Towards chiral polystyrene based materials: controlled polymerization of p-(2,2′-diphenylethyl)styrene
Christiane
Hohberger
,
Klaus
Beckerle
and
Jun
Okuda
*
, Landoltweg 1, D-52074, Aachen, Germany. E-mail: Jun.okuda@ac.rwth-aachen.de; Fax: +0049-2418092644
First published on 21st January 2010
Abstract
p-(2,2′-Diphenylethyl)styrene (DPES) was polymerized in an atactic, syndiotactic, and isospecific fashion. Atactic polymerization was initiated by 2,2′-azobis(2-methylpropionitrile) (AIBN). Syndiotactic polymer was obtained using the catalyst system TiCp*Cl3/MAO. Isospecific polymerization was performed with the homochiral postmetallocene catalystdichloro{trans-1,2-dithiocyclohexanediyl-2,2′-bis(4,6-di-tert-butylphenolato)}titanium/MAO. Optically active isotactic polymers were obtained by a controlled reduction of the molecular weight, employing two different chain transfer methodologies. In addition to 1-hexene as a chain transfer agent (CTA), the use of diethylzinc as a CTA for DPES oligomerization was introduced. Polymers with molecular weights below Mn of 50,000 g mol−1 showed specific rotation values ([α]23D) between ±0.2 and 2.2.
Introduction
Optically active polymeric materials consisting exclusively of hydrocarbons are not easily accessible.1 Their inertness should allow for their use as auxiliaries in asymmetric reactions,2 providing chiral reaction space, or as stationary phases in chiral chromatography.3Polystyrene is a widely accessible polymer with many potential applications. Isotactic polystyrene crystallizes in a helix and therefore exhibits a chiral secondary structure. However it suffers from a long induction period for crystallization.4 The recent introduction of structurally well-defined isospecific catalysts for styrene5,6 now allows a better control of the chain length and endgroups.7,8Due to the phenomenon of cryptochirality, high molecular weight homochiral isotactic poly(α-olefins) showing optical activity are not accessible.9 It is therefore important to find suitable chain transfer agents to achieve a controlled reduction of molecular weight. We have recently shown that it is possible to reduce the molecular weight of isotactic polystyrenes in a controlled fashion by using 1-hexene as a chain transfer agent during catalyticpolymerization, resulting in optically active materials.7 An alternative method for a controlled reduction of the molecular weight of polymers involves diethylzinc as a chain transfer agent. In contrast to oligoethylenes10 and oligopropylenes11 there have so far only been few reports on molecular weight control of polystyrenes using diethylzinc.12 Compared to 1-hexene as chain transfer agent,7,8diethylzinc offers some advantage. Oligomer chain ends can be directly functionalized by quenching with various electrophiles11 to give terminally functionalized polystyrene derivatives.
To influence the crystallization behavior of polystyrene, we synthesized a styrene derivative which contains a 2,2′-diphenylethylgroup in para position of the phenyl ring. When polymerized in an isospecific fashion, the additional phenyl groups could favor the formation of a stable helical structure due to π–π stacking of the substituents. By employing a homochiral catalyst, the helical structure should result in optical activity.
Results and discussion
Synthesis and polymerization of p-(2,2′-diphenylethyl)styrene
The new functionalized styrene monomer was prepared by a nucleophilic substitution of the chlorine atom in p-vinylbenzylchloride (pVBC). After the lithiation of diphenylmethane (DPM) with n-butyllithium (LinBu), pVBC was slowly added to the reaction mixture and p-(2,2′-diphenylethyl)styrene (DPES) was isolated in good yields as colorless needles (Scheme 1).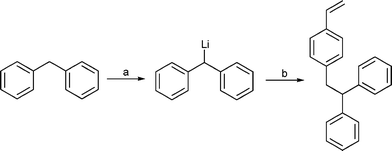 | ||
| Scheme 1 Synthesis of DPES. Reagents and conditions: (a) LinBu, toluene, thf, −70 to +70 °C; (b) pVBC, −70 to +25 °C. | ||
Due to the presence of additional aromatic groups DPES shows fluorescence emission both in solution (0.1 mM, CH2Cl2) and in the solid state. These do not differ from each other. After excitation at 277 nm the monomer exhibits in addition to the typical excimer fluorescence of styrene13 (313 nm) the fluorescence of the diphenylmethyl fragment14 (625 nm) (see Fig. 1).
 | ||
Fig. 1
Fluorescence emission spectra of p-(2,2′-diphenylethyl)styrene (![[dash dash, graph caption]](https://www.rsc.org/images/entities/char_e091.gif) ) and the corresponding isotactic polymer ( ) and the corresponding isotactic polymer (![[thick line, graph caption]](https://www.rsc.org/images/entities/char_e117.gif) ), (both in 0.1 mM CH2Cl2). ), (both in 0.1 mM CH2Cl2). | ||
This combination leads to a material with a fluorescence gap between 400 and 600 nm which may be interesting for optical applications.
DPES was polymerized in atactic, syndiotactic, and isotactic fashion. The tacticity of the resulting polymers is analyzed by 13C NMR spectroscopy (see Fig. 2). The ipsocarbon atom of the aromatic ring exhibits a characteristic chemical shift for the three possible stereoisomers. Table 1 summarizes the polymerization results including the chemical shifts of the ipsocarbon atom, Tg, Tm and Tdec. Comparison of the chemical shift of the ipsocarbon atoms reveals high tacticity of the isotactic polymer, with no detectable stereoerrors.
| Tacticity | δipsoa | T g | T m | T dec |
|---|---|---|---|---|
| a Chemical shift of the ipsocarbon atom in ppm. b In °C. c Initiated by AIBN, T = 60 °C, t = 2 h. d Catalyst system: [TiCp*Cl3]/MAO, T = 60 °C, t = 3 h. e Catalyst system: rac-2/MAO, T = 40 °C, t = 3 h. | ||||
| Atacticc | 141.20–144.08 | 89 | — | > 350 |
| Syndiotacticd | 142.18 | 99 | — | > 350 |
| Isotactice | 144.10 | 86 | — | > 350 |
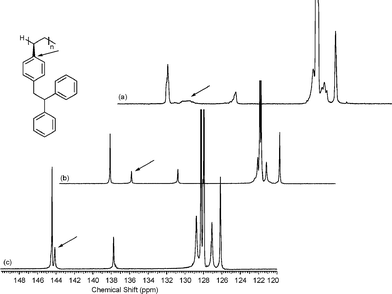 | ||
| Fig. 2 13C NMR spectra of atactic (a), syndiotactic (b), isotactic poly(DPES) (c) The arrow indicates the ipsocarbon atom and the related signal. | ||
The isospecific polymerization of DPES was carried out in toluene at 40 °C using the previously described precatalyst dichloro{trans-1,2-dithiocyclohexanediyl-2,2′-bis(4,6-di-tert-butylphenolato)}titanium rac-2 (Fig. 3, Scheme 2) activated by MAO.6 After 3 h a highly isotactic polymer with a number average molecular weight (Mn) of 120,000 g mol−1 and a narrow molecular weight distribution (Mw/Mn = 1.6) was obtained. The polymer was isolated as a colorless solid, soluble in common organic solvents, including tetrahydrofuran, toluene, chloroform, and dichloroethane. The polymer shows good thermal stability and decomposes only above 350 °C. It exhibits the same fluorescence properties and emission wavelength as the monomer (Fig. 1).
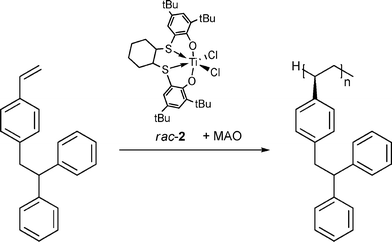 | ||
| Scheme 2 Isospecific polymerization of DPES. | ||
 | ||
| Fig. 3 Racemic and chiral isospecific bis(phenolato) titaniumcatalysts containing an (OSSO)-type ligand. | ||
Polymerizations using the enantiopure catalyst precursors 2a and 2b (Fig. 3) led to the homochiral isotactic polymers, which do not exhibit optical activity. This is due to the phenomenon of cryptochirality, which renders high molecular weight polymers of prochiral α-olefin units optically inactive.
Oligomerization using 1-hexene
Previous work has shown that it is possible to control the molecular weight of polystyrenes during polymerization reactions by using 1-hexene as a chain transfer agent.7,8 This methodology has been adopted here for the case of DPES. A series of oligo(DPES)s with different molecular weight distributions were synthesized by variation of the 1-hexene to DPES ratio. Increasing the 1-hexene/DPES ratio to a value of 2 decreased Mn from 120,000 g mol−1 to 2300 g mol−1, corresponding to 8 monomer units. The oligomer obtained consists of an isotactic oligo(DPES) unit, which is terminated by a regio- and stereoirregular oligo(1-hexene) tail (one to five 1-hexene units). This irregular oligo(1-hexene) tail does not exhibit any optical activity.15Chiral oligo(DPES)s were obtained, when the enantiomerically pure resolved precatalysts 2a or 2b were employed in the oligomerization reactions. Oligomerizations conducted with the (–)-(Λ,R,R)-2a precatalyst led to an oligomer with a negative sign in optical rotation. Table 2 summarizes the oligomerization results including the 1-olefin ratios, yields, Mn and optical rotation values.
As depicted in Table 2 and in agreement with previous results, the (–)-(Λ,R,R)-titanium precatalyst 2a produces only oligomers with the same sign of optical rotation, corroborating that enantiomorphic site control is operative. Increasing the amount of 1-hexene leads to a slight decrease in the polymer yield. This is most probably due to a combination of several effects: Augmentation of the 1-hexene concentration leads to a decreased local DPES concentration which evokes a slower coordination/insertion mechanism of DPES and therefore leads to a decelerated polymerization kinetic. Additionally due to the increased 1-hexene concentration oligo(1-hexene)s are produced as byproduct.
Oligomerization using diethylzinc
In addition to the oligomerization reactions using 1-hexene as transfer agent, the control of the molecular weight with diethylzinc was of great interest (Scheme 3). In comparison to the chain transfer with α-olefins, the use of diethylzinc allows further functionalization of the obtained oligomers by post-polymerization reactions. These are easily accessible, since zinc capped polymer chains are obtained, which can undergo further reactions with electrophiles. | ||
| Scheme 3 Synthesis of isotactic DPES oligomer. | ||
The number average molecular weight of poly(DPES) could be controlled by a variation of the ratio of diethylzinc to [Ti]-precatalyst 2 (Table 3). The reduction of the monomer units in the polymer from 200 to 20 could be achieved in a controlled fashion by increasing the ratio of diethylzinc to the [Ti]-precatalyst 2 from ∼150 to ∼2500.
| Runa | ZnEt2/ [Ti] | Cat.b | Yieldc | M n | M w/Mnd | [α]23De | [Φ]23D |
|---|---|---|---|---|---|---|---|
| a Reaction time for run 1–8: 24 h, for run 9–13: 40 h. b Activated by 1400 eq. MAO. c In (%). d Determined by GPC using polystyrene standard.16 e c = 0.06 g mL−1 CH2Cl2. | |||||||
| 0 | — | rac-2 | 62 | 120,000 | 1.37 | — | — |
| 1 | 143 | 2a | 61 | 58,140 | 1.79 | 0.00 | 0.00 |
| 2 | 215 | 2a | 60 | 49,910 | 1.53 | +0.20 | +17 |
| 3 | 287 | 2a | 55 | 45,380 | 1.76 | +0.16 | +13 |
| 4 | 520 | 2a | 39 | 28,830 | 1.70 | +0.34 | +17 |
| 5 | 520 | 2b | 23 | 29,310 | 1.55 | –0.49 | –26 |
| 6 | 780 | 2a | 19 | 15,560 | 1.70 | +0.79 | +22 |
| 7 | 1040 | 2a | 14 | 11,150 | 1.61 | +1.30 | +25 |
| 8 | 1040 | 2b | 17 | 15,540 | 1.53 | –1.27 | –34 |
| 9 | 1443 | 2a | 14 | 8380 | 1.60 | +1.22 | +18 |
| 10 | 1793 | 2a | 29 | 8950 | 1.68 | +1.64 | +26 |
| 11 | 2160 | 2a | 16 | 5770 | 1.91 | +1.67 | +17 |
| 12 | 2160 | 2b | 7 | 4030 | 2.20 | –2.27 | –16 |
| 13 | 2519 | 2a | 13 | 5790 | 1.65 | +2.17 | +22 |
Increase of the diethylzinc to [Ti]-precatalyst ratio led to a decrease in the yield. This was probably due to a coordination of the zinc alkyl to the free coordination site at the titanium center, inhibiting DPES coordination and insertion into the growing polymer chain. Increasing the reaction time did not dramatically improve the yield. To prove the action of diethylzinc as chain transfer agent, the catalyst efficiency in these oligomerization reactions was calculated (catalyst efficiency is defined here as the percentage of active initiators, calculated from the observed number of polymer chains relative to the theoretical ones). Without the addition of a chain transfer agent the catalyst efficiency is 2, i.e. each catalyst center produced two polymer chains. By employing diethylzinc in the oligomerization reaction an increase of the catalyst efficiency can be observed. At higher concentrations of the chain transfer agent a catalyst efficiency of ∼10 could be achieved (Fig. 4).
 | ||
| Fig. 4 Catalyst efficiency of 2 as a function of the diethylzinc to titanium precatalyst ratio (catalyst efficiency = MWtheor./MWobs., MWtheor. = MWmonomer conversion (nmonomer/ncatalyst)). | ||
To obtain optically active materials a series of oligomers was synthesized using both enantiomers of the precatalyst, 2a and 2b, in toluene. The dependence of the specific optical rotation values on the number average molecular weight is depicted in Fig. 5. It is evident that above Mn = 50,000 (Table 3, Run 2) no optical activity could be detected, but below this threshold specific rotation values [α]23D of ±0.2 to ±2.2 could be measured reproducibly.
![Dependence of the specific rotation [α]23D (▲) and molar rotation [Φ]D23 (○) as a function of the number average molecular weight of isotactic poly(DPES) terminated by diethylzinc.](/image/article/2010/PY/b9py00286c/b9py00286c-f5.gif) | ||
| Fig. 5 Dependence of the specific rotation [α]23D (▲) and molar rotation [Φ]D23 (○) as a function of the number average molecular weight of isotactic poly(DPES) terminated by diethylzinc. | ||
In contrast to the chain transfer using 1-hexene, each enantiomeric titanium catalyst produced exclusively oligomers with the opposite sign in optical rotation (Fig. 5, Table 3). Oligomerizations of styrene with diethylzinc led to the same results.
The values of optical rotation obtained for oligo(DPES)s are in the same range as obtained for the chiral styrene oligomers.7 This indicates that helix formation does not occur in solution, even though the p-(2,2′-diphenylethyl) substituents would suggest some sort of intra- or inter-chain interaction.17
Experimental section
Materials
All operations were performed under an inert atmosphere of argon using standard Schlenk-line or glovebox techniques. Diethyl ether, thf, and toluene were distilled from sodium benzophenone ketyl. Alternatively, the above solvents were dried using an MBraun SPS-800solventpurification system. Methylaluminoxane (MAO, 10 wt-% solution in toluene) and LinBu (2.5 M in hexane) were purchased from Aldrich and used as received. Diphenylmethane and p-vinylbenzylchloride were treated with CaH2 for 12 h and distilled under reduced pressure before use. The bis(phenolato) precatalysts 2 were synthesized according to the procedures reported in the literature.5Instrumentation
NMR spectra were recorded at room temperature on a Bruker DRX 400 spectrometer (1H, 400.1 MHz; 13C, 100.6 MHz). Chemical shifts for 1H and 13C NMR spectra are referenced internally using the residual solvent resonances. Elemental analyses were performed by the Microanalytical Laboratory of this department. Molecular weights of the oligomers were measured using gel permeation chromatography (GPC) at 30 °C in thf and calibrated with respect to polystyrene standards. Specific rotation was measured using a Perkin Elmer 241 Digital Polarimeter. Standard solutions prepared in 2 mL of appropriate solvent as indicated below were used for measuring the specific rotations (path length 10 cm, volume 1 mL, λ = 589.3 nm at 23 °C).Synthesis of p-(2,2′-diphenylethyl)styrene
To a solution of 10 mL of thf and 100 mL of toluene were added 5.9 g diphenylmethane (35 mmol). After cooling to −80 °C, 14 mL of LinBu (2.5 M in hexane, 35 mmol) were slowly added. The reaction mixture was kept at this temperature for 10 min. After warming to room temperature the solution was refluxed for 1 h, cooled to −70 °C and 5.5 mL of p-vinylbenzylchloride (35 mmol) were added. The solution was allowed to warm to room temperature and was kept stirring for additional 2 h. Then 50 mL of brine were added and the reaction mixture was extracted with diethylether and water. The organic phases were collected, dried over Na2SO4 and the solvent was evaporated in vacuo. The resulting yellowish solid was recrystallized twice in methanol (–30 °C) to give colorless needles (5.26 g, 18.5 mmol, 52%). 1H NMR (400 MHz, CDCl3) δ = 3.07 (d, 2 H, –CH2–, 3JHH = 7.57 Hz), 3.92 (t, 1 H, 3JHH = 7.81 Hz), 4.87 (d, 1 H, vinyl, 3JHH = 10.74 Hz), 5.35 (d, 1 H, vinyl, 2JHH = 17.58 Hz), 6.30 (q, 1 H, vinyl, 3JHH = 10.99 Hz), 6.67 (d, 2 H, arom, 3JHH = 8.06 Hz), 6.85–7.02 ppm (m, 12 H, arom). 13C NMR (400 MHz, CDCl3) δ = 41.85, 53.01, 112.89, 125.88, 126.12, 127.95, 128.27, 129.13, 135.13, 136.57, 139.92, 144.28 ppm. Anal. Calcd for C22H20: C, 92.91; H, 7.09. Found: C, 92.43; H, 7.08.Isotactic polymerization
A Schlenk tube was charged with toluene (calculated for a total volume of 7 mL), 1.2 mL of a MAO solution in toluene (10 wt%) and 0.51 g of p-(2,2′-diphenylethyl)styrene (1.8 mmol). This mixture was stirred for 20 min. at 40 °C, followed by the addition of 0.5 mL of a 0.25 μM stock solution of the titanium complex rac-2 (17 mg (25 μmol) in 10 mL of toluene). The reaction mixture was stirred at 40 °C for 3 h, quenched by the addition of 0.5 mL of isopropanol and the polymer was precipitated from 100 mL of acidified methanol. The polymer was dissolved in chloroform and precipitated again from acidified methanol. After repeating this procedure twice the colorless oligomer was dried in vacuo overnight. Mn = 120,000, Mw/Mn = 1.37.Atactic polymerization
A Schlenk tube was charged with 0.5 g of p-(2,2′-diphenylethyl)styrene (1.8 mmol) and 10 mL toluene. After addition of 20 mg 2,2′-azobis(2-methylpropionitrile) (AIBN, 0.12 mmol) the reaction mixture was heated at 60 °C for 2 h, then cooled to room temperature and the polymer was precipitated from 100 mL of acidified methanol. The polymer was dissolved in chloroform and precipitated again from acidified methanol. After repeating this procedure twice, the polymer was dried in vacuo overnight. Mn = 6590, Mw/Mn = 1.79.Syndiotactic polymerization
A Schlenk tube was charged with toluene (calculated for a total volume of 7 mL), 1.2 mL of a MAO solution in toluene (10 wt%) and 0.51 g of p-(2,2′-diphenylethyl)styrene (1.8 mmol). This mixture was stirred for 20 min. at 60 °C, followed by the addition of 1 mL of a 3.8 μM stock solution of [TiCp*Cl3] (5 mg (19 μmol) in 5 mL of toluene). The reaction mixture was stirred at 60 °C for 3 h, quenched by addition of 0.5 mL of isopropanol and the polymer was precipitated from 100 mL of acidified methanol. The product was dissolved in chloroform and precipitated again from acidified methanol. After repeating this procedure twice, the polymer was dried in vacuo overnight. Mn = 40,547, Mw/Mn = 1.31.Oligomerization using ZnEt2
A Schlenk tube was charged with toluene (calculated for a total volume of 15 mL) and 0.5 g of p-(2,2′-diphenylethyl)styrene (1.8 mmol). Afterwards 1.2 mL of a MAO solution (10 wt%) and the appropriate amount of diethylzinc were added. After 20 min. at 40 °C the reaction was activated by adding 0.5 mL of a catalyst solution (2, 18 mg in 10 mL toluene, 1.33 μmol). The resulting reaction mixture was kept stirring at 40 °C and was quenched after the appropriate time by adding 0.5 mL of isopropanol. The obtained solution was poured into acidified methanol in order to precipitate the oligomer. The resulting colorless solid was dissolved in CHCl3 and precipitated in acidified methanol. The oligomer was redissolved in CHCl3, filtered through silica and precipitated into acidified methanol. After filtration and washing with methanol, it was dried overnight in vacuo.Oligomerization using 1-hexene
A Schlenk tube was charged with toluene (calculated for a total volume of 15 mL) and 0.5 g of p-(2,2′-diphenylethyl)styrene (1.8 mmol) and the appropriate amount of 1-hexene. After stirring for 5 min 1.2 mL of a MAO solution (10 wt%) were added. After 20 min. at 40 °C the reaction was activated by adding 0.5 mL of a catalyst solution (2, 18 mg in 10 mL toluene, 1.33 μmol). The resulting reaction mixture was kept stirring at 40 °C and was quenched after the appropriate time by adding 0.5 mL of isopropanol. The obtained solution was poured into acidified methanol in order to precipitate the oligomer. The resulting colorless solid was dissolved in CHCl3 and precipitated in acidified methanol. The oligomer was redissolved in CHCl3, filtered through silica and precipitated into acidified methanol. After filtration and washing with methanol, it was dried overnight in vacuo.Conclusion
We report here the synthesis of a new optically active polystyrene derivative, isotactic poly(p-(2,2′-diphenylethyl)styrene). The isotactic polymerization using a homochiral catalyst led to a chiral polymer which did not exhibit optical activity. The synthesis of an optically active polymer could be achieved by a controlled reduction of the molecular weight during isospecific polymerization using two different chain transfer agents. Oligomerizations conducted with 1-hexene led to oligomers which showed the same sign in optical activity as the employed homochiral catalyst. Additionally we could show that diethylzinc is an effective chain transfer agent for DPES. It is possible to reduce the molecular weight of the polymer by increasing the diethylzinc to [Ti]-precatalyst ratio. Oligomers consisting of only 20 units were obtained. The oligomers showed the opposite sign of optical rotation to the homochiral (OSSO)-type catalyst employed. This allows the synthesis of a chiral material with a desired sign and degree of optical rotation.Notes and references
- (a) L. Guadagno, M. Raimondo, C. Silvestre, I. Immediata, P. Rizzo and G. Guerra, J. Mater. Chem., 2008, 18, 567 RSC; (b) A. M. Buono, I. Immediata, P. Rizzo and G. Guerra, J. Am. Chem. Soc., 2007, 129, 10992 CrossRef CAS; (c) A. L. Segre, M. Delfini, M. Paci, A. M. Raspolligalletti and R. Solaro, Macromolecules, 1985, 18, 44 CrossRef CAS; (d) E. Chiellini, A. M. Raspolligalletti and R. Solaro, Macromolecules, 1984, 17, 2212 CrossRef CAS; (e) C. Carlini and E. Chiellini, Makromol. Chem., Macromol. Chem. Phys., 1975, 176, 519 Search PubMed.
- (a) T. Kawasaki, C. Hohberger, Y. Araki, K. Hatase, K. Beckerle, J. Okuda and K. Soai, Chem. Commun., 2009, 5621 RSC; (b) C. Girard and H. B. Kagan, Angew. Chem., Int. Ed., 1998, 37, 2923 CrossRef CAS.
- (a) Y. Okamoto and T. Ikai, Chem. Soc. Rev., 2008, 37, 2593 RSC; (b) T. E. Hopkins and K. B. Wagener, Adv. Mater., 2002, 14, 1703 CrossRef CAS.
- (a) A. Mamun, S. Umemoto, N. Okui and N. Ishihara, Macromolecules, 2007, 40, 6296 CrossRef CAS; (b) F. Auriemma, C. De Rosa and P. Corradini, Macromol. Chem. Phys., 2004, 205, 390 CrossRef CAS; (c) J. Zhang, Y. Duan, D. Shen, S. Yan, I. Noda and Y. Ozaki, Macromolecules, 2004, 37, 3292 CrossRef CAS; (d) G. Xue, J. Zhang, J. L. Chen, Y. Q. Li, J. B. Ma, G. C. Wang and P. C. Sun, Macromolecules, 2000, 33, 2299 CrossRef CAS; (e) J. Brandrup and E. H. Immergut, Polymer Handbook, John Wiley & Sons, 3rd edn, 1995 Search PubMed; (f) M. Tsuji, S. K. Roy and R. S. J. Manley, Polymer, 1984, 25, 1573 CrossRef CAS; (g) P. Blah and R. S. J. Manley, J. Polym. Sci., Part A-2, 1966, 4, 1022 CrossRef.
- (a) J. Li, M. Li, S. Li, L. Shi, C. Ren, D. Cui, Y. Wang and T. Tang, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 1240 CrossRef CAS; (b) K. Michiue, M. Onda, H. Tanaka, H. Makio, M. Mitani and T. Fujita, Macromolecules, 2008, 41, 6289 CrossRef CAS; (c) A. S. Rodrigues, E. Kirillov and J. F. Carpentier, Coord. Chem. Rev., 2008, 252, 2115 CrossRef CAS; (d) R. J. Kern, H. G. Hurst and W. J. Richard, J. Polym. Sci., 1960, 45, 195 CrossRef CAS; (e) C. Overberger and H. Mark, J. Polym. Sci., 1959, 35, 381 CrossRef CAS; (f) G. Natta and P. Corradini, Makromol. Chem., 1955, 16, 77 CrossRef CAS; (g) G. Natta, P. Pino, P. Corradini, F. Danusso, E. Mantica, G. Mazzanti and G. Moraglio, J. Am. Chem. Soc., 1955, 77, 1708 CrossRef CAS; (h) Isopropylidene-bridged ansa-zirconocenecatalysts were reported to produce isotactic polystyrene: T. Arai, S. Suzuki, and T. Ohtsu, Olefin Polymerization, ACS Symposium Series, American Chemical Society, Washington, DC, 2000, 749, 66 Search PubMed; (i) T. Arai, T. Ohtsu and S. Suzuki, Polym. Prepr., 1998, 39, 220 CAS; (j) For anionic polymerization to give isotactic polystyrene, see: T. Makino and T. E. Hogen-Esch, Macromolecules, 1999, 32, 5712 Search PubMed; (k) L. Cazzaniga and R. E. Cohen, Macromolecules, 1989, 22, 4125 CrossRef CAS.
- (a) K. Beckerle, R. Manivannan, T. P. Spaniol and J. Okuda, Organometallics, 2006, 25, 3019 CrossRef CAS; (b) C. Capacchione, R. Manivannan, M. Barone, K. Beckerle, R. Centore, L. Oliva, A. Proto, A. Tuzi, T. P. Spaniol and J. Okuda, Organometallics, 2005, 24, 2971 CrossRef CAS; (c) K. Beckerle, C. Capacchione, H. Ebeling, R. Manivannan, R. Mülhaupt, A. Proto, T. P. Spaniol and J. Okuda, J. Organomet. Chem., 2004, 689, 4636 CrossRef CAS; (d) C. Capacchione, A. Proto, H. Ebeling, R. Mülhaupt, K. Möller, T. P. Spaniol and J. Okuda, J. Am. Chem. Soc., 2003, 125, 4964 CrossRef CAS.
- (a) G. J. M. Meppelder, K. Beckerle, R. Manivannan, B. Lian, G. Raabe, T. P. Spaniol and J. Okuda, Chem.–Asian J., 2008, 3, 1312 CrossRef CAS; (b) K. Beckerle, R. Manivannan, B. Lian, G. J. M. Meppelder, G. Raabe, T. P. Spaniol, H. Ebeling, F. Pelascini, R. Mülhaupt and J. Okuda, Angew. Chem., Int. Ed., 2007, 46, 4790 CrossRef CAS.
- B. T. Gall, F. Pelascini, H. Ebeling, K. Beckerle, J. Okuda and R. Mülhaupt, Macromolecules, 2008, 41, 1627 CrossRef CAS.
- (a) P. Pino, M. Galimberti, P. Prada and G. Consiglio, Makromol. Chem., Macromol. Chem. Phys., 1990, 191, 1677 Search PubMed; (b) P. Pino, P. Cioni and J. Wei, J. Am. Chem. Soc., 1987, 109, 6189 CrossRef CAS.
- (a) P. D. Hustad, R. L. Kuhlman, E. M. Carnahan, T. T. Wenzel and D. J. Arriola, Macromolecules, 2008, 41, 4081 CrossRef CAS; (b) J. O. Ring, R. Thomann, R. Mülhaupt, J. M. Raquez, P. Degee and P. Dubois, Macromol. Chem. Phys., 2007, 208, 896 CrossRef CAS; (c) M. van Meurs, G. J. P. Britovsek, V. C. Gibson and S. A. Cohen, J. Am. Chem. Soc., 2005, 127, 9913 CrossRef CAS; (d) G. J. P. Britovsek, S. A. Cohen, V. C. Gibson and M. van Meurs, J. Am. Chem. Soc., 2004, 126, 10701 CrossRef CAS; (e) M. Mitani, J. Mohri, R. Furuyama, S. Ishii and T. Fujita, Chem. Lett., 2003, 32, 238 CrossRef CAS; (f) E. Agouri, C. Parlant, P. Mornet, J. Rideau and J. F. Teitgen, Makromol. Chem., 1970, 137, 229 CrossRef.
- (a) W. Zhang and L. R. Sita, J. Am. Chem. Soc., 2008, 130, 442 CrossRef CAS; (b) T. Shiono, H. Kurosawa and K. Soga, Macromolecules, 1995, 28, 437 CrossRef CAS; (c) H. Kurosawa, T. Shiono and K. Soga, Macromol. Chem. Phys., 1994, 195, 3303 CrossRef CAS; (d) H. Kurosawa, T. Shiono and K. Soga, Macromol. Chem. Phys., 1994, 195, 1381 CrossRef CAS; (e) T. Shiono, H. Kurosawa and K. Soga, Makromol. Chem., Macromol. Chem. Phys., 1992, 193, 2751 Search PubMed; (f) T. Shiono, K. Yoshida and K. Soga, Makromol. Chem. Rapid Commun., 1990, 11, 169 CrossRef CAS; (g) D. R. Burfield, Polymer, 1984, 25, 1817 CrossRef CAS.
- (a) H. Teshima and N. Tomotsu, US 5739227, 1998; (b) A. B. Deshpande, R. V. Subramanian and S. L. Kapur, J. Polym. Sci., Part A-1, 1969, 7, 3437 CrossRef CAS; (c) L. C. Anand, A. B. Deshpande and S. L. Kapur, J. Polym. Sci., Part A-1, 1967, 5, 665 CrossRef CAS; (d) A. B. Deshpande, R. V. Subramanian and S. L. Kapur, J. Polym. Sci., Part A-1, 1966, 4, 1799 CrossRef.
- (a) H. Itagaki and I. Takahashi, Chem. Phys. Lett., 1993, 205, 446 CrossRef CAS; (b) Y. C. Wang and H. Morawetz, Makromol. Chem., 1975, 1, 283 CrossRef; (c) J. W. Longworth, Biopolymers, 1966, 4, 1131 CrossRef; (d) S. S. Yanari, R. Lumry and F. A. Bovey, Nature, 1963, 200, 242 CAS.
- M. Fujiwara and A. Yamasaki, J. Chem. Soc., Faraday Trans., 1998, 94, 2525 RSC.
- (a) B. Lian, K. Beckerle, T. P. Spaniol and J. Okuda, Eur. J. Inorg. Chem., 2009, 311 CrossRef CAS; (b) B. Lian, K. Beckerle, T. P. Spaniol and J. Okuda, Angew. Chem., Int. Ed., 2007, 46, 8507 CrossRef CAS.
- Maldi-tof measurements of low molecular weight oligomers revealed no significant divergence from the results obtained by GPC measurements against PS standards. Therefore in terms of comparability all molecular weights of oligomers and polymers were determined by GPC.
- E. Yashima, K. Maeda, H. Iida, Y. Furusho and K. Nagai, Chem. Rev., 2009, 109, 6102 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |