Organic support for ethylene polymerization based on the self-assembly in heptane of end-functionalized polyisoprene
Bertrand
Heurtefeu
ab,
Jan
Merna
c,
Emmanuel
Ibarboure
ab,
Éric
Cloutet
ab and
Henri
Cramail
*ab
aUniversité de Bordeaux, Laboratoire de Chimie des Polymères Organiques, IPB-ENSCBP, 16, Avenue Pey Berland, Pessac Cedex, F-33607, France. E-mail: cramail@enscbp.fr; Fax: +335 40 00 84 87; Tel: +335 40 00 84 86
bCNRS, Laboratoire de Chimie des Polymères Organiques, Pessac Cedex, F-33607, France
cInstitute of Chemical Technology, Prague, Department of Polymers, Technická 5, 166 28, Prague 6, Czech Republic
First published on 28th May 2010
Abstract
In this work, we report how micellar structures obtained by the self-assembly in heptane of polyisoprene end-capped with a hindered tertiary alcohol moiety (PI-φ2OH) can be efficiently used as organic supports for olefin polymerization single-site catalysts. PI-φ2OH has been synthesized by end-capping living polyisoprenyllithium with an excess of benzophenone. Dynamic light scattering analysis indicates a self-assembly of PI-φ2OH in heptane, a good solvent for polyisoprene and a poor one for the polar end-group. The so-formed micelle-like nanoparticles, composed of a di-phenyl alcohol group core and a polyisoprene corona were used as organic supports for catalytic system composed of aluminic activators, trimethylaluminium (TMA) or methylaluminoxane (MAO), and metallocene or post-metallocene catalysts, to produce micrometric polyethylene beads.
Introduction
Metallocenes and late transition metal complexes are effective catalysts for the homogeneous polymerization of olefins, enabling the production of polyolefins with a tunable microstructure and molecular weight distribution. In order to control the polyolefin morphology (formation of beads) and also to prevent fouling of large scale reactors in industry, fixing the catalytic system on a support is required. The immobilization of catalytic systems is generally performed with a solid inorganic carrier1,2 such as silica,3 alumina4 and magnesium chloride.5,6 After polymerization, some residual inorganic support can remain in the polyolefin which affects the optical and mechanical properties of the final materials.7,8 To avoid such a drawback, one solution is to replace the inorganic supports with organic templates. Most of the investigations carried out have been on the development of functional organic supports essentially based on polystyrene (PS).9 For instance, a metallocene catalyst can be attached to a PS backbone through its aromatic ligands.10–13 Another strategy is to anchor the activator, generally methylaluminoxane (MAO) or trimethylaluminium (TMA), on a polar organic support thanks to weak oxygen-aluminium interactions. Typically, PS nanogel with a small number of peripheral ethylene oxide units was described as carrier for MAO/2,6-bis{1-2,6}-(diisopropylphenyl)imino]ethylpyridynyl iron dichloride, [MeDIP(2,6-iPrPh)2FeCl2], a catalytic system towards ethylene polymerization.14 However in all cases, a multistep synthesis of these organic templates is required that can be seen as a limitation for the industrialization.A more straightforward strategy to design organic support is based on the self-assembly of end-functionalized polymers or block-copolymers.15 Bouilhac et al. have synthesized linear PS end-functionalized by a benzoic acid or a benzophenone moiety16 and investigated the self-assembly in toluene of these functional PS into micellar aggregates. The purpose was the encapsulation of Fe-based catalytic system to produce millimetric polyethylene (PE) beads with a high catalytic activity. Similarly, polystyrene-block-poly(4-vinyl benzoic acid) copolymers were synthesized and their ability to control the PE morphology was also proven.17 This new concept of support for the production of PE beads has been essentially applied on the self-assembly, in toluene, of PS-based supports with MeDIP(2,6-iPrPh)2FeCl2 as a catalyst.
Following this strategy, we describe in this paper the use of micellar structures based on the self-assembly in heptane of polyisoprene end-capped with a diphenyl-tertiary-alcohol moiety, as nanoreactors for the production of polyolefin beads. Diphenyl-tertiary-alcohol end-group has been chosen for its polarity which induces micellization in aliphatic solvent and also for the capacity of tertiary alcohol to entrap free TMA leading to an increase of the catalytic activity.18 MeDIP(2,6-iPrPh)2FeCl2, N,N′-bis(2,6-diethylphenyl)-2,3-(naphthalene-1,8-diyl)-1,4-diazabuta-1,3-diene) nickel(II) dibromide, [(α-diimine)nickel(II)] and bis(indenyl)zirconium dichloride [Ind2ZrCl2] (Scheme 1), were tested as catalysts in the presence of MAO or TMA as activators.
 | ||
| Scheme 1 Catalysts used with PI-φ2OH as support for the polymerization of ethylene. | ||
Results and discussion
Synthesis of PI-Φ2OH
The anionic polymerization of styrene, ended by the addition of benzophenone to quench the active species has been reported by Quirk and colleagues.19 On this basis, we performed the anionic polymerization of isoprene in cyclohexane, initiated by s-butyllithium, and quenched the reaction by adding an excess of benzophenone in order to obtain low molar mass diphenylhydroxy-terminated polyisoprene (PI-ϕ2OH, see Scheme 2).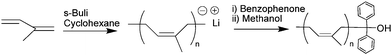 | ||
| Scheme 2 Synthesis of polyisoprene diphenyl alcohol, PI-ϕ2OH, by anionic polymerization. | ||
As illustrated in Table 1, anionic polymerization in apolar solvent leads to a very good control over the polymer dimensions together with the advantage of working above room temperature.
| Run | Mn,SECb/g mol−1 | D | Mn,NMRc/g mol−1 | f (%) |
|---|---|---|---|---|
| a Polymerization in cyclohexane at 50 °C. b Determined by SEC with THF as eluent (RI detector). c Determined by 1H NMR. | ||||
| 1 | 2515 | 1.08 | 1900 | 100 |
| 2 | 5843 | 1.05 | 3900 | 100 |
The chain-end funtionnalization has been checked by 1H and 13C NMR. As it can be seen in the 1H NMR spectrum (Fig. 1), the apparition of peaks at 7.2 and 7.5 ppm are representative of the aromatic protons and the one at 3.06 ppm of the proton of the alcohol function.
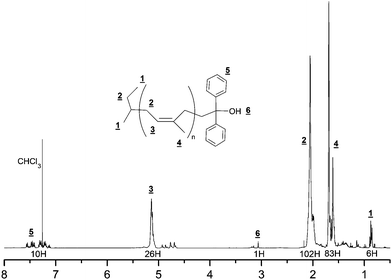 | ||
| Fig. 1 1H NMR spectra (400 MHz, CDCl3) of diphenyl alcohol terminated polyisoprene (run 1). | ||
Properties of PI-Φ2OH solubilized in heptane studied by dynamic light scattering
All the PI-Φ2OH dispersions in heptane used for the dynamic light scattering (DLS) analysis were initially heated at 65 °C and then cooled down to 30 °C to favor the micellization process. The same methodology was followed in the presence of the aluminic activator (see later in the text).First, DLS experiments were performed on a PI-Φ2OH (Mn = 2515 g mol−1) dispersion at 1 mg ml−1 in heptane at 30 °C.
Collected data revealed the existence of a self assembly of PI-Φ2OH in heptane into micelle-like structures that likely consist of an alcohol core and a polyisoprene corona (heptane being a good solvent for polyisoprene). The values of the hydrodynamic radii (RH) of the different nanoparticles were calculated from the decay times using the Stokes–Einstein equation. These relaxation times were determined by applying the CONTIN analysis20 to the autocorrelation functions; an example is given in Fig. 2 for PI-Φ2OH of Mn = 2515 g mol−1.
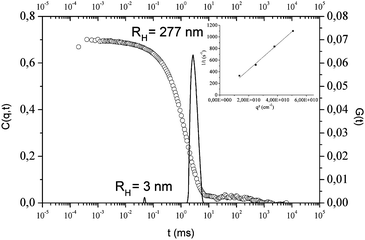 | ||
| Fig. 2 Relaxation curve and corresponding Contin fit obtained by DLS analysis (θ = 50°) of PI-Φ2OH (Mn = 2515 g mol−1) in heptane (c = 1 mg mL−1, T = 30 °C). The inset shows the dependency of the decay rate Γ on the square scattering vector q2. | ||
The DLS analysis of higher molar mass PI-Φ2OH (Mn = 5843 g mol−1) in heptane at 1 mg ml−1 and 30 °C showed a very low diffusion intensity indicating the presence of a main population composed of free chains (2 nm size) together with a minor second population attributed to aggregates (125 nm). This result clearly proves that such PI is not capable to form micellar structures due to a too long size of the PI chain. For this reason, lower molar mass PI-Φ2OH (Mn = 2515 g mol−1) was only used as template for the synthesis of PE beads.
Effect of MAO addition onto PI-Φ2OH self-assembly
The effect of MAO addition onto PI-Φ2OH self-assembly process in heptane was investigated by DLS. For this purpose, MAO was added to PI-Φ2OH dispersion and the whole system was maintained for 24 h at 65 °C then cooled down at 30 °C. The addition of MAO (Al/OH = 75) into PI-Φ2OH dispersion (Mn = 2515 g mol−1, c = 1 mg mL−1) leads to an increase of the particle radius (from an average value of 290 nm to 530 nm), see Fig. 3. This phenomenon would indicate that some MAO is encapsulated within the aggregates.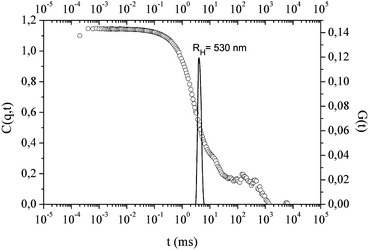 | ||
| Fig. 3 Autocorrelation function and relaxation times at θ = 90° of PI-Φ2OH (Mn = 2515 g mol−1) in heptane (c = 1 mg ml−1, T = 30 °C) with MAO: Al/OH = 75. | ||
The signal increase observed in the correlation function at high relaxation times also indicates the formation of aggregates which can be explained by the presence of non-encapsulated MAO. It is worth noting that in the absence of PI-Φ2OH, the MAO is not dispersable in heptane at all. The encapsulation phenomenon is supported by the visual aspect of the dispersion which remains homogeneous and stable at least 2 h (no sedimentation observed). This means that MAO is fully dispersed in heptane thanks to the presence of PI-Φ2OH.
Consequently, the decay rate versus squared scattering vector plot could not be fitted with a linear curve, explained by the presence of different populations of aggregates with a non-spherical shape (data not shown).
Effect of TMA addition onto PI-Φ2OH self-assembly
TMA is soluble in heptane and purchased in solution (2 mol L−1) in this solvent. The effect of TMA addition (Al/OH = 25) on PI-Φ2OH self-assembly has also been checked by DLS (see Fig. 5). As for MAO, TMA was added to PI-Φ2OH dispersions and the whole system was maintained for 24 h at 65 °C then cooled down at 30 °C. As expected, the addition of TMA onto PI-Φ2OH dispersion (Mn = 2515 g mol−1, c = 1 mg mL−1) leads to an increase of the particle size with a RH value of 551 nm without formation of larger aggregates as it was observed for MAO. In the case of TMA addition, the dispersion remains homogeneous and the decay rate versus squared scattering vector plot can be fitted with a linear curve, proof of the formation of spherical particles (inset in Fig. 4).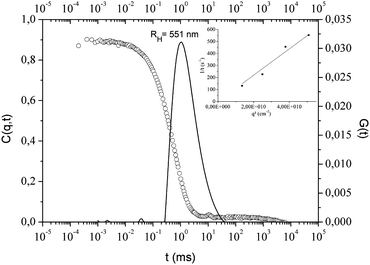 | ||
| Fig. 4 Relaxation curve and corresponding Contin fit obtained by DLS analysis (θ = 50°) of PI-Φ2OH (Mn = 2515 g mol−1) in heptane (c = 1 mg mL−1, T = 30 °C) with TMA: AL/OH = 25. The inset shows the decay rate Γ dependency to the square scattering vector q2. | ||
 | ||
| Fig. 5 SEC traces (viscosimeter) of PEs made with PI-Φ2OH and MeDIP(2,6iPrPh)2FeCl2 as catalyst. | ||
Polymerization of ethylene using MeDIP(2,6-iPrPh)2FeCl2
The micellar structures obtained from the self-assembly of PI-Φ2OH in heptane were subsequently used to support the tridentate bis(imino) pyridinyl iron catalyst, MeDIP(2,6-iPrPh)2FeCl2. The polymerizations were then performed at 30 °C under 1 bar of ethylene pressure for 1 h.The influence of the support on the production of PE using MeDIP(2,6-iPrPh)2FeCl2 was investigated; data are collected in Table 2.
| Run | n(cata)/μmol | Activator | Al/Fe | Activity/kg mol(C)−1 h−1bar−1 | Al/OH | Tm/°Cc | χcc | M w /kg mol−1 | D | Bead size/μm |
|---|---|---|---|---|---|---|---|---|---|---|
| a Experimental conditions: heptane, 30 mL; reaction time = 1 h; T = 30 °C; Pethylene = 1 bar. b Experimental conditions: heptane, 30 mL; reaction time = 1 h; T = 30 °C; Pethylene = 1 bar; reaction time before polymerization = 24 h at 65 °C under stirring; PI-Φ2OH: Mn = 2515 g mol−1; [PI-Φ2OH] = 1 mg ml−1. c Determined by DSC. d Determined by SEC (1,2,4-trichlorobenzene at 150 °C). | ||||||||||
| blank 1a | 0.6 | MAO | 1500 | 2413 | — | 131 | 62% | 290 | bimodal | — |
| 1b | 0.6 | MAO | 1500 | 2987 | 75 | 134 | 65% | 760 | 6.5 | 0.5 |
| blank 2a | 0.6 | TMA | 500 | 1753 | — | 139 | 61% | 530 | bimodal | — |
| 2b | 0.6 | TMA | 500 | 1965 | 25 | 133 | 28% | 980 | 5 | 0.5–2 |
| 3b | 0.6 | TMA | 400 | 280 | 20 | 140 | 62% | 1114 | 15.7 | 1–10 |
| 4b | 0.6 | TMA | 300 | 162 | 15 | 139 | 62% | 831 | 9 | 1–4 |
With MAO, an increase of the catalytic activity in the presence of the support compared to homogeneous conditions is observed (2987 kg PE/(mol Fe h bar, run 1, vs. 2413 kg PE/(mol Fe h bar), run blank 1). This increase can be explained by the entraping of the free TMA, present in the MAO solution, through the alcohol function of the support.
The use of TMA as a cocatalyst at a ratio Al/Fe = 500, leads to slightly higher values of catalytic activity in the presence of PI-Φ2OH in comparison to homogeneous conditions (1753 kg PE/(mol Fe h bar), run blank 2, vs. 1965 kg PE/(mol Fe h bar), run 2). This can be reasonably explained by a local increase of the cocatalyst concentration thanks to the micellar support. Reduction of Al/Fe ratio below 500 leads to a sharp decrease of the catalytic activity. According to SEC analysis, oligomers were never formed (Fig. 5), which is in agreement with a suppression/reduction of transfer reaction to TMA, proving the efficiency of the support to trap the activator within the micelles. The reduction of the Al/Fe ratio also leads to a diminution of the molar masses (run 3 and run 4).
Additionally, the morphology of the PE beads was investigated by scanning electron microscopy.
In the absence of PI-Φ2OH support, the so-formed PE does not exhibit any specific morphology. In contrast, both MAO and TMA activated catalytic systems supported with PI-Φ2OH formed spherical PE particles (Fig. 6).
 | ||
| Fig. 6 SEM pictures of PE prepared with MeDIP(2,6iPrPh)2FeCl2 under homogeneous conditions (a) and prepared in the presence of PI-Φ2OH (b) and (c). | ||
MAO-based catalytic system enables the formation of rather well-defined PE spherical particles with a sizes of 0.5 μm range. In the case of TMA, the size of the PE particles remains in the micron range but varies upon the experimental conditions (Al/Fe ratio) as shown in Table 2. A better control of the particle size is observed with respect to the higher Al/Fe ratio which corresponds to the higher catalytic activity. The higher the catalytic activity, the smaller the PE beads.
Concerning the polymerization kinetic, the decrease of ethylene pressure was followed by connecting the schlenk to a guard bottle charged with 1 bar of monomer. The pressure drop in homogeneous conditions (blank 1) and in the presence of PI-Φ2OH (run 1) are represented Fig. 7.
 | ||
| Fig. 7 Pressure decrease during ethylene polymerization with MeDIP(2,6iPrPh)2FeCl2/MAO under homogeneous conditions (Blank 1) and with PI-Φ2OH (Run 1). | ||
As can be observed, the decrease of ethylene pressure in both conditions follows the same profile, showing that PI-Φ2OH does not modify the kinetic of ethylene polymerization. Further analyses dealing with kinetic studies in various experimental conditions are currently in progress.
Polymerization of ethylene using (α-diimine)nickel(II)
In order to extend this concept of supports to a large range of catalytic systems for olefin polymerization, micellar-like structures obtained from PI-Φ2OH self-assembly have been tested as supports of nickel-based catalyst. (α-diimine)nickel(II) is known to produce branched PE (chain-walking mechanism) for which the number of branches can be tuned by catalyst structure, ethylene pressure and temperature.21–23 Less bulky catalysts are more resistant to chain-walking process and produce less branched PE. Therefore, nickel catalyst with relatively small ethyl substituents in ortho positions of aryl rings was chosen. The influence of the support and the temperature on the efficiency of (α-diimine)nickel(II) have been studied; data are collected in Table 3.| Run | n(cata)/μmol | Activator | Al/Ni | T/°C | Activity kg/mol(C)−1 h−1 bar−1 | Al/OH | Tm/°Cc | χcc | M w /kg mol−1 | D | Bead size/μm |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a Experimental conditions: heptane, 30 mL; reaction time = 1 h; T = 30 °C; Pethylene = 1 bar. b Experimental conditions: heptane, 30 mL; reaction time = 1 h; T = 30 °C; Pethylene = 1 bar; reaction time before polymerization = 24 h at 65 °C under stirring; PI-Φ2OH: Mn = 2515 g mol−1; [PI-Φ2OH] = 1 mg ml−1. c Determined by DSC. d Determined by SEC (1,2,4-trichlorobenzene at 135 °C). e Bimodal distribution. | |||||||||||
| Blank 1a | 5 | TMA | 200 | 0 | 208 | — | 131 | 52% | 413 | 7 | — |
| 1b | 5 | TMA | 200 | 0 | 11 | 84 | 133 | 34% | 406 | 3.5e | 1–4 |
| Blank 2a | 5 | MAO | 200 | 30 | 344 | — | 69 | 1% | 156 | 2.6 | — |
| 2b | 5 | MAO | 200 | 30 | 317 | 84 | 63 | 1% | 242 | 1.5 | 5–80 |
| Blank 3a | 5 | MAO | 200 | 0 | 525 | — | 126 | 38% | 433 | 4.8 | — |
| 3b | 5 | MAO | 200 | 0 | 61 | 84 | 122 | 31% | 192 | 1.4 | 0.5 |
| Blank 4a | 5 | MAO | 200 | −10 | 324 | — | 128 | 44% | 490 | 3.5 | — |
| 4b | 5 | MAO | 200 | −10 | 90 | 84 | 126 | 40% | 2 440 | 1.8 | 0.5 |
| Blank 5a | 1 | MAO | 460 | −10 | 1341 | — | 138 | 61% | 773 | 5.8 | — |
| 5b | 1 | MAO | 460 | −10 | 402 | 39 | 135 | 60% | 1 720 | 1.4 | 0.5 |
Catalytic activities of (α-diimine)nickel(II) in the presence of PI-Φ2OH support vary from 11 to 402 kg PE/(mol Ni h bar) with respect to the activator and the temperature. Polymerizations activated by TMA showed generally lower activity compared to MAO activated ones.
Unlike polymerizations performed at 30 °C, for which the catalytic activities are comparable whatever the conditions, at low temperature (−10/0 °C), the catalytic activity was found to be much lower in the presence of PI-Φ2OH support.
This behavior can be explained by the difficulty encountered by the catalyst to penetrate the micellar structure, thus impeding the effective formation of the active species.
As expected, the catalytic activity increased with the Al/Ni ratio, whatever the experimental conditions (activity = 402 kg PE/(mol Ni h bar) for MAO/Ni = 460, run 5, and activity = 90 kg PE/(mol Ni h bar) for MAO/Ni = 200, run 4). With MAO as an activator (runs 2–5), PEs with monomodal molar mass distributions and narrow dispersities, D, were obtained while PE with a bimodal distribution is obtained with TMA (run 1) (Fig. 8). As it could be expected, a decrease in the reaction temperature yields PEs with a higher crystallinity ratio (higher melting temperature) in agreement with a lower chain-walking rate and the formation of high molecular weight linear polyethylene (run 4).
 | ||
| Fig. 8 GPC chromatograms (refractive index) of PE produced with (α-diimine)nickel(II) and PI-Φ2OH as support. | ||
In all cases, scanning electron microscopy revealed the formation of spherical PE beads (Fig. 9). Interestingly, the size and size distribution of the PE particles depend on the experimental conditions.
 | ||
| Fig. 9 SEM pictures of PE prepared with (α-diimine)nickel(II) under homogeneous conditions (a) and prepared in the presence of PI-Φ2OH (b) and (c). | ||
While PE particles with a broad size distribution of between 5 μm and 80 μm are obtained at 30 °C (run 2), small and narrow distributed PE particles (0.5 μm) are formed at low temperature (−10 °C, run 4). These results clearly show that the PE particle size is strongly affected by the polyethylene microstructure (linear or branched).
The formation of highly linear polyethylene (high crystalline ratio) leads to the formation of a large number of particle nuclei yielding small PE particles with a narrow size distribution. Conversely, the formation of highly branched amorphous polyethylene favor the formation of large PE particles. This phenomenon can be reasonably supported by the aggregation of growing elementary particles explained by the amorphous state of the branched polyethylene.
Polymerization of ethylene using Ind2ZrCl2
This concept of supporting single-site catalysts was finally extended to classical metallocene. It is worth mentioning that previous attempts to trap metallocene/MAO catalytic system within PS-based micellar structures in toluene16,17 lead to systems that revealed unsuccessful to produce PE beads with a controlled morphology, probably, because only few MAO was trapped by the support. In this study, Ind2ZrCl2 was tested with MAO as an activator.The two PI-Φ2OH supports (Mn = 2515 g mol−1 and Mn = 5843 g mol−1) were used for the polymerization of ethylene using the conditions mentionned in Table 4.
| Run | n(cata)/μmol | Al/Zr | M n (PI-Φ2OH)/g mol−1 | Activity/kg mol(C)−1 h−1 bar−1 | Al/OH | Tm/°Cb | χcc | M w /kg mol−1 | D | Bead size/μm |
|---|---|---|---|---|---|---|---|---|---|---|
| a Experimental conditions: hetpane, 60 mL; reaction time = 1 h; Pethylene = 1 bar. b Experimental conditions: hetpane, 60 mL; reaction time = 1 h; Pethylene = 1 bar; Reaction time before polymerization = 24 h at 65 °C under stirring [PI-Φ2OH] = 1mg ml−1. c Determined by DSC. d Determined by SEC (1,2,4-trichlorobenzene at 150 °C). | ||||||||||
| Blank 0a | 3 | 500 | — | 337 | — | 135 | 64% | n.d. | n.d. | — |
| 1b | 3 | 500 | 2515 | 261 | 63 | 135 | 66% | 306 | 4 | 1 |
| 2b | 3 | 500 | 5843 | 517 | 146 | 136 | 68% | 306 | 3.3 | 1 |
Surprisingly, the catalytic activities obtained in the presence of PI-Φ2OH supports are comparable to the one obtained in homogeneous conditions. An activity increase is even noticed in the presence of the highest molar mass PI. The molar mass and molar mass distribution, D, of the so-formed PE remain the same, irrespective of the experimental conditions.
As it can be seen in Fig. 10, PE beads could be readily obtained in the presence of PI-Φ2OH supports. The size of the PE beads stays around 1 μm irrespective of the support molar mass. This last result proves the validity of the concept that consist in using micellar structures as organic supports for metallocene catalysts.
 | ||
| Fig. 10 SEM pictures of PE prepared with Ind2ZrCl2 under homogeneous conditions (a) and prepared in the presence of PI-Φ2OH (b). | ||
Role of PI-Φ2OH support in the production of PE beads
In order to check the presence of polyisoprene support in the so-formed PE beads, differential scanning calorimetry (DSC) was first carried out. This technique did not reveal the presence of a PI phase. Dynamic mechanical analysis (DMA) was therefore performed on PE samples prepared in both homogeneous and heterogenous conditions (Fig. 11). | ||
| Fig. 11 DMA analysis of PE produced with Ind2ZrCl2 on which the Tg of polyisoprene is visible. | ||
Such an analysis was done on PEs produced with Ind2ZrCl2. The tan δ versus temperature plot shows a first peak at −114 °C, usually denoted γ, which corresponds to the Tg of the PE. A second peak at −55 °C is present only for the PE sample produced in presence of PI-Φ2OH. This transition corresponds to the polyisoprene Tg proving that some PI is trapped within PE beads and thus that PI-Φ2OH supports play a role in the formation of PE beads. The last transition, at 60 °C, denoted α, is related with vibrational motion and reorientation within the PE crystal.24
Such an analysis was also performed on PE samples produced with MeDIP(2,6iPrPh)2FeCl2 but the presence of PI phase was not revealed in the PE beads because of the high catalytic activity (Table 2, runs 1 and 2).
Experimental section
Materials
Isoprene, sec-butyllithium, benzophenone, MAO, TMA were purchased from Aldrich. Cyclohexane and heptane were dried over polystyryl-lithium and distilled under vacuum. Isoprene was dried over di-n-butylmagnesium and distilled under vacuum and kept at 5 °C. Sec-butyllithium was filtered prior to use. Benzophenone was sublimated and kept under vacuum at room temperature. TMA (2 M solution in heptane), MAO (10%w in toluene) were used as received. MeDIP(2,6-iPrPh)2FeCl2 was synthesized as reported in the literature25 and kept in a glovebox under argon atmosphere. Ind2ZrCl2 was purchased from Strem and kept in a glovebox under argon atmosphere. (α-diimine)nickel(II) was synthesized as reported in the literature26 and kept in a glovebox under argon atmosphere.Synthesis of polyisoprene diphenyl alcohol terminated (PI-Φ2OH)
A dry round bottom flask equipped with a magnetic stirrer was charged with 40 mL of cyclohexane, s-butyllithium (1.67 mL; 0.002 mol) and isoprene (5.88 mL). The flask was immersed in an oil bath at 50 °C for 2h. A solution (1M) of benzophenone in cyclohexane was then added (4 mL; 0.004 mol) and the mixture was stirred at 50 °C overnight. The polymer was precipitated into an excess of methanol, then an excess of ethanol at 30 °C to remove unreacted benzophenone and finally dried under vacuum at room temperature. Mn = 2515 g mol−1, Mw/Mn = 1.07, 1H NMR (400 MHz, CDCl3): δ (ppm) = 1.64 (CH3 of PI, b), 2.05 (CH2 of PI, m), 3.06 (H of OH, s), 5.1 (H of PI, b), 7.5–7.2 (aromatic, 10H, m).Polymerization of ethylene
The desired quantity of PI-Φ2OH support was introduced in a Schlenk tube and lyophilized during a night with dioxane. Dry heptane and activator (TMA or MAO) was added. The reactor mixture was heated to 65 °C for 24h. The reaction mixture was then connected to a 1 bar ethylene gas outlet using a rubber tube. Schlenk tube was purged by ethylene for 20 min to remove argon. Polymerization was initiated by injection of the required amount of catalyst in toluene via a syringe. After 1 h of polymerization, the vessel was disconnected from the ethylene outlet, and the polymer was precipitated by addition of ethanol containing 10% HCl. The precipitated polymer was filtered, washed, and dried to constant weight.Analysis
1H NMR spectra were recorded using Bruker AC-400 NMR spectrometer using CDCl3 at room temperature. Size exclusion chromatography (SEC) was performed in THF at 40 °C at a flow rate of 1 mL min−1 using a differential refractometer (Varian) and a UV-visible spectrophotometer (Varian) operating at 254 nm and 4 TSK columns (G5000HXL (9 μm), G4000HXL (6 μm), G3000HXL (6 μm), and G2000HXL (5 μm)). Calibration was performed using linear (1,4)-polyisoprene standards.The PE weight average molar mass (Mw) and molar mass distribution of (α-diimine)nickel(II) produced PEs were determined using a Waters GPCV2000 SEC instrument, equipped with two PLgel (300 × 7.5 mm) columns, and an online viscometer and refractive index detector, at 135 °C in 1,2,4-trichlorobenzene as solvent at a flow rate of 0.9 mL min−1.
The PE weight average molar mass (Mw) and molar mass distribution of MeDIP(2,6-iPrPh)2FeCl2 and Ind2ZrCl2 produced PEs were determined using a Waters GPCV2000 SEC instrument, equipped with three columns PLgel Olexis Guard (300 × 7.5 mm), and an online viscometer and refractive index detector, at 150 °C in 1,2,4-trichlorobenzene as a solvent at a flow rate of 1 mL min−1.
PE beads were observed under scanning electronic microscopy (SEM) analyses on a JEOL JSM 2500 apparatus.
Dynamic light scattering (DLS) measurements were performed using an ALV Laser goniometer, which consists of a 22 mW HeNe linear polarized laser with 632.8 nm wavelength and an ALV-5000/EPP multiple tau digital correlator with 125 ns initial sampling time. The samples were kept at constant temperature (30 °C) during all the experiments. The accessible scattering angle range is from 10° up to 150°. However, due to difficulties in removing dust, most of the dynamic measurements were done at diffusion angles higher than 40°. The dispersions were introduced into 10 mm diameter glass cells. Dynamic light scattering measurements were evaluated by fitting the measured normalized time autocorrelation function of the scattered light intensity with the help of the constrained regularization algorithm (CONTIN), which provides the distribution of relaxation times τ, A(τ), as the inverse Laplace transform of g(1)(t) function:
 | (1) |
The apparent diffusion coefficient D was obtained by plotting the relaxation frequency, Γ (Γ = τ−1) versus q2 where q is the wavevector defined as
 | (2) |
 | (3) |
The DMA analysis measurements were performed with a Thermal Analysis RSA 3 instrument using a cylindrical geometry over a temperature range of −140 to 150 °C at a rate of 10 °C min−1 under nitrogen flow. All samples were scanned at a frequency of 1Hz.
Conclusions
In this paper, we report the use of polyisoprene end-capped with a diphenyl alcohol, PI-Φ2OH, as support for ethylene polymerization in an aliphatic solvent, i.e., heptane. The self-assembly of these end-functionalized PI oligomers in heptane allowed us to use the formed micellar structures as organic supports for both post-metallocene and metallocene catalytic systems. Ethylene polymerizations performed in the presence of such nanoreactors lead to the formation of PE beads with a micrometric size. Remarkably, this paper shows, for the first time, that the polyethylene microstructure (linear or branched) affects the size of the so-formed PE beads (in the case of Ni catalyst) and also that such concept of organic templates can be effectively extended to metallocene catalysts, which generally require a quite high concentration of aluminic activator.Acknowledgements
The present research was financially supported by the French Ministry of Research and Education, the Centre National de la Recherche Scientifique, the Institut Universitaire de France and the Aquitaine Council; the Grant Agency of the Czech Republic (grant No. 104/07/P264) and Ministry of Education, Youth and Sports (program MSM 6046137302).Notes and references
- J. K. F. Langhauser, M. Kersting, P. Kölle, D. Lilge and P. Müller, Angew. Makromol. Chem., 1994, 223, 155–164 CrossRef.
- M. R. Ribeiro, A. Deffieux and M. F. Portela, Ind. Eng. Chem. Res., 1997, 36, 1224–1237 CrossRef CAS.
- T. Uozumi, T. Toneri, K. Soga and T. Shiono, Macromol. Rapid Commun., 1997, 18, 9–15 CrossRef CAS.
- S. Collins, W. M. Kelly and D. A. Holden, Macromolecules, 1992, 25, 1780–1785 CrossRef CAS.
- K. Soga, T. Uozumi, M. Saito and T. Shiono, Macromol. Chem. Phys., 1994, 195, 1503–1515 CrossRef CAS.
- S. S. Sarma and S. Sivaram, Macromol. Chem. Phys., 1997, 198, 495–503 CrossRef CAS.
- G. Fink, B. Steinmetz, J. Zechlin, C. Przybyla and B. Tesche, Chem. Rev., 2000, 100, 1377–1390 CrossRef CAS.
- M. A. Ferrero and M. G. Chiovetta, Polym. Eng. Sci., 1987, 27, 1436–1447 CrossRef CAS.
- J. R. Severn, J. C. Chadwick, R. Duchateau and N. Friederichs, Chem. Rev., 2005, 105, 4073–4147 CrossRef CAS.
- H. Nishida, T. Uozumi, T. Arai and K. Soga, Macromol. Rapid Commun., 1995, 16, 821–830 CrossRef CAS.
- T. Kitagawa, T. Uozumi, K. Soga and T. Takata, Polymer, 1997, 38, 615–620 CrossRef CAS.
- S. C. Hong, H. T. Ban, N. Kishi, J. Jin, T. Uozumi and K. Soga, Macromol. Chem. Phys., 1998, 199, 1393–1397 CrossRef CAS.
- A. G. M. Barrett and Y. R. De Miguel, Chem. Commun., 1998, 2079–2080 RSC.
- C. Bouilhac, E. Cloutet, H. Cramail, A. Deffieux and D. Taton, Macromol. Rapid Commun., 2005, 26, 1619–1625 CrossRef CAS.
- Centre National de la Recherche Scientifique, France Pat., WO2009007544, 2008.
- C. Bouilhac, E. Cloutet, D. Taton, A. Deffieux, R. Borsali and H. Cramail, Macromolecules, 2008, 41, 7321–7329 CrossRef CAS.
- C. Bouilhac, E. Cloutet, D. Taton, A. Deffieux, R. Borsali and H. Cramail, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 197–209 CrossRef CAS.
- V. Busico, R. Cipullo, F. Cutillo, N. Friederichs, S. Ronca and B. Wangt, J. Am. Chem. Soc., 2003, 125, 12402–12403 CrossRef CAS.
- R. P. Quirk, T. Takizawa, G. Lizarraga and L.-F. Zhu, Journal of Applied Polymer Science: Applied Polymer Symposium, Kurume, Jpn, 1992 Search PubMed.
- S. W. Provencher, Comput. Phys. Commun., 1982, 27, 213–227 CrossRef.
- D. P. Gates, S. A. Svejda, E. Onate, C. M. Killian, L. K. Johnson, P. S. White and M. Brookhart, Macromolecules, 2000, 33, 2320–2334 CrossRef.
- F. Alobaidi, Z. Ye and S. Zhu, Polymer, 2004, 45, 6823–6829 CrossRef CAS.
- Fangming Zhu, Wei Xu, Xinxing Liu and Shangan Lin, J. Appl. Polym. Sci., 2002, 84, 1123–1132 CrossRef CAS.
- U. Hippi, J. Mattila, M. Korhonen and J. Seppälä, Polymer, 2003, 44, 1193–1201 CrossRef CAS.
- B. L. Small, M. Brookhart and A. M. A. Bennett, J. Am. Chem. Soc., 1998, 120, 4049–4050 CrossRef CAS.
- C. M. Killian, D. J. Tempel, L. K. Johnson and M. Brookhart, J. Am. Chem. Soc., 1996, 118, 11664 CrossRef.
| This journal is © The Royal Society of Chemistry 2010 |