Dynamic supramacromolecular self-assembly: deformable polymer fabricated nanostructures through a host-controlled approach†
Qiang
Yan
,
Jinying
Yuan
*,
Yan
Kang
and
Yingwu
Yin
Key Lab of Organic Optoelectronics & Molecular Engineering, Department of Chemistry, Tsinghua University, Beijing, 100084, People's Republic of China. E-mail: yuanjy@mail.tsinghua.edu.cn; Fax: +86-10-62771149; Tel: +86-10-62783668
First published on 16th February 2010
Abstract
Host molecules (α-CD) hierarchically thread onto the double grafts of the comb-copolymerEC-g-PCL-b-PEO from the outer block to the inner block in aqueous solution. Adjusting the molar ratio between α-CD and copolymer can induce these supramacromolecular complexes to dynamically and reversibly self-tune their fabrication from micelles (0-D) to cylinders (1-D) to vesicles (3-D) to sheets (2-D) like “living” assemblies.
Reversible control over “supramacromolecular” structures is a challenging topic in polymer chemistry.1 Deformable polymernanostructures exhibit a certain dynamic feature, which is similar to “living” assemblies.2 In this respect, stimulus-response polymers play a key role in realizing reversibility.3 An exciting example is that a terpolymer can switch from micelles to vesiclesvia temperature stimuli.4 However, these strategies are limited to finite stimuli-types and static self-assembly principles, and do not match the regulation means of biological systems. To this end, dynamic self-assembly, as a ubiquitous phenomenon in the organism, may be a promising way to address above issues.5 Prior studies on small molecular deformable fabrications have made preliminary progress,6 however, utilizing natural or artificial macromolecules for the simulation of dynamic process is still a challenge.
Herein, we develop a host-controlled approach to accomplish supramacromolecular self-assembly through a dynamic host–guest interaction, and reversibly fabricate various polymer hierarchical nanostructures. In our study, the copolymer (as macromolecular guest) can continuously trap a host molecule (α-cyclodextrin, α-CD) on its side chains to trigger a kinetic fabricated evolution. In contrast, competitive guests (phenol) strip α-CDs away from the polymer stepwise, leading to a hierarchical diassembly. This dynamic-equilibrium facilitates the complexes forming bottom-up deformable supramolecular objects (Fig. 1).
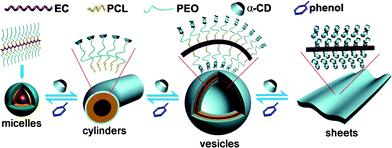 | ||
| Fig. 1 A schematic illustration of the dynamic supramacromolecular self-assembly transformation for a series of “living” polymernanostructures from micelles (zero-dimension, 0-D) to cylinders (1-D) to vesicles (3-D) to sheet superstructures (2-D). | ||
To realize the above dynamic self-assembly, we designed and synthesized a comb-copolymer (Scheme 1), ethylcellulose-g-poly(caprolactone)-b-poly(ethylene oxide) (EC-g-PCL-b-PEO, 1), viaring-opening polymerization and an efficient coupling reaction. PCL and PEO double side chains are selected based on their different capabilities of association with α-CD species,7 which may provide an opportunity to drive the dynamic phase transitions. Fully structural characterization demonstrates that the copolymer possess a high-molecular-weight (Mn = 327.8 kg mol−1 and PDI = 1.22) with weight fraction of PEO segments (wEO) to be 0.62 (wEO = MEO/Mn, Fig. S1–S3, Scheme S1, and Table S1 in ESI†).
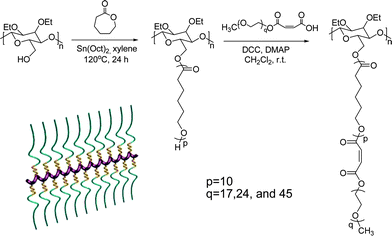 | ||
| Scheme 1 A schematic illustration of synthetic process and topological structure of comb-copolymer1. | ||
Without α-CD, copolymer1 in THF–H2O solution can self-assemble into sphercial micelles consisting of PCL cores and PEO hydrophilic coronas. The critical micelle concentration (CMC) is determined to be 1.43 × 10−6 M by turbidity studies and the probe fluorescence technique (Fig. S4–S5 in ESI†). Dynamic light scattering (DLS) reveals that these micellar aggregates can maintain an almost constant hydrodynamic radius, ‘Rh’, of 12–18 nm at a wide concentration range above the CMC (Fig. S6 in ESI†), presenting a fixed phase state.
Interestingly, when α-CD was added to the solution, a dynamic supramacromolecular self-assembly transition takes place. In the presence of 3.6 × 10−4 M α-CDs, the Rh of polymersomes increases to ∼51 nm with a smaller aggregate coexisting (5–20 nm), suggesting a new intermediate phase. Upon successively increasing α-CD concentration to 2.3 × 10−3 M, much larger aggregates probably vesicles with Rh ∼69 nm were observed. Most notably, giant planar assemblies (>400 nm) with broad size-distribution can be obtained while α-CD is up to 8.7 × 10−3 M (Fig. 2a). The host-dependent aggregated phenomenon indicates that the fabricated evolutions require an inexhaustible matter-flow supply.
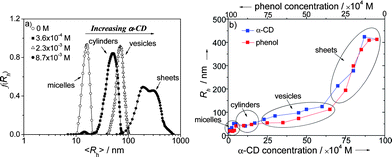 | ||
| Fig. 2 (a) The dynamic shift of the hydrodynamic radius distribution, f(Rh), obtained for binary assembly system of polymer-α-CD through successively adding α-CD to the polymer solution, (○) without α-CD inducing micelles, (■) 3.6 × 10−4 M α-CDs inducing cylinders, (△) 2.3 × 10−3 M α-CDs inducing vesicles, and (●) 8.7 × 10−3 M α-CDs inducing sheet superstructures; (b) the size-variation curves of the dynamic self-assembly process: reversible nanomorpholgoical deformation via the molar variation of host (α-CD) and guest (phenol) trigger. | ||
To elucidate the aforementioned deformable nanostructures, transmission electron microscopy (TEM) was employed to visualize the successive transformation process. Indeed, polymer1 in solution forms spherical micelles of 29 nm in diameter. With continual injection of host molecules, as expected, it is demonstrated that the assemblies transform hierarchically from cylinders through vesicles to irregular sheet superstructures (Fig. 3a–d). The sizes of the three types of geometries in the TEM images are 105 ± 15, 140 ± 30, and > 420 nm, respectively, which is in line with the DLS results. The incredible jump in scales and morphologies shows that host plays a crucial role in manipulating the dynamic behavior of the polymerassemblies. Moreover, atomic force microscopy (AFM) images also support these results, exhibiting a “living” nanostructural transition from spherical micelles to cylinders to vesicles to sheet superstructures (Fig. 4a–d). Notably, the thickness of the supramolecular sheets is ca. 7–15 nm, which is agreement with the molecular whole length of the double grafts of 1, indicating that the sheet adopts planar monolayer structure.
 | ||
| Fig. 3 TEM micrographs of the dynamically supramacromolecular morphological transition through addition of α-CD successively into the polymer solutions: (a) without α-CD (bar = 100 nm), (b) 3.6 × 10−4 M α-CD (bar = 100 nm), (c) 2.3 × 10−3 M α-CD (bar = 200 nm), and (d) 8.7 × 10−3 M α-CD (bar = 500 nm). | ||
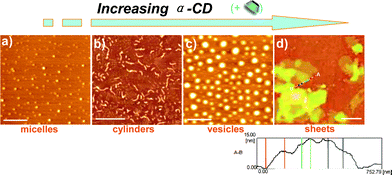 | ||
| Fig. 4 AFM micrographs of the dynamic supramacromolecular morphological transition, obtained by a binary assembly system polymer-α-CD upon continuously adding α-CD to the polymer solution. (a) Pure polymerassemblies without α-CD (micelles), (b) with 3.6 × 10−4 M α-CDs (cylinders), (c) with 2.3 × 10−3 M α-CDs (vesicles), and (d) with 8.7 × 10−3 M α-CDs (sheets); bar = 500 nm. Inset: the height changes of the supramolecular sheet. | ||
Our next interest focuses upon whether the host-controlled objects are reversible. For this purpose, phenol, as a stronger guest of α-CD than 1, was introduced into the system to compete with the polymer.8 As a result, the kinetic equilibrium of ternary system polymer-α-CD-phenol results in a series of top-down disassemblies (Fig. 5a–d). The addition of 1.2 × 10−3 M phenols causes the fusion and erosion of these sheet-edge, which illustrates that zero-curvature superstructures cannot sustain after removing partial α-CDs from the side chains of the polymer. With continuous increasing phenol concentration to 4.7 × 10−3 M, the sheetlike objects are entirely disrupted, whereas smaller vesicles (20–90 nm) are reconstructed. It seems that vesicular geometry is a stable phase, because it can exist at a broad concentration range of both α-CD and phenol. In contrast, cylinders are in a metastable transition, and only narrow guest range (8.1 × 10−3–8.7 × 10−3 M) can retune this morphology. Finally, complete removal of α-CDs from the polymers when the phenol concentration is increased to 9.5 × 10−3 M reverts back to initial micellar morphologies (14–25 nm). In situ DLS measurements verified that the process is reversible and repeatable (Fig. S7 in ESI†).
 | ||
| Fig. 5 TEM micrographs of the dynamic-hierarchical disassembly of the ternary system of polymer-α-CD-phenol through adjusting phenol concentration: (a) 1.2 × 10−3 M phenol (bar = 250 nm), (b) 4.7 × 10−3 M phenol (bar = 200 nm), (c) 8.6 × 10−3 M phenol (bar = 100 nm), and (d) 9.5 × 10−3 M phenols (bar = 150 nm). | ||
This dynamic self-assembly mechanism is reasoned as follows: the host-controlled matter-flow incessantly changes the entropies and interfacial energies of assembly system, which readjusts the amphiphilicity of the polymer, thus resulting in a continuous phase transition. Generally, for covalent block copolymers, when the different blocks are chemically immiscible, amphiphilic balance drives phase separation and chemical bonding constrains the ordered domains.9 In particular, for PEO-containing aggregates, global micelles predominate if wEO > 0.55, whereas cylinders form when wEO = 0.50–0.55, and wEO < 0.45 favors vesicles with the lowest energy.10 According to the theories for our non-covalently bound host–guest polymer system, therefore, 1 (wEO = 0.62 higher than the threshold) self-assembles naturally into spheres. Upon adding a small quantity of α-CD, they can nest into PEO blocks to slightly inhibit the hydrophilicity of the copolymer.11 This is equivalent to reducing wEO indirectly for the cylindrical micelles. Upon sequential injection to the assemblies, more α-CDs are captured by per PEO graft and their hydrophobic cavities further suppress interfacial energy, turning the aggregates into superior architectures with lower curvature such as vesicles. As α-CD inclusion is up to a maximum, they move from outer PEO blocks to both inner PEO and PCL segments,12 and closed-packed PEO/PCL polyrotaxane palisades are formed, which extremely reduce the hydrophilicity and mobility of polymerassemblies; furthermore, these polyrotaxanes tend to adopt a parallel arrangement on the copolymer backbones due to α-CD crystalline induced effect.13 This effect promotes vesicles changing into planar superstructures. 1D-NMR, 2D-NMR, and XRD results are used to further confirm the dynamic host–guest interplay variation between α-CD, phenol, and polymer (Fig. S8–S10 in ESI†). For the opposite disassociation process, phenol dethreads α-CDs from the polymer chains stepwise, depending on its amount in the system. Kinetic studies on noncovalent interactions of the ternary system by 1H NMR again elucidate the hierarchical disassembly principle (Fig. S11 in ESI†). One can tune phenol and α-CD pairs to precisely manipulate the scales and shapes of the self-assemblies as one requires (Fig. 2b).
In summary, a host-controlled dynamic supramacromolecular self-assembly approach has been developed to build reversible polymer fabricated architectures. Adjusted by the host–guest pairs, the copolymer fabrication shows smart self-rearrange and deformable behavior. We envision that this self-assembly model will open up a facile strategy for “living” assemblies.
We gratefully acknowledge the financial support of the National Natural Science Foundation of China (No. 20836004, No. 20974058), and the National Basic Research Program (2009CB930602).
Notes and references
- (a) J. M. Lehn, Science, 2002, 295, 2400–2403 CrossRef CAS; (b) D. E. Discher and A. Eisenberg, Science, 2002, 297, 967–973 CrossRef CAS; (c) Z. L. Zhou, Z. B. Li, Y. Ren, M. A. Hillmyer and T. P. Lodge, J. Am. Chem. Soc., 2003, 125, 10182–10183 CrossRef CAS; (d) H. Xiang, K. Shin, T. Kim, S. I. Moon, T. J. McCarthy and T. P. Rusell, Macromolecules, 2005, 38, 1055–1056 CrossRef CAS; (e) H. G. Cui, Z. Y. Chen, S. Zhong, K. L. Wooley and D. J. Pochan, Science, 2007, 317, 647–650 CrossRef CAS; (f) J. T. Zhu and R. C. Hayward, J. Am. Chem. Soc., 2008, 130, 7496–7052 CrossRef CAS.
- (a) S. J. Rowan, Nat. Mater., 2009, 8, 89–91 CrossRef CAS; (b) Y. F. Zhou and D. Y. Yan, J. Am. Chem. Soc., 2005, 127, 10468–10469 CrossRef CAS; (c) J. W. Szostak, D. P. Bartel and P. L. Luisi, Nature, 2001, 409, 387–390 CrossRef CAS; (d) T. Zhu and J. W. Szostak, J. Am. Chem. Soc., 2009, 131, 5705–5713 CrossRef CAS.
- (a) pH-responsive: J. Rodriguez-Hernandez and S. J. Lecommandoux, J. Am. Chem. Soc., 2005, 127, 2026–2027 Search PubMed; (b) pH-responsive: J. Z. Du, Y. P. Tang, A. L. Lewis and S. P. Armes, J. Am. Chem. Soc., 2005, 127, 17982–17983 Search PubMed; (c) Thermal-responsive: A. J. Convertine, B. S. Lokitz, L. J. Myrick, C. W. Scales, A. B. Lowe and C. L. McCormick, Macromolecules, 2006, 39, 1724–1730 Search PubMed; (d) Thermal-responsive: Y. T. Li, B. S. Lokitz and C. L. McCormick, Angew. Chem., Int. Ed., 2006, 45, 5792–5795 Search PubMed; (e) Light-responsive: X. K. Liu and M. Jiang, Angew. Chem., Int. Ed., 2006, 45, 3846–3850 Search PubMed; (f) Redox-responsive: A. Napoli, M. Valentin, N. Tirelli, M. Müller and J. A. Hubbell, Nat. Mater., 2004, 3, 183–189 Search PubMed.
- A. Sundararaman, T. Stephan and R. B. Grubbs, J. Am. Chem. Soc., 2008, 130, 12264–12265 CrossRef CAS.
- (a) B. A. Grzybowski, H. A. Stone and G. M. Whitesides, Nature, 2000, 405, 1033–1036 CrossRef CAS; (b) B. A. Grzybowski and C. J. Campbell, Chem. Eng. Sci., 2004, 59, 1667–1676 CrossRef CAS; (c) M. F. Copeland and D. B. Weibel, Soft Matter, 2009, 5, 1174–1187 RSC.
- (a) S. Yagai, S. Kubota, H. Saito, K. Unoike, T. Karatsu, A. Kitamura, A. Ajayaghosh, M. Kanesato and Y. Kikkawa, J. Am. Chem. Soc., 2009, 131, 5408–5409 CrossRef CAS; (b) C. Wang, S. C. Yin, S. L. Chen, H. P. Xu, Z. Q. Wang and X. Zhang, Angew. Chem., Int. Ed., 2008, 47, 9049–9052 CrossRef CAS; (c) C. Wang, Y. S. Guo, Y. P. Wang, H. P. Xu and X. Zhang, Chem. Commun., 2009, 5380–5382 RSC.
- (a) A. Harada and M. Kamachi, Macromolecules, 1990, 23, 2821–2823 CrossRef CAS; (b) Y. Kawaguchi, T. Nishiyama, M. Okada, M. Kamachi and A. Harada, Macromolecules, 2000, 33, 4472–4477 CrossRef CAS.
- H. Q. Dong, Y. Y. Li, S. J. Li, J. Cai, R. X. Zhuo, X. Z. Zhang and L. J. Liu, Angew. Chem., Int. Ed., 2008, 47, 5573–5576 CrossRef CAS.
- (a) T. Smart, H. Lomas, M. Massignani, M. V. Flores-Merino, L. R. Perez and G. Battaglia, Nano Today, 2008, 3, 38–46 CrossRef CAS; (b) J. N. Israelachvili, D. J. Mitchell and B. W. Ninham, J. Chem. Soc., Faraday Trans. 2, 1976, 72, 1525–1568 RSC.
- (a) S. Jain and F. S. Bates, Science, 2003, 300, 460–464 CrossRef CAS; (b) P. Dalhaimer, F. S. Bates and D. E. Discher, Macromolecules, 2003, 36, 6873–6877 CrossRef CAS.
- R. Breslow and S. D. Dong, Chem. Rev., 1998, 98, 1997–2011 CrossRef CAS.
- W. Z. Yuan and J. Ren, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 2754–2762 CrossRef CAS.
- (a) C. Travelet, G. Schlatter, P. Hébraud, C. Brochon, A. Lapp, D. V. Anokhin, D. A. Ivanov, C. Gaillard and G. Hadziioannou, Soft Matter, 2008, 4, 1855–1860 RSC; (b) L. X. Ren, F. Y. Ke, Y. M. Chen, D. H. Liang and J. Huang, Macromolecules, 2008, 41, 5295–5300 CrossRef CAS; (c) C. C. Rusa, J. Fox and A. E. Tonelli, Macromolecules, 2003, 36, 2742–2747 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and characterization details. See DOI: 10.1039/c0py00024h |
| This journal is © The Royal Society of Chemistry 2010 |