 43.9↓ (NHCH(CH2)2).
MS ES+ TOF: (calculated mass 323.14) 346.131 (100% MNa+), 214.068 (59% arylCH(OH)2+), 197.079 (47% arylCH2O+).
:5, v/v). A pale yellow solid was obtained, 7.05 g, 90% yield. Characterisation data agreed with that in the prior report.53
IR: νmax (KBr)/cm−1: 3307 (OH), 2951, 2921 and 2889 (CH), 1655 (CO), 1607 (–CC–).
1H NMR δH (399.8 MHz, DMSO-d6) 7.32 (1H, br d, NH), 5.66 (1H, m, CHH), 5.62 (1H, m, CHH), 4.61 (2H, t, OH, 3J 5), 3.78 (1H, dtt, NHCH(CH2)2, 3J 5.8, 2.2, 8.12), 3.44 (4H, m, CH(CH2)2(OH)2), 1.86 (3H, dd, CCH34J 1, 1.5).
13C NMR δC (100.5 MHz, CDCl3 77.07) 167.97 (CO), 140.54 (COC(CH3)), 119.44 (H2C), 60.68 (2(CH2OH)), 53.76 (NHCH(CH2)2), 19.11 (CH3). 13C DEPT NMR (DMSO-d6) 119.44↑ (H2C), 60.68↑ (2(CH2OH)), 53.76↓ (NHCH(CH2)2), 19.11↓ (CH3).
Synthesis of polymers
IR: νmax (KBr)/cm−1: 3436br and 3305br (NH), 2973, 2935 and 2876 (CH), 1651 (CO), 1546 (NH), 1459 (CH), 1387 and 1368 (CH3).
1H NMR δH (399.8 MHz, DMSO-d6) 7.87–6.74 (1H, NH), 4.05–3.68 (1H, CH(CH3)2), 3.02–2.91 (2H, SCH2CH2NH2), 2.75–2.64 (2H, SCH2CH2NH2), 2.28–1.82 (1H, CH backbone), 1.71–1.27 (2H, CH2 backbone), 1.18–0.92 (6H, br, CH(CH3)2).
13C NMR δC (100.5 MHz, D2O) 175.4, 42.2, 41.8, 34.4, 21.6.
Diagnostic peak positions were very similar for polymers P2, P3 and P5 as these differed only in co-monomer content, with only variations in integral ratios in 1H NMR for the respective co-monomer ratios.
Polymer P2: IR: νmax (KBr)/cm−1: 3433 and 3313 (NH), 2971, 2933 and 2833 (CH), 1650 (CO), 1541 (NH), 1459 (CH2), 1386 and 1366 (CH3), 1229 (CO).
1H NMR δH (399.8 MHz, DMSO-d6) 7.55–7.01 (1H, NH), 6.19–6.15 (2H, 2CH aromatic), 5.10–4.7 (2H, 2OH (partial hydrolysis of polymer)), 4.05–3.95 (1H, NHCH(CH2)2), 3.94–3.7 (1H, CH(CH3)2), including peaks for 6H, 2(COCH3) and 3H, OCH3 (too small to detect), 2.9–2.8 (2H SCH2CH2NH2), 2.25–1.8 (1H, CH backbone), 1.65–1.25 (2H, CH2 backbone), 1.15–0.86 (6H, CH(CH3)2).
Polymer P3: IR: νmax (KBr)/cm−1: 3303 (NH), 2972, 2935 and 2876 (CH), 1648 (CO), 1544 (NH), 1459 (CH2), 1387 and 1367 (CH3), 1229 (CO), 1057 (CO).
1H NMR δH (399.8 MHz, DMSO-d6) 7.75–7.0 (1H, NH), 6.25–6.08 (2H, 2CH aromatic), 5.95–5.85 (1H, OCHO), 5.75–5.68 (1H, OCHO), 5.05–4.6 (2H, 2OH (partial hydrolysis of polymer)), 4–3.95 (1H, NHCH(CH2)2), 3.95–3.6 (1H, CH(CH3)2), 3.76 (6H, 2(COCH3)–polymer) 3.73 (3H, COCH3–polymer), 3.11–3.06 (2H, SCH2CH2NH2), 2.87–2.8 (2H, SCH2CH2NH2), 2.2–1.8 (1H, CH, backbone), 1.7–1.25 (2H, CH2 backbone), 1.15–0.88 (3H, CH(CH3)2).
Polymer P4: synthesis and characterisation. N-Isopropylacrylamide (0.497 g, 4.39 mmol) was added to DMSO (3 mL), followed by N-(2-(2,4,6-trimethoxyphenyl)-1,3-dioxane-5-yl)acrylamide (0.198 g, 0.61 mmol) and N-(1,3-dihydroxypropan-2-yl)methacrylamide (0.101 g, 0.64 mmol). Aminoethanethiol hydrochloride (9 mg, 0.08 mmol) was then added as chain transfer agent, followed by the initiator, AIBN (11 mg, 0.07 mmol). The solution was purged with argon for 10 minutes, then sealed and heated to 65 °C for 24 hours. The mixture was then allowed to cool to room temperature, before being added dropwise to a large excess of diethyl ether (400 mL). The solvent was decanted and the remaining oily gum was dissolved in THF (5 mL) and added dropwise to diethyl ether (400 mL). The precipitated material was collected by filtration before redissolving in THF and reprecipitating in diethyl ether two further times. The product was isolated as a pale yellow solid 0.230 g, yield 30%.
Polymer P4: IR: νmax (KBr)/cm−1: 3316 (NH), 2972, 2934 and 2875 (CH), 1651 (CO), 1544 (NH), 1459 (CH2), 1386 and 1367 (CH3), 1229 (CO), 1059 (CO).
1H NMR δH (400 MHz, DMSO-d6) 10.24 (s, 1H, COH (residual aldehyde)), 7.96–6.86 (1H, NH), 6.27 (2H, CH aromatic-residual 2,4,6-trimethoxybenzaldehyde), 6.26–6.12 (2H, 2CH aromatic), 5.94–5.84 (1H, OCHO), 5.79–5. 67 (1H, OCHO), 5–4.64 (2H, 2OH (partial hydrolysis of polymer)), 4.03–3.98 (1H, NHCH(CH2)2), 3.98–3.64 (1H, CH(CH3)2), 3.79–3.74 (6H, 2(COCH3)), 3.74–3.7 (3H, OCH3), 3.0–2.91 (2H, SCH2CH2NH2), 2.77–2.67 (2H, SCH2CH2NH2), 2.17–1.76 (1H, CH backbone), 1.76–1.23 (2H, CH2 backbone), 1.18–0.88 (3H, CH(CH3)2).
δ C (100.5 MHz, DMSO-d6) 173.81, 159.73, 56.31, 55.67, 35.9, 22.74.
Polymer P5: IR: νmax (KBr)/cm−1: 3301br (NH), 2972, 2935, 2876 (CH), 1649 (CO), 1545 (NH), 1459 (CH), 1387 and 1368 (CH3), 1230 (CO), 1059 (CO).
1H NMR δH (399.8 MHz, DMSO-d6), 7.91–6.54 (br s, 1H, NH), 6.24–6.07 (2H, 2CH aromatic), 5.95–5.81 (1H, OCHO), 5.78–5.68 (1H, OCHO), 5.01–4.6 (2H, 2OH (partial hydrolysis of polymer)), 4.03–3.96 (1H, NHCH(CH2)2), 3.96–3.63 (1H, CH(CH3)2), 3.79–3.74 (6H, 2(COCH3)), 3.74–3.7 (3H, OCH3), 3.1–3 (2H, SCH2CH2NH2), 2.9–2.8 (2H SCH2CH2NH2), 2.28–1.79 (1H, CH backbone), 1.73–1.23 (2H, CH2 backbone), 1.18–0.82 (6H, CH(CH3)2).
13C NMR (100.5 MHz, DMSO-d6) 186.1, 173.8, 166.5, 163.9, 159.7, 108.4, 91.2, 60.8, 56.5, 55.7, 42.0, 34.5, 22.7.
In some NMR spectra of polymers on standing or after dialysis for prolonged periods against distilled water, small peaks at 11.1, 10.24, 6.2–6.4 and 3.82–3.85 ppm were apparent indicative of aldehyde protons arising from hydrolysis of the acetal side-chain and generation of 2,4,6-trimethoxybenzaldehyde.
Polymer P1-g-PEI: yield 202 mg. IR νmax (KBr)/cm−1: 3419br (NH), 2971, 2935 and 2841 (CH), 1651 (CO), 1557 (NH), 1470, 1387 and 1367 (CH).
1H NMR δH (399.8 MHz, D2O) 7.95–7.84 (1H, NH), 3.84–3.72 (1H, CH(CH3)2), 3.19–3.00 (2H, NH2CH2CH2NH–), 3.00–2.5 (581H, CH2CH2, PEI), 2.15–1.82 (1H, CH, backbone), 1.67–1.2 (2H, CH2 backbone), 1.15–0.95 (6H, CH(CH3)2).
13C NMR δC (100.5 MHz, D2O) 175.3, 52.5, 51, 47.0, 45.5, 41.8, 38.8, 37.3, 23.4, 21.6.
Spectroscopic data for P4-g-PEI and P5-g-PEI were very similar, with IR bands and NMR resonances varying only in differential intensities, as expected based on their close structural similarity. The presence of the extra methyl groups in the polymer backbone from DHPMA were overlapped by other backbone protons, but the raised cloud point of the polymer indicated the presence of polymerised DHPMA in the structure.
Polymer P4-g-PEI: yield 286 mg. IR νmax (KBr)/cm−1: 3392br (NH), 2964 and 2838 (CH), 1645 (CO), 1552 (NH), 1469, 1386 and 1367 (CH), 1305 and 1071 (CO).
1H NMR δH (600 MHz, D2O, 10 °C), 7.80–7.66 (1H, NH), 6.07–5.85 (2H, CH, aromatic), 5.85–5.75 (1H, OCHO), 5.75–5.63 (1H, OCHO), 3.7–3.4 (1H CH(CH3)2 and 9H, 3(COCH3)), 3.13–2.9 (2H, NH2CH2CH2NH–), 2.85–1.9 (581H, CH2CH2, PEI), 1.85–1.5 (1H, CH, backbone), 1.5–1.02 (2H, CH2 backbone), 1.0–0.6 (6H, CH(CH3)2).
13C NMR δC (500.132 MHz, D2O, 10 °C) 174.9, 164.2, 158.9, 125.13, 104.94, 104.05, 96.76, 91.03, 69.1, 60.11, 55.77, 55.46, 55.07, 54.59, 52.56, 50.67, 49.44, 49.27, 48.34, 47.29, 45.23, 41.51, 39.94, 39.13, 38.15, 37.18, 34.45, 27.37, 21.25.
Polymer P5-g-PEI: yield 108 mg (21%). IR νmax (KBr)/cm−1: 3423 (NH), 2941 and 2837 (CH), 1647 (CO), 1557 (NH), 1464, 1385, and 1367, 1306 (CH).
1H NMR δH (500.132 MHz, D2O, 10 °C) 7.8–7.68 (1H, NH), 6.02–5.86 (2H, CH, aromatic), 5.86–5.73 (1H, OCHO), 3.73–3.61 (1H, OCH2CHCH2CO), 3.61–3.34 (1H CH(CH3)2 and 9H, 3(COCH3)), 2.88–2.7 (2H, NH2CH2CH2NH–), 2.7–1.95 (CH2CH2 PEI), 1.81–1.54 (1H, CH, backbone), 1.54–1.45 (2H, CH2 backbone), 0.98–0.6 (6H, CH (CH3)2).
13C NMR δC (125.772 MHz, D2O, 10 °C) 176.6 (CO), 164.2 (COCH3, aromatic), 158.9 (2C(OCH3) aromatic), 96.8 (CH aromatic), 91.03 (2(O)CHC), 69.9 (NHCH(CH2)2), 60.1 (2(CH2)OCH), 55.7 and 55.1 3(OCH3), 52.3 (![[double bond splayed left]](https://www.rsc.org/images/entities/char_e009.gif) NCH2CH2NH–), 50.6 (NCH2CH2N
NCH2CH2NH–), 50.6 (NCH2CH2N![[double bond splayed right]](https://www.rsc.org/images/entities/char_e00a.gif) ), 48.3 (–NHCH2CH2NH2), 47.1 (–NHCH2CH2NH–), 45.1 (–NHCH2CH2N), 41.5 (CH(CH3)2), 38.7 (NH2CH2CH2NH–), 36.9 (NH2CH2CH2N), 35 (CH2 backbone), 21.2 (2(CH3)).
), 48.3 (–NHCH2CH2NH2), 47.1 (–NHCH2CH2NH–), 45.1 (–NHCH2CH2N), 41.5 (CH(CH3)2), 38.7 (NH2CH2CH2NH–), 36.9 (NH2CH2CH2N), 35 (CH2 backbone), 21.2 (2(CH3)).
Evaluation of lower critical solution temperatures (LCSTs) via cloud point
Cloud points of all the polymers (P1–P5) were measured at pH 7.4 and 5.6. The polymers were dissolved in PBS solutions (adjusted to pH 7.4 and 5.6) to a concentration of 5 mg mL−1. Solutions of grafted copolymers (P1-g-PEI, P4-g-PEI and P5-g-PEI) were adjusted to pH 7.4 or 5.6 using aliquots of HCl (1 M). These solutions were heated from about 10 °C to about 65 °C at a rate of 0.5 °C min−1 in a UV spectrometer. The UV absorbance at 550 nm was measured at every 0.5 °C increase in temperature against the blank. The LCST was taken to be the temperature at which a sharp increase in absorption at 550 nm occurred.Dynamic light scattering measurements
Stock solutions of polymers P1-g-PEI, P4-g-PEI and P5-g-PEI (5 mg mL−1) in PBS solution (adjusted to pH 7.4 or pH 5.6 with HCl (1 M)) were diluted to 0.88 mg mL−1. Sample solutions were allowed to equilibrate to the temperature of measurement for 4 minutes prior to commencing measurement. Hydrodynamic radii of the polymeric species were measured via scattered light recorded at 90° angle to incident radiation in a Viscotek 802 dynamic light scattering (DLS) instrument equipped with a 50 mW internal laser operating at a wavelength of 830 nm. Particle sizes reported are averages from 10 measurements per sample. Each measurement lasted 3 seconds.Change in LCST with acetal hydrolysis
Polymers P2–P5 were dissolved in PBS solution (adjusted to pH 7.4 and 5.6) to a concentration of 1 mg mL−1. Immediately after dissolution, the polymer solutions were heated starting at 15 °C at a rate of 1 °C min−1 in a UV spectrometer. The UV absorbance at 550 nm was measured at every 1 °C increase against the blank. The solutions were incubated at 37 °C, in between measurements.Instrumentation
944000.
Acknowledgements
We thank the Biotechnology and Biological Sciences Research Council (BBSRC, Grant BB/C515855/1) and the University of Nottingham for financial support. We also thank Dr Steve Holding, Smithers-RAPRA UK for GPC, Marie Smith, School of Biomedical Sciences, University of Nottingham and Christine Grainger-Boultby, School of Pharmacy, for technical assistance.Notes and references
- D. Roy, J. N. Cambre and B. S. Sumerlin, Chem. Commun., 2009, 2106–2108 RSC.
- C. D. H. Alarcon, S. Pennadam and C. Alexander, Chem. Soc. Rev., 2005, 34, 276–285 RSC.
- Y. Lee, S. Fukushima, Y. Bae, S. Hiki, T. Ishii and K. Kataoka, J. Am. Chem. Soc., 2007, 129, 5362–5363 CrossRef CAS.
- T. M. Fulghum, N. C. Estillore, C. D. Vo, S. P. Armes and R. C. Advincula, Macromolecules, 2008, 41, 429–435 CrossRef CAS.
- G. B. Webber, E. J. Wanless, S. P. Armes and S. Biggs, Faraday Discuss., 2005, 128, 193–209 RSC.
- G. Chen, P. M. Wright, J. Geng, G. Mantovani and D. M. Haddleton, Chem. Commun., 2008, 1097–1099 RSC.
- E. Wischerhoff, N. Badi, J.-F. Lutz and A. Laschewsky, Soft Matter, 2010, 6, 705–713 RSC.
- M. A. C. Stuart, W. T. S. Huck, J. Genzer, M. Muller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsurkruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101–113 CrossRef.
- M. E. H. El-Sayed, A. S. Hoffman and P. S. Stayton, Expert Opin. Biol. Ther., 2005, 5, 23–32 Search PubMed.
- B. Twaites, C. D. Alarcon and C. Alexander, J. Mater. Chem., 2005, 15, 441–455 RSC.
- R. V. Ulijn, J. Mater. Chem., 2006, 16, 2217–2225 RSC.
- E. S. Place, J. H. George, C. K. Williams and M. M. Stevens, Chem. Soc. Rev., 2009, 38, 1139–1151 RSC.
- N. Matsuda, T. Shimizu, M. Yamato and T. Okano, Adv. Mater., 2007, 19, 3089–3099 CrossRef CAS.
- Y. Y. Li, X. Z. Zhang, G. C. Kim, H. Cheng, S. X. Cheng and R. X. Zhuo, Small, 2006, 2, 917–923 CrossRef CAS.
- D. J. Gan and L. A. Lyon, J. Am. Chem. Soc., 2001, 123, 7511–7517 CrossRef CAS.
- F. Kohori, M. Yokoyama, K. Sakai and T. Okano, J. Controlled Release, 2002, 78, 155–163 CrossRef CAS.
- M. Yokoyama, Drug Discovery Today, 2002, 7, 426–432 CrossRef CAS.
- H. S. Bisht, D. S. Manickam, Y. Z. You and D. Oupicky, Biomacromolecules, 2006, 7, 1169–1178 CrossRef CAS.
- B. R. Twaites, C. D. H. Alarcon, M. Lavigne, A. Saulnier, S. S. Pennadam, D. Cunliffe, D. C. Gorecki and C. Alexander, J. Controlled Release, 2005, 108, 472–483 CrossRef CAS.
- E. Piskin, Int. J. Pharm., 2004, 277, 105–118 CrossRef CAS.
- M. Kurisawa, M. Yokoyama and T. Okano, J. Controlled Release, 2000, 69, 127–137 CrossRef CAS.
- P. C. Griffiths, C. Alexander, R. Nilmini, S. S. Pennadam, S. M. King and R. K. Heenan, Biomacromolecules, 2008, 9, 1170–1178 CrossRef CAS.
- M. D. Lavigne, S. S. Pennadam, J. Ellis, L. L. Yates, C. Alexander and D. C. Górecki, J. Gene Med., 2007, 9, 44–54 CrossRef CAS.
- S. S. Pennadam, J. S. Ellis, M. D. Lavigne, D. C. Gorecki, M. C. Davies and C. Alexander, Langmuir, 2007, 23, 41–49 CrossRef CAS.
- J.-L. Zhu, H. Cheng, Y. Jin, S.-X. Cheng, X.-Z. Zhang and R.-X. Zhuo, J. Mater. Chem., 2008, 18, 4433–4441 RSC.
- L. Yu and J. D. Ding, Chem. Soc. Rev., 2008, 37, 1473–1481 RSC.
- G. Pasparakis, M. Vamvakaki, N. Krasnogor and C. Alexander, Soft Matter, 2009, 5, 3839–3841 RSC.
- M. A. Cole, N. H. Voelcker, H. Thissen and H. J. Griesser, Biomaterials, 2009, 30, 1827–1850 CrossRef CAS.
- Y. Lee and K. Kataoka, Soft Matter, 2009, 5, 3810–3817 RSC.
- C. LoPresti, H. Lomas, M. Massignani, T. Smart and G. Battaglia, J. Mater. Chem., 2009, 19, 3576–3590 RSC.
- V. Knorr, L. Allmendinger, G. F. Walker, F. F. Paintner and E. Wagner, Bioconjugate Chem., 2007, 18, 1218–1225 CrossRef CAS.
- P. Erbacher, T. Bettinger, E. Brion, J. L. Coll, C. Plank, J. P. Behr and J. S. Remy, J. Drug Targeting, 2004, 12, 223–236 Search PubMed.
- J. L. Coll, P. Chollet, E. Brambilla, D. Desplanques, J. P. Behr and M. Favrot, Hum. Gene Ther., 1999, 10, 1659–1666 CrossRef CAS.
- D. Neradovic, W. L. J. Hinrichs, J. J. Kettenes-Van Den Bosch, C. F. Van Nostrum and W. E. Hennink, J. Controlled Release, 2001, 72, 252–253.
- D. Neradovic, W. L. J. Hinrichs, J. J. Kettenes-Van Den Bosch and W. E. Hennink, Macromol. Rapid Commun., 1999, 20, 577–581 CrossRef CAS.
- J. Rickerby, R. Prabhakar, M. Ali, J. Knowles and S. Brocchini, J. Mater. Chem., 2005, 15, 1849–1856 RSC.
- J. Rickerby, R. Prabhakar, A. Patel, J. Knowles and S. Brocchini, J. Controlled Release, 2005, 101, 21–34 CrossRef CAS.
- M. J. Vicent, R. Tomlinson, S. Brocchini and R. Duncan, J. Drug Targeting, 2004, 12, 491–501 Search PubMed.
- R. Tomlinson, M. Klee, S. Garrett, J. Heller, R. Duncan and S. Brocchini, Macromolecules, 2002, 35, 473–480 CrossRef CAS.
- E. R. Gillies and J. M. J. Frechet, Chem. Commun., 2003, 1640–1641 RSC.
- F. Ganachaud, M. J. Monteiro, R. G. Gilbert, M. A. Dourges, S. H. Thang and E. Rizzardo, Macromolecules, 2000, 33, 6738–6745 CrossRef CAS.
- Cloud points were measured at a concentration of 1 mg mL−1 and the temperatures obtained at pH 7.4 can be seen to be slightly higher compared to those in Table 1 (measured at a higher concentration of 5 mg mL−1). A small increase (∼1 °C) in cloud-point temperature of aqueous PNIPAm solutions with decrease in polymer concentration has also been observed by Furyk et al.43 for polymers with low Mw (below 50 kDa).
- S. Furyk, Y. J. Zhang, D. Ortiz-Acosta, P. S. Cremer and D. E. Bergbreiter, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 1492–1501 CrossRef CAS.
- Ratios of acetal-containing polymers to PEI determined by NMR in D2O were determined by integrals for peaks of isopropyl methyl groups on the PNIPAm components to PEI methylene groups and via the TNBS assay. Some peak broadening was apparent for the PNIPAm isopropyl groups indicating partial collapse of the side-chains even at 25 °C for P4-g-PEI and P5-g-PEI, thus the apparent ratios for these polymers likely underestimated graft content compared to TNBS. We estimate one P1 chain to between 1 and 2 PEI chains in P1-g-PEI, one P4 chain to 2–4 PEI chains in P4-g-PEI and one P5 chain to 2–5 chains in P5-g-PEI.
- E. R. Gillies, A. P. Goodwin and J. M. J. Frechet, Bioconjugate Chem., 2004, 15, 1254–1263 CrossRef CAS.
- The 2 nm species were most likely single polymer chains from the lower molar mass range of the polydisperse samples, but it should be noted that particle sizing at this length scale was close to the limits of accurate sizing by our DLS instrumentation.
- T. Govender, S. Stolnik, C. D. Xiong, S. Zhang, L. Illum and S. S. Davis, J. Controlled Release, 2001, 75, 249–258 CrossRef CAS.
- M. Nakayama and T. Okano, J. Drug Delivery Sci. Technol., 2006, 16, 35–44 Search PubMed.
- J. Fahrmeir, M. Gunther, N. Tietze, E. Wagner and M. Ogris, J. Controlled Release, 2007, 122, 236–245 CrossRef CAS.
- N. Cheng, W. G. Liu, Z. Q. Cao, W. H. Ji, D. C. Liang, G. Guo and J. Y. Zhang, Biomaterials, 2006, 27, 4984–4992 CrossRef CAS.
- H. Vihola, A. Laukkanen, L. Valtola, H. Tenhu and J. Hirvonen, Biomaterials, 2005, 26, 3055–3064 CrossRef CAS.
- M. Thomas, Q. Ge, J. J. Lu, J. Z. Chen and A. M. Klibanov, Pharm. Res., 2005, 22, 373–380 CrossRef CAS.
- X. Huang, F. Du, R. Ju and Z. Li, Macromol. Rapid Commun., 2007, 28, 597–603 CrossRef CAS.
- Dialysis was conducted with a membrane MWCO (50 kDa) which was approximately twice the molecular weight of the un-reacted excess bPEI and un-reacted linear copolymers we intended to remove. The Mn values for P1-g-PEI and P5-g-PEI were 23.57 kDa and 21.51 kDa, respectively; however the Mw values for these polymers were calculated to be 68.55 kDa and 57.52 kDa, respectively. Accordingly, some loss of the desired co-polymers at their lower molecular weight fractions was expected during dialysis. This loss was found to be reduced by employing temperature cycling during dialysis, i.e. for 50% of the dialysis time the polymer solutions were maintained at a temperature above the polymer cloud point. This aggregated the thermoresponsive polymers, while allowing the excess PEI to pass through the membrane.
Footnote |
| † Electronic supplementary information (ESI) available: Proton NMR spectra of polymers P5 and P1-g-PEI and 13C NMR of P5-g-PEI. Also TEM images of P4-g-PEI are shown under neutral pH and acidic pH conditions. See DOI: 10.1039/c0py00080a |
| This journal is © The Royal Society of Chemistry 2010 |