Performance polymers from renewable monomers: high molecular weight poly(pentadecalactone) for fiber applications
Matthijs
de Geus
a,
Inge
van der Meulen
a,
Bart
Goderis
b,
Kristof
van Hecke
c,
Marko
Dorschu
d,
Harm
van der Werff
d,
Cor E.
Koning
a and
Andreas
Heise
*ae
aTechnische Universiteit Eindhoven, Laboratory of Polymer Chemistry, Den Dolech 2, P.O. Box 513, 5600 MB, Eindhoven, The Netherlands. E-mail: a.heise@tue.nl; Fax: +0031-(0)40 2463966; Tel: +0031-(0)40 2473012
bKatholieke Universiteit Leuven, Chemistry Department, Molecular and Nanomaterials, Celestijnenlaan 200F, 3001, Heverlee, Belgium
cKatholieke Universiteit Leuven, Chemistry Department, Biomolecular Architecture, Celestijnenlaan 200F, 3001, Heverlee, Belgium
dDSM Research, P.O. Box 18, 6160 MD, Geleen, The Netherlands
eDublin City University, School of Chemical Sciences, Glasnevin, Dublin 9, Ireland
First published on 20th January 2010
Abstract
Enzymatic ring-opening polymerization was applied to synthesize high molecular weight polypentadecalactone (PPDL). The synthetic procedure was optimized on a small-scale and subsequently transferred to 30 g scale to yield sufficient material for fiber spinning. Molecular weights (Mw) of 143 000 g mol−1 were obtained. Mechanical and thermal properties of the non-oriented, high molecular weight PPDL were determined and are largely in agreement with the literature data. The high molecular weight PPDL was melt-processed into fibers, which were further elongated to about 9–10 times their original length. Analysis of the fibers revealed differences in crystal orientation as a function of the processing conditions. Preliminary fiber tensile measurements confirm a high strength of up to 0.74 GPa for the fiber with the highest crystal orientation.
Introduction
There is an increasing interest in sustainable materials and production methods in materials science. Under legislative, environmental and economic pressure the implementation of eco-friendly production methods and raw materials have received academic and industrial attention. However, new materials face the challenge that they have to compete with current materials on performance. In particular polymers from bio-derived resources often have inferior physical properties when compared to synthetic performance materials, which limits their applicability in more demanding applications. One particular class of renewable polymers is aliphatic polyesters, of which poly(hydroxyalkanoates) and poly(lactic acid) are prominent examples. Increasing attention was also recently given to polymers derived from long chain ω-hydroxy fatty acids.1 Primarily materials with various substitutions along the polymer chain, obtained by polycondensation processes, have been reported. We are particularly interested in the corresponding unsubstituted polymers obtained by ring-opening polymerization (ROP) of cyclic ω-hydroxy fatty acids like pentadecalactone (PDL). PDL belongs to the class of naturally occurring macrocyclic musks and is used in the fragrance industry. Its eco-friendliness led to an increased demand and the development of improved synthetic routes, which makes PDL commercially available in larger quantities.2Poly(pentadecalactone) (PPDL) is a semi-crystalline polymer with a melting point around 100 °C and a glass transition temperature far below room temperature (−27 °C).3–5 The crystallization behavior and the crystal structure of PPDL reveal large similarities with polyethylene (PE).6–8 End-functionalized low molecular weight PPDL was recently applied for coating applications.9 While often referred to as a degradable PE, we have recently shown that PPDL is neither enzymatically nor hydrolytically degradable in buffer solutions.10 Nevertheless, under harsher hydrolytic conditions as often found in the environment (e.g. soil) and in recycling processes, PPDL can be expected to degrade. Availability of PPDL of high molecular weight could open high performance application areas typically occupied by PE for example in tape and fiber products. The goal of our research is to develop a synthetic route, which gives access to larger quantities of high molecular weight PPDL (Mw > 100,000 g mol−1) and its application in fibers.
High molecular weight polyesters can only be realized by ring opening polymerization (ROP) of the corresponding macrocyclic lactones (Fig. 1). Several groups have reported on the polymerization of PDL, via chemical as well as via biocatalytic methods. It was observed that PDL and other macrolides behave differently from smaller lactones (e.g.δ-valerolactone and ε-caprolactone) with respect to their polymerization behavior.11 Lebedev and Yevstropov reported the chemical ROP using diethyl zinc as a catalyst but no details about the synthesis nor the polymer were given.12 Duda et al. used zinc octoate/butyl alcohol for the ROP and reported low activity of the catalyst towards PDL, resulting in low monomer conversion after long reaction times at elevated temperatures (26% after 7 days at 100 °C).13 In addition, the molecular weight of the obtained PPDL was low (∼1,350 g mol−1). To date, the best results for a chemical ROP of PDL were obtained by Feijen and co-workers, who reported high monomer conversion and reasonable molecular weights (Mn = 30 kg mol−1) by using yttrium isopropoxide.14 Significantly higher molecular weight PPDL was reported by enzymatic ROP using lipases. Employing Novozym 435 (Candida AntarticaLipase B immobilized on macroporous resin), number average molecular weights (Mn) of up to 86 kg mol−1 (PDI = 2.37) were reported by Gross and co-workers after precipitation in methanol.15 Even high molecular weights were reported by the enzymatic polymerization of PDL in miniemulsion with Pseudomonas Cepacia lipase, giving PPDL with Mn of 200 kg mol−1.16 However, these reported molecular weights have to be interpreted with great care as they were measured by size exclusion chromatography (SEC) using chloroform as the eluent. Our own analysis showed irreproducible molecular weights and often broad molecular weight distributions under these conditions. A closer look at the system revealed a large pressure built up during SEC-analysis of PPDL-samples above a molecular weight of ca. 10,000 g mol−1. Although apparently transparent polymer solutions were obtained in chloroform, this indicates that the polymer did not dissolve properly in chloroform at 25 °C even after a prolonged dilution time. This is not surprising since not only the solid state, but also the solution properties are similar to PE. Only Palmans and co-workers reported on the poor solubility of PPDL in chloroform,17 after which they decided to use elevated temperature (80 °C) and o-dichlorobenzene as an eluent for SEC analysis.11
 | ||
| Fig. 1 The enzymatic synthesis of poly(ω-pentadecalactone) (PPDL) by ring-opening polymerization of pentadecalactone (PDL). | ||
This prompted us to carefully re-examine the route to and characterization of high molecular weight PPDL on small scale (1–2 g of product) with the goal to push the molecular weight of PPDL as high as possible. The obtained information was then applied to produce high molecular weight PPDL on a larger scale (30 g product), which allowed for the melt-spinning and investigation of PPDL fibers.
Experimental procedures
Materials
All chemicals were purchased from Aldrich, stored over molecular sieves and used without further purification unless otherwise noted. Toluene (Biosolve, AR-grade) was dried over alumina and stored over molecular sieves. Novozym 435 was obtained from Novozyme AS and stored over P2O5 in a desiccator. Molecular sieves (3 Å) were dried in an oven at 420 °C prior to use. Para-oxon was dissolved in toluene before use as enzyme-inhibitor to instantly stop the reaction.Synthesis
Methods
| Fiber code | PPDL a | Processing conditions |
|---|---|---|
| a Numbers refer to entries in Table 3. | ||
| F1-1 | 3 | Melt pushed trough nozzle, stretched over hotplate |
| F2-1 | 4 | Ram extrusion: T = 110 °C; W = 15 kg; WS = 0 |
| F2-2 | 4 | Ram extrusion: T = 110 °C; W = 15 kg; WS = 1 m min−1. |
| F2-3 | 4 | Ram extrusion: T = 120 °C; W = 17 kg; WS = 1 m min−1. |
| F2-4 | 4 | Ram extrusion: T = 120 °C; W = 17 kg; WS = 10 m min−1. |
Determination of filament mechanical properties were carried out on a semi-automatic, microprocessor controlled tensile tester (the Favimat from Textechno Herbert Stein GmbH & Co. KG, Mönchengladbach, Germany), which works according to the principle of constant rate of extension (ISO 5079). The Favimat tester was equipped with a 1200 cN balance. Gauge length was 50 mm and rate of extension was 25 mm min−1. Filament linear density (i.e. mass/length) was determined by weighing of the filaments on a μ-balance. Tensile strengths in cN/dtex were converted to GPa assuming a density of PPDL of 1 gr/cm3.
The thermal behaviour and crystallinity of the reaction powders and resulting fibers was obtained via specific heat capacity, cp(T), measurements. The instrument involved was a Perkin-Elmer Pyris-1Differential Scanning Calorimeter with the block surrounding the measuring unit thermostated at −10 °C and the measuring unit being flushed with dry nitrogen. Typically, a 5 mg sample was presented in standard aluminium solid state pans, except for sample F2-4 where only 1 mg was used. This particular fiber sample was very thin and could not easily be compacted into a DSC pan. As can be expected, the lower sample mass resulted in somewhat sharper melting and crystallization transitions. Calibration was done at 10 °C min−1 with benzophenone and indium for the temperature and with indium for the enthalpy. The thermal profile was composed of a heating–cooling–heating sequence between 20 and 170 °C at 10 °C min−1 with 10 min waiting each time at 20 or 170 °C. From the measured heat flow an empty pan measurement was subtracted and after dividing by the sample mass and scanning rate cp(T) data were obtained (Jg−1K−1), which in turn were used to calculate the sample weight fraction crystallinity, wc(T), as a function of temperature according to:
 | (1) |
 | (2) |
Single fibers were selected and mounted perpendicular to an X-ray beam (Cu-Kα radiation, 500 μm across) for the collection of 2D X-ray fiber diffraction patterns at room temperature in view of retrieving information on the polymer chain orientation within the fiber crystals as a function of the elongation. A SMART 6000 diffractometer was used, equipped with 2D CCD detector. The sample-to-detector distance was 45 mm and an irradiation time of 15 min/image was used.
According to the literature, the orientation of a crystallographic plane can be extracted from a radial scan of the appropriate diffraction peak using eqn (3):21
 | (3) |
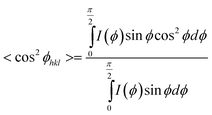 | (4) |
With ϕc being the angle between the crystallographic c-axis and the stretch direction, and g being the cosine of the angle between the normal to the hkl plane and the crystallographic c-axis. Gazzano et al. reported a pseudo orthorhombic crystal structure for PPDL, which results in g = 0 when considering an hk0 plane.7 In this case the Hermans orientation value f for the molecular orientation within the crystals is given by:
 | (5) |
 | ||
| Fig. 2 Corrected 2D scattering pattern of PPDL powder using a logarithmic gray scale for the intensity, # detector counts (left) and corresponding azimuthally averaged 1D scattering pattern with the Miller indices at the most important reflections (right). | ||
The corrected azimuthal 110 intensities were fitted to the sum of a constant value and a Gaussian. The fitted data were used as input for eqn (5). Processing of the data was performed with homemade scripts running under V for Windows (version 3.5b, Digital Optics Ltd).
Results and discussion
A prerequisite for mechanically strong fibers is a maximum of lateral interactions between the polymer chains in the fiber. In the case of PE this is achieved by the orientation of the chains in crystallites. The crystallinity and thus the mechanical strength can be further increased by a post-spinning drawing process, which aligns the polymer chains and crystallites. Similar to PE, the long linear aliphatic carbon chain of PPDL is responsible for its high crystallinity. It is thus reasonable to expect good fiber properties from this material similar to those of PE. Important material parameters in that respect are the molecular weight of the polymer, which should be as high as possible for maximum lateral chain interaction and for limiting the number of chain ends. Moreover, short plasticizing polymer chains should be absent as they reduce the mechanical properties of the bulk material.Synthesis of high molecular weight PPDL
The approach that can be followed to obtain high molecular weight polymers in enzymatic ROP is similar to the approach that is followed in chain polymerization by using a very high monomer to initiator ratio. It has to be noted, though, that enzymatic ROP is not a controlled polymerization and inter-chain and intra-chain transesterification reactions occur throughout the polymerization process and become dominant at high monomer conversion. In our approach, water, which is intrinsically present in the reaction medium, acts as a very efficient nucleophile (initiator). We followed an earlier reported optimized drying protocol to minimize the water concentration in the solvent, monomer and the enzyme but leaving traces of water for the enzyme to retain its activity (Fig. 1).18,24 This drying protocol was applied to all experiments to ensure the critical water concentration to be similar within experimental errors.Initially the enzymatic ROP of PDL to high molecular weight was performed on a small scale (1–2 g of product) at 70 °C in order to gain information on polymerization characteristics and product analysis. Immediately noticeable was the high viscosity of the polymerization mixture at longer reaction times due to the high molecular weight and crystallinity of PPDL. The impact this has on scaling-up will be discussed later but even on a small scale this causes difficulties as representative sampling from the reaction proved impossible. Therefore, parallel reactions were performed and stopped at various times so as to obtain insight into the rate of polymerization and the development of molecular weight during the reaction. While special care was taken to ensure that all reactions were run under the same conditions, one has to be aware that small variations in reaction composition (like water concentration) can have an effect on the obtained results. Fig. 3 depicts the monomer conversion of a typical polymerization reaction series. It is noticeable that the reaction is relatively slow in comparison to literature reports, reaching only about 40% conversion within the first hour. This suggests a low initial concentration of nucleophiles in the reaction medium due to the successful drying process of the reaction components. After a reaction time of about 1440 min (24 h) full monomer conversion was reached. However, the exact point of quantitative conversion cannot be concluded from these data.
 | ||
| Fig. 3 Monomer conversion in enzymatic ROP of PDL determined by 1H-NMR. All data points were obtained from single reactions at 70 °C in toluene. | ||
The molecular weights of all reaction samples were initially determined by SEC in chloroform as suggested in the literature.14–16 However, due to the large pressure build-up and the irreproducible results in chloroform, all SEC analyses in this study were performed in trichlorobenzene (TCB) at 160 °C, i.e., under common conditions for polyolefines. Inspection of Fig. 4 reveals an almost linear increase of the molecular weight (Mw is reported here as this is common in fiber applications), reaching a maximum of about 305,000 g mol−1 (PS standards) at 95% conversion. When the reactions were continued beyond that point a slight drop in the molecular weight was observed. We speculate that this is due to enzyme catalyzed chain scission, which is becoming more frequent at strongly reduced monomer concentration. Due to the high viscosity of the reaction medium and the inefficient agitation at this stage this water could locally result in enzyme-catalyzed esterhydrolysis and cause significant chain degradation. This is also supported by an increase of the PDI to about 3 at high monomer conversion, while the PDI of all other samples is between 2 and 2.5. The PPDL molecular weights obtained in this study are the highest reported so far. However quantitative comparison with literature data is not possible since in previous reports SEC analysis was performed in chloroform. For the scaled-up reaction it can be concluded that the polymerization should be stopped at a maximum of 95% monomer conversion in order to produce polymer with a maximum molecular weight.
 | ||
| Fig. 4 Weight average molecular weight (Mw) and polydispersity index (PDI) of PPDL as a function of monomer conversion for the small-scale reaction (1–2 g). Data were obtained from SEC in TCB at 160 °C (polystyrene standards). Dashed lines are added to guide the eye. | ||
In order to obtain enough polymer for fiber spinning, the enzymatic polymerization was then conducted on a larger scale. To obtain a high molecular weight polymer with a relatively narrow molecular weight distribution (polydispersity ∼2.0), mass transport limitations, both in the enzyme particle (internal) as well as in an outer stagnant film (external), should be avoided. Two approaches can be followed to achieve this: (1) decreasing the particle size and (2) ensure good mixing. Intra-particle mass transport limitation depends strongly on the size of the catalyst particle, as the molar flux of a compound in a catalyst particle is inversely correlated with the size of the particle.25–27 The average particle size of Novozym 435 is ∼400 μm (varying from 100–1000 μm), with the enzyme located in the outer shell of the particle (∼80 μm).28 Therefore, it is not possible to reduce the (effective) particle size without grinding the particles. In this study, this has not been performed, as it was believed that this reduces the enzyme activity.
A Teflon overhead anchor stirrer was used to ensure good agitation and mixing throughout the entire reaction even at high conversion (high viscosity). Moreover, since the obtained PPDL was difficult to dissolve, a relatively large reactor volume (250–300 mL) was used for the reaction. This allowed for the addition of extra solvent after the reaction, so that the polymer and enzyme could subsequently be separated by filtration. In addition, a double-wall glass reactor vessel connected to a thermostatic oil bath was chosen to ensure an optimal reactor heating. An enzyme to monomer ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 (w/w-%) and an equal monomer to solvent (toluene) weight ratio were used. The total reaction volume was around 70 mL. In all polymerizations nearly complete monomer conversion was obtained after ∼70 h of reaction as determined by 1H-NMR spectroscopy. Table 2 gives three examples of results obtained from the large scale ROP of PDL (PPDL-2–PPDL-4). Samples PPDL-3 and PPDL-4 were obtained under identical conditions, which confirms the reproducibility of the synthesis.
:20 (w/w-%) and an equal monomer to solvent (toluene) weight ratio were used. The total reaction volume was around 70 mL. In all polymerizations nearly complete monomer conversion was obtained after ∼70 h of reaction as determined by 1H-NMR spectroscopy. Table 2 gives three examples of results obtained from the large scale ROP of PDL (PPDL-2–PPDL-4). Samples PPDL-3 and PPDL-4 were obtained under identical conditions, which confirms the reproducibility of the synthesis.
| Entry | T R/°C | M n/g mol−1 | M w/g mol−1 | PDI |
|---|---|---|---|---|
| a Small scale reaction, 10% enzyme. b Molecular sieves added to reaction; crystallization of polymer at the stirrer during polymerization, 10% enzyme. c Molecular sieves added to reaction; no crystallization of polymer during polymerization, 1% enzyme. | ||||
| PPDL-1a | 70 | 143,000 | 305,000 | 2.1 |
| PPDL-2b | 80 | 83,000 | 136,000 | 1.9 |
| PPDL-3c | 85 | 80,000 | 143,000 | 2.0 |
| PPDL-4c | 85 | 77,000 | 143,000 | 2.0 |
Initially all large-scale polymerizations were conducted at 70 °C similar to the small-scale reaction. However, polymercrystallization was observed on the stirrer shaft and only low molecular weight polymers were isolated. Therefore, the reaction temperature was increased to 80 °C and molecular sieves were added to the reaction mixture in order to further reduce the participation of water in the reaction (PPDL-2, Table 2). While crystallization was still observed at this temperature, a sharp increase in molecular weight was noted (Mw = 136 kg mol−1). For PPDL-3, only 1 w/w% of Novozym 435 was used with respect to monomer in addition to the molecular sieves. Moreover, the reaction temperature was further increased to 85 °C and thus crystallization was avoided. The reaction yielded white polymer with a weight average molecular weight of 143 kg mol−1 after precipitation. The SEC traces of all polymers were monomodal (Fig. 5). Noticeable is that the maximum molecular weights obtained in the large-scale reactions are only half of the molecular weight of the small-scale reaction (PPDL-1, Table 2). Although the same drying protocol was followed as in the small-scale reaction, the larger reaction volume presents more challenges for efficient drying. It is reasonable to assume that the lower molecular weight as compared to the small-scale reactions is thus due to a higher water concentration. By further optimization and specialized equipment it should be possible to further increase the PPDL molecular weight even on large scale.
 | ||
Fig. 5
SEC traces of polymer obtained from the small-scale reaction at 98% monomer conversion (![[dash dash, graph caption]](https://www.rsc.org/images/entities/char_e091.gif) ; PPDL-1; Mw: 305 kg mol−1) and the scaled-up reaction ( ; PPDL-1; Mw: 305 kg mol−1) and the scaled-up reaction (![[thick line, graph caption]](https://www.rsc.org/images/entities/char_e117.gif) ; Mw: 143 kg mol−1; PPDL-3, Table 2). Samples were measured in TCB at 160 °C. ; Mw: 143 kg mol−1; PPDL-3, Table 2). Samples were measured in TCB at 160 °C. | ||
Properties of high molecular weight PPDL
The DSCcp(T) based melting point (95 °C) and the degree of crystallinity of 67% (Table 4 and Fig. 8) of the high molecular weight PPDL-4 (Table 2) is in agreement with reported literature values.4 From dynamic mechanical analysis (DMA) a Tg of −25 °C was determined.The mechanical properties of PPDL have hardly been investigated. Only Scandola and co-workers have reported very briefly on these properties.4 In Table 3, the tensile properties are collected as they were observed in both the study of Scandola and co-workers and in the present study. The tensile modulus, E, is around 420 MPa for the compression molded films, which is in the same order as reported in literature. The stress at break (σbreak), which had not been reported before, was observed to be as high as 38 MPa. Typically, the strain at break of the PPDL synthesized in this study, being εbreak > 1200% (Fig. 6), is much higher than the previously reported value (εbreak = 100–200%). This could be an effect of the higher molecular weight of our samples, as low molecular weight fractions act as plasticizer and have a deleterious effect on the elongation at break. However, it is difficult to draw a final conclusion since different molecular weight determination methods were used. On the other hand, we also observed that already small defects (air bubbles, etc.) in the PPDL-film could cause a lowering of the elongation at break.
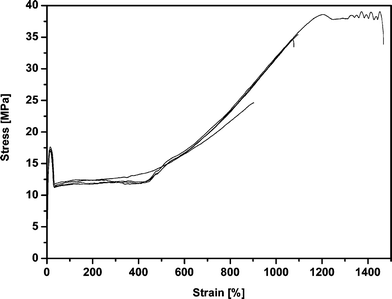 | ||
| Fig. 6 Stress-strain behavior of dumbbells obtained from a compression molded film from PPDL-4 (Table 2). | ||
PPDL fibers
PPDL fibers were first produced by melt-spinning through a nozzle but this method resulted in very inhomogeneous fibers. Better results were obtained by using Ram-extrusion following the principle of a melt indexer in which a polymer is molten in a thermostated chamber (slightly) above the melting temperature. Using a certain load on top of the chamber, the molten polymer is pushed through a dye (1.05 mm) and collected on a godet roll after quenching in a water bath (Fig. 7). The thickness of the PPDL fiber was varied by changing the winding speed of the godet roll. The PPDL fibers were further elongated over a hot plate to about 9–10 times their original length. | ||
| Fig. 7 PPDL-4 powder and examples of PPDL fibers spun from the melt by Ram extrusion. | ||
Fig. 8 illustrates the evolution of cp(T) and wc(T) during a heating–cooling–heating run at 10 °C min−1 for sample PPDL-4, representing an original powder after synthesis, and F2-3 and F1-1, representing typical fiber materials (Table 1). In Table 4 the crystallinity values at room temperature prior to first (wc1(25 °C)) and second heating (wc2(25 °C)) are listed for all samples investigated together with the crystallization (Tc) and melting peak temperatures during first (Tm1) and second heating (Tm2). Clearly, the crystallinity of the starting powders is higher than that of the fibers. However, after a heating-cooling cycle, the crystallinity of these samples reduces to that found for the fibers that were melt processed. The crystallinity of all fibers is comparable (0.54 on average) and does not alter after a heating-cooling cycle. The melting points, too, do not seem to depend on the fiber processing protocol and do not shift after a heating-cooling cycle.
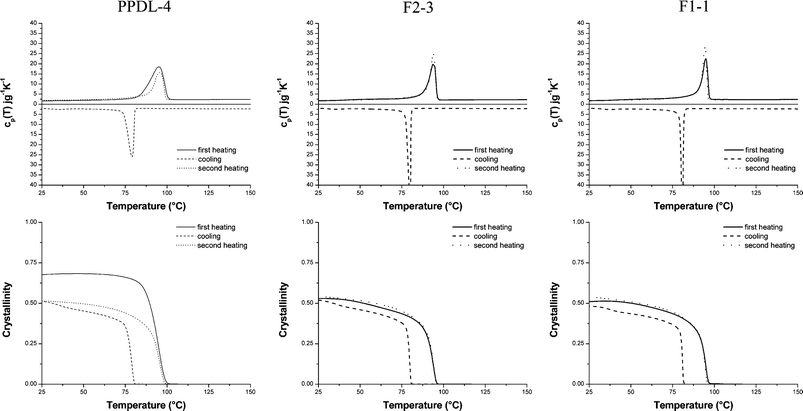 | ||
| Fig. 8 Evolution of cp(T) and wc(T) during a heating–cooling–heating run at 10 °C min−1 for three representative samples (fibre codes refer to Table 1). | ||
| Entry | T m1 /°C | T c /°C | T m2 /°C | w c1 (25 °C) | w c2 (25 °C) | f |
|---|---|---|---|---|---|---|
| a DSC melting peak first heating cycle. b DSC crystallization peak. c DSC melting peak peak second heating cycle. d Crystallinity fraction prior to first heating cycle. e Crystallinity fraction prior to second heating cycle. f Crystal orientation factor. | ||||||
| PPDL-4 | 95 | 79 | 96 | 0.68 | 0.51 | 0 |
| F2-1 | 96 | 79 | 95 | 0.56 | 0.54 | 0.12 |
| F2-2 | 95 | 80 | 95 | 0.52 | 0.54 | 0.39 |
| F2-3 | 94 | 80 | 94 | 0.53 | 0.54 | 0.26 |
| F2-4 | 94 | 80 | 93 | 0.54 | 0.55 | 0.85 |
| PPDL-3 | 96 | 80 | 96 | 0.62 | 0.53 | 0 |
| F1-1 | 95 | 81 | 95 | 0.53 | 0.52 | 0.48 |
PPDL crystal molecular orientation
In Fig. 9, the 2D scattering patterns of three representative fiber samples are illustrated. The anisotropy in the scattering pattern is most obvious in the pattern of sample F2-4, where the 110 arcs are in an equatorial position with respect to the fiber axis. The other features in this particular scattering pattern are rather weak because the fiber was extremely thin. The arcs of the (small angle) 001 reflection are better visible in the other scattering patterns and are positioned meridionally, as expected. | ||
| Fig. 9 Corrected 2D scattering pattern of three representative fiber samples using the same logarithmic gray scale as in Fig. 2. The fiber axis lays approximately parallel to the beam stop axis. | ||
Fig. 10 displays the fits through the corrected 110 reflections as a function of the azimuthal angle ϕ, which is defined as zero in the direction of the fiber axis. The integral of the displayed intensity is normalized so as to have an enclosed area in the ϕ range between 0 and 180° equal to 1 for easy comparison. The corresponding values for the crystal orientation function f (eqn (5)) are listed in Table 4. A zero value is obtained when the crystals are oriented randomly with respect to the fiber direction and a value of 1 corresponds to a perfect alignment with respect to the fiber axis. The highest degree of orientation was reached for the sample that experienced the highest winding speed (Table 1). Although the crystals and hence the molecules in the crystals are partially oriented with respect to the fiber axis, it seems that the majority of the amorphous material in between the crystallites is randomly oriented. Azimuthal scans through 2θ values where only amorphous material contributes to the scattering pattern (see experimental section) do not reveal any anisotropy.
 | ||
| Fig. 10 Fits through the azimuthal intensity distribution of the 110 reflections of the different investigated samples. | ||
PPDL fiber mechanical properties
Unfortunately it was not possible to monitor the tensile properties for the entire range of elongated fibers reported in Table 4. Since some fibers proved to be too thin to be measured, preliminary tensile test were conducted on selected PPDL fibers, i.e. F1-1, F2-2 and F2-1. As the crystallinity and the melting points of all fibers are identical, only the degree of crystal (and thus molecular) orientation can explain the differences in mechanical behavior. Fig. 11 clearly shows that there is a significant increase of the fiber strength with the degree of crystal orientation. A maximum value of 0.74 GPa is reached for F1-1 with an orientation factor of 0.48. Furthermore, the strength of the fibers is significantly higher than that of the isotropic films, which confirms that the elongation and orientation of the PPDL crystals and thus the molecules in the fiber during and after the spinning process is a crucial factor. On the other hand, a decrease of the elongation at break with increasing degree of orientation is evident due to the fact that a maximum orientation in these fibers has been reached during processing, not or hardly allowing for any further elongation in a tensile experiment.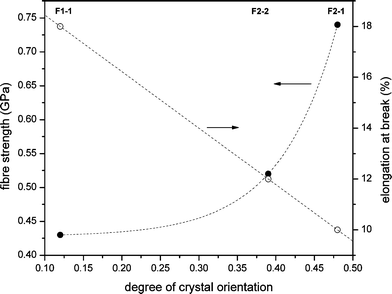 | ||
| Fig. 11 Fiber tensile strength and elongation at break of three PPDL fibers as a function of their degree of crystal orientation. Dashed lines are added to guide the eye. | ||
Conclusions
We have synthesized PPDL with the highest molecular weight reported to date. Optimization of the SEC analysis revealed that reliable molecular weight data could only be obtained by high temperature SEC similar to polyethylene. By scaling-up the polymerization larger amounts of PPDL were produced. Despite preliminary optimization of the reaction conditions the molecular weights obtained in the scaled-up process were lower than those obtained on smaller scale. This was clearly due to mass transport limitations caused by the high viscosity of the reaction medium. Further optimization and the use of specialized equipment are expected to circumvent these issues.Mechanical and thermal properties of the non-oriented, high molecular weight PPDL were determined and are largely in agreement with literature data. The high molecular weight PPDL was melt-processed into fibers, which were further elongated. Analysis of the fibers revealed differences in crystal orientation as a function of the processing conditions. Preliminary fiber tensile measurements confirm a high strength of up to 0.74 GPa for the fiber with the highest crystal orientation.
Acknowledgements
This work forms part of the research program of the Dutch Polymer Institute, project no. 381. A.H. thanks the Science Foundation Ireland (SFI) for financial support.References
- J. P. Jain, M. Sokolski, N. Kumar and A. J. Domb, Polym. Rev., 2008, 48, 156 Search PubMed.
- J. Panten, H. Surburg and B. Hölscher, Chem. Biodiversity, 2008, 5, 1011 CrossRef CAS.
- B. Lebedev and A. Yevstropov, Makromol. Chem., 1984, 185, 1235 CrossRef CAS.
- M. L. Focarete, M. Scandola, A. Kumar and R. A. Gross, J. Polym. Sci., Part B: Polym. Phys., 2001, 39, 1721 CrossRef CAS.
- P. Skoglund and A. Fransson, Polymer, 1998, 39, 3143 CrossRef CAS.
- P. Skoglund and A. Fransson, Polymer, 1998, 39, 1899 CrossRef CAS.
- M. Gazzano, V. Malta, M. L. Focarete, M. Scandola and R. A. Gross, J. Polym. Sci., Part B: Polym. Phys., 2003, 41, 1009 CrossRef CAS.
- J. Cai, B. S. Hsiao and R. A. Gross, Polym. Int., 2009, 58, 944 CrossRef CAS.
- N. Simpson, M. Takwa, K. Hult, M. Johansson, M. Martinelle and E. Malmstrom, Macromolecules, 2008, 41, 3613 CrossRef CAS.
- I. van der Meulen, M. de Geus, H. Antheunis, R. Deumens, E. A. J. Joosten, C. E. Koning and A. Heise, Biomacromolecules, 2008, 9, 3404 CrossRef CAS.
- L. Van Der Mee, F. Helmich, R. de Bruijn, J. A. J. M. Vekemans, A. R. A. Palmans and E. W. Meijer, Macromolecules, 2006, 39, 5021 CrossRef CAS.
- B. Lebedev and A. Yevstropov, Makromol. Chem., 1984, 185, 1235 CrossRef CAS.
- A. Duda, A. Kowalski, S. Penczek, H. Uyama and S. Kobayashi, Macromolecules, 2002, 35, 4266 CrossRef CAS.
- Z. Zhong, P. J. Dijkstra and J. Feijen, Macromol. Chem. Phys., 2000, 201, 1329 CrossRef CAS.
- A. Kumar, B. Kalra, A. Dekhterman and R. A. Gross, Macromolecules, 2000, 33, 6303 CrossRef CAS.
- A. Taden, M. Antonietti and K. Landfester, Macromol. Rapid Commun., 2003, 24, 512 CrossRef CAS.
- L. Van Der Mee, J. Antens, B. Van De Kruijs, A. R. A. Palmans and E. W. Meijer, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 2166 CrossRef CAS.
- M. De Geus, J. Peeters, M. Wolffs, T. Hermans, A. R. A. Palmans, C. E. Koning and A. Heise, Macromolecules, 2005, 38, 4220 CrossRef CAS.
- http://athas.prz.rzeszow.pl/ .
- Calorimetry and Thermal Analysis of Polymers, ed. V. B. F. Mathot, Hanser Publishers, New York, 1994, ch. 5 Search PubMed.
- L. E. Alexander: X-Ray Diffraction Methods in Polymer Science, Wiley Interscience, New York, 1969 Search PubMed.
- Z. W. Wilchinsky, Adv. X-Ray Anal., 1963, 6, 231 CAS.
- C. J. Gommes, B. Goderis, J. Appl. Crystallogr., submitted Search PubMed.
- M. de Geus, L. Schormans, A. R. A. Palmans, C. E. Koning and A. Heise, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 4290 CrossRef CAS.
- H. Scott Fogler, Elements of Chemical Reaction Engineering, Prentice Hall PTR, New Jersey, 3 edn, 1999, p 967 Search PubMed.
- B. J. Kline, S. S. Lele, E. J. Beckman and A. J. Russell, AIChE J., 2001, 47, 489 CrossRef CAS.
- S. V. Kamat, E. J. Beckman and A. J. Russell, Enzyme Microb. Technol., 1992, 14, 265 CrossRef CAS.
- Y. Mei, L. Miller, W. Gao and R. A. Gross, Biomacromolecules, 2003, 4, 70 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |