Elastomeric polypeptide-based biomaterials
Linqing
Li†
,
Manoj B.
Charati†
and
Kristi L.
Kiick
*
Department of Materials Science and Engineering, University of Delaware, Newark, DE 19716, USA. E-mail: kiick@udel.edu.; Fax: +302-831-4545; Tel: +302-831- 0201
First published on 2nd June 2010
Abstract
Elastomeric proteins are characterized by their large extensibility before rupture, reversible deformation without loss of energy, and high resilience upon stretching. Motivated by their unique mechanical properties, there has been tremendous research in understanding and manipulating elastomeric polypeptides, with most work conducted on the elastins but more recent work on an expanded set of polypeptide elastomers. Facilitated by biosynthetic strategies, it has been possible to manipulate the physical properties, conformation, and mechanical properties of these materials. Detailed understanding of the roles and organization of the natural structural proteins has permitted the design of elastomeric materials with engineered properties, and has thus expanded the scope of applications from elucidation of the mechanisms of elasticity to the development of advanced drug delivery systems and tissue engineering substrates.
 Linqing Li | Linqing Li is currently a Ph.D. candidate in the Department of Materials Science and Engineering at the University of Delaware. He received his B.S. degree in Materials Chemistry from the University of Science and Technology of Beijing, China in 2007. He started his doctoral program in 2007 at UD under the instruction of Prof. Kristi L. Kiick. His Ph.D. project is mainly focused on generating and characterizing a library of resilin-based polypeptides to create elastomeric hydrogels with necessary bioresponsiveness for engineering mechanically active tissues such as vocal folds, heart, and blood vessels. |
 Kristi L. Kiick | Kristi Kiick is an Associate Professor of Materials Science and Engineering at the University of Delaware. She received a BS in Chemistry from the University of Delaware in 1989, and an MS in Chemistry as an NSF Predoctoral Fellow from the University of Georgia in 1991. She began doctoral studies in 1996 under the direction of David Tirrell, graduating with a PhD in Polymer Science and Engineering from the University of Masschusetts Amherst in 2001 after completing her research as an NDSEG Fellow at the California Institute of Technology. Her current research programs are focused on combining biosynthetic techniques, chemical methods, and bioinspired assembly strategies in the production of polymeric materials with novel multifunctional behavior. |
Introduction
Elastomeric proteins are present in a wide range of biological systems, where they have evolved to meet specific functional needs. In general, they exhibit rubber-like elasticity, undergo high deformation under stress without rupture, and recover to their original state when the stress is removed (Fig. 1). The ability of any protein to exhibit elasticity lies in its molecular and structural organization; individual components must be conformationally free so that they can respond quickly to the applied stress and they must form an inter-connected network of chains through covalent or non-covalent crosslinks, to effectively distribute the applied stress throughout the structure. The elastic properties of such proteins depend on the nature of elastomeric domains, the size of the domain and the degree of crosslinking. Interests in employing elastomeric proteins, particularly elastin and resilin, in high performance applications has remained of significant research interest owing to the biological properties and potential medical value of these polymers. In addition, investigation of the basic mechanisms underlying materials properties have continued, owing to the relevance of defects in the elastomeric proteins in human diseases (e.g., understanding the role of aortic elastin in atherosclerosis or use of elastin and collagen to study platelet interaction and prevent tissue adhesion in wound healing). The development of sophisticated spectroscopic techniques has enabled investigation of the structural and biomechanical properties of these proteins at the molecular level, and has provided valuable clues to inform both research approaches.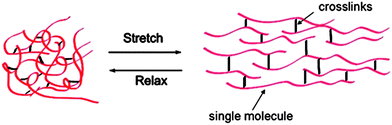 | ||
| Fig. 1 A general schematic of elastomeric proteins. | ||
The elastomeric proteins are extremely diverse in their mechanical properties (Table 1) that are in many cases crucial to their functions. Not all elastomeric proteins are exceptional in their material properties, but all represent good designs because the properties that define their function are well matched to relevant environmental conditions; indeed, elastomeric proteins are known to play unique and unusual roles in living animals and insects and have evolved accordingly. For example, elastin and resilin work well as strain energy storage devices in environments where high resilience and excellent fatigue resistance are required. Collagen has unmatched capacity for storage of strain energy and function in strong tendons that provide links between muscle and bone; whereas spider silks have unmatched toughness that is necessary in energy-dissipating devices. In other organisms, abductin ligaments in clams (bivalve molluscs) open the shell when the muscles relax, generating a primitive swimming action; byssus threads bind mussels to rocks and comprise elastic and rigid domains to resist wave action. Elastomeric proteins also endow organisms from fleas to large mammals with the ability of power amplification via a catapult mechanism enabling them to jump further than what would otherwise be possible. The best known examples of the use of catapult mechanism in jumping are observed in insects such as flea,1 froghoppers,2 locusts3 and beetles4,5 It was discovered that the springs responsible for the catapult mechanism contain a specialized rubbery protein, namely resilin, that mainly serves as an energy store.
| Material | Modulus (GPa) | Extensibility ε (max) | Resilience (%) | |
|---|---|---|---|---|
| Elastin (bovine ligament) | 0.0011 | 1.5 | 90 | |
| Resilin (dragonfly tendon) | 0.002 | 1.9 | 92 | |
| Collagen (mammalian tendon) | 1.2 | 0.13 | 90 | |
| Dragline silk | 10 | 0.3 | 35 | |
| Kevlar | 130 | 0.027 | ||
| Carbon fiber | 300 | 0.013 | ||
Despite the wealth of information on the probable roles of the elastomeric proteins, only a few have been characterized in detail due to difficulties in their isolation from natural sources and the complexity of their chemical and physical properties (e.g., their non-globular nature and insolubility of crosslinked networks). In conjunction with spectroscopic characterization, recombinant approaches have thus also been valuable in the investigation of these proteins, and they have been developed for many potential uses in biomaterials and tissue engineering. Various processing techniques such as photolithography, chemical etching, polymer demixing, electrospinning, molecular self-assembly, as well as thermally induced phase separation, have been employed extensively to process polypeptidic and polymeric biomaterials. These processing approaches have been reviewed elsewhere.6–12 Here, we focus on recent developments in the recombinant synthesis of elastomeric polypeptides of desired compositions and properties; brief descriptions of select elastomeric proteins are thus given below.
Naturally occuring elastomeric proteins
Elastin
Elastin is a major extracellular protein that is found in connective tissues, lungs, and blood vessels and is known to provide elasticity to these tissues/organs both statically and dynamically. One of the most extensively studied elastomeric proteins, elastin is secreted as a soluble precursor tropoelastin with alternating hydrophobic domains of variable length and alanine-rich lysine-containing cross-linking domains. Elastin consists of putative repeats of VPGXG that mainly impart rubber-like properties to the protein.15 This domain also contains other hydrophobic amino acids such as alanine, leucine, and isoleucine, which imparts to the protein an inverse transition (much like a lower critical solution temperature), in which the protein solution phase separates into polymer-rich and polymer-poor phases (also called coacervation) upon an increase in temperature.16,17 This coacervation is thought to play an important role in the assembly of tropoelastin and to affect the mechanical properties of the elastomeric protein.13,18,19 After hydroxlation and cross-linking, elastin forms an insoluble mature elastin network.Previous work, including the circular dichroic characterization of synthetic peptides and fragments of elastin, has clearly revealed the presence of folded conformations such as type II β-turns. Based on these results, Urry proposed that elastins adopt a β-spiral structure (Fig. 2) that consists of multiple repeats of type II β-turns.19,20 In particular, on the basis of extensive studies on synthetic poly(VPGVG) (Table 2), a structural model was introduced as a mechanism of elasticity. In the polypentapeptide repeats, the β-turns are repeated regularly and act as spacers between the turns of the spiral, suspending the chain segments in a kinetically free state. On stretching, the conformational freedom is reduced, resulting in a decrease in conformational entropy; this loss of entropy provides the restoring force. Studies by Tamburro and coworkers21,22 put forward a new model based on CD and NMR studies performed on short peptides in different solvents and at different temperatures. These studies revealed the presence of two different types of conformers; folded or semi-folded conformation consisting of β-turns and extended conformations such as polyproline-II.23 The presence of folded and extended conformers giving rise to dynamic β-turns that slide along the chain. The high entropy of the relaxed state is thought to originate from the general equilibrium of the folded state in addition to the sliding β-turns. On stretching, the equilibrium is shifted toward the open or extended conformations, resulting in decrease in the conformational entropy and providing the force for elastic recoil. Although the details of the exact mechanism for elastic recoil in elastin is still a source of investigation, the driving force for recovery in elastin is considered to be mainly entropic in origin.
| Protein | Consensus repeat sequence |
|---|---|
| Abductin | GGFGGMGGGX |
| Elastin | VPGVG,VPGIG,VPGG,APGVGV |
| Flagelliform Silk | GPGGX |
| HMW subunit of gluten | PGQGQQ, GYYPTSPQQ |
| Resilin | GGRPSDSYGAPGGGN |
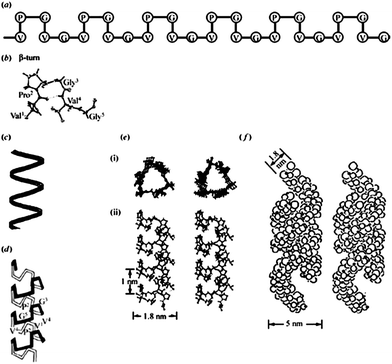 | ||
| Fig. 2 A description of the proposed molecular structure of poly(GVGVP). (a) A schematic representation of the repeating Pro-Gly-containing sequence in the β-turn structure, with crystallographic detail in part (b). (c) A schematic helical representation of the structure that forms on raising the temperature above the inverse transition temperature, upon which an increase in order of the polypeptide chain is observed. (d) A schematic representation of the helical structure, called a β-spiral, but with the β-turns included and functioning as spacers between turns of the helix. (e) Stereo pairs of the β-spiral structure in atomic detail, (i) end view and (ii) side view. (f) Associated β-spirals having formed multi-stranded twisted filaments, which is the more accurate description of the hydrophobically folded and assembled state. Reproduced with permission from ref. 14 © 2002 Philosophical Transactions of the Royal Society B: Biological Sciences. | ||
Biomaterials based upon natural elastin have been suggested for a massive scope of potential applications. Some of the well-known examples are split-skin autografts for burn wounds, purified porcine elastin biomaterials as gastrointestinal patches in the esophagus,24 and decellularized heart valves and vasculature containing elastin for heart valve replacement and vascular grafts.25–27 However, due to its poorly defined purification, batch-to-batch variations, high propensity to calcify upon cardiovascular prosthetic implants,28,29 as well as the fact that cross-linked elastin may serve as a nucleation site for mineralization,25,30 natural elastin is not used that often as a tissue engineering scaffold. Some of these issues, such as calcification, can be minimized by aluminium chloride treatment,31 ethanol/EDTA treatment,32 the presence of glycosaminoglycans,33 or the presence of basic fibroblast growth factor.34 Therefore, interest in further understanding the properties of natural elastin and in emulating its highly elastomeric behavior has motivated the investigation of elastin-like polypeptide-based biomaterials. These recombinant polypeptides exhibit promising biological, biomechanical, biochemical and biophysical properties and have been employed in the design of thermally reversible nanoparticles, drug-targeting cancer cells, spinal cord repair materials and tissue substitutes.35–37 The readers are also referred to several recent reviews for more information on elastin-based biomaterials.38–42
Wheat gluten
Wheat gluten is the main elastomeric protein in plants. The high molecular weight (HMW) subunits of wheat gluten are seed storage proteins, for storage of essential nutrients such as carbon, nitrogen and sulfur for growth of seedlings. The hexa-amino-acid repeats PGQGQQ and the nona-amino-acid repeats GYYPTSPQQ (Table 2) in the protein are considered to be mainly responsible for the elasticity of wheat gluten. The repetitive sequences are rich in glycine and proline, but in contrast to other known elastomeric proteins, the remaining amino acid residues are hydrophilic; i.e. mainly glutamine-rich.43 CD and FTIR spectroscopy on the short peptides based on the repeat motifs of the HMW subunits of gluten suggested the presence of β-turns between the repeats.44 Characterization of a recombinant polypeptide corresponding to the HMW subunit also showed the presence of β-turns in equilibrium with a polyproline II-like structure in solution, however less β-turn and a predominantly β-sheet-like structure was observed in hydrated solids.45 Interestingly, Tatham and coworkers have shown that while the elastic modulus of HMW wheat gluten subunits crosslinked by γ-irradiation is similar to that of the crosslinked elastin polypentapeptide poly(VPGVG), the mechanism of elasticity is likely to be different.43 The gluten contains repetitive β-turns which could lead to formation of β-spirals as hypothesized for elastin, but in addition to the entropic component, enthalpic contributions related to the extensive hydrogen bonding within and between the subunits is also proposed to contribute to the total elastomeric force.Abductin
Abductin, a natural elastomer found in the bivalve mollusc shell, is located in the inner hinge region between the shell junction and acts as an elastic pivot that antagonizes the action of the abductor muscle, thus permitting the opening of the shell upon relaxation. This action provides the swimming mechanism in scallops, allowing them to swim a couple of meters at a time by opening and closing their shells (at a low frequency of approximately 4 Hz) in order to escape slow moving predators such as starfish.46 Abductin is a unique protein as it is the only elastomer discovered in nature that exhibits compressible elasticity as the main mode of action.47 The N-terminal domain of abductin is characterized by the presence of alanine-rich sequences containing two conserved cysteine residues that are involved in inter- and intramolecular disulfide formation. The C-terminal domain comprises a glycine- and methionine-rich decapeptide sequence;47 the methionine residues have been reported to be converted to methionine sulfoxide in surf clams.48 The high glycine content is likely to account for the reported lack of secondary structure, although structural organization may be imparted by the well conserved, repeating residue pattern in the C-terminal part of protein (Table 2).49 The N-terminal domain is cleaved during the secretion process, thus the probable candidates for crosslinking sites could be the two conserved lysine residues and one conserved tyrosine residue in the C-terminal domain.48As far as structure-elasticity relationships are concerned, the predicted secondary structure of abductin indicates that the repetitive C-terminal domain adopts a random coil conformation.47 Using molecular dynamics and solid phase peptide synthesis, Bochicchio et al. designed and synthesized a peptide mimetic of abductin that self-assembles into a biphasic aggregate with rubber-like properties even without covalent cross-links.50 It is suggested that hydrophobic interactions result in physical cross-linking, and act as a driving force for compressive elasticity. Accordingly, it is reasonable to suggest that abductin shares a similar elasticity mechanism with elastin, however, emphasis should be made on the fact that two proteins express their elastic recoil based on completely opposite forces, that is a stretching force in the case of elastin and a compressive force in the case of abductin.
Flagelliform silk
Spiders are known to produce a variety of structural silk proteins, which have evolved to function in air rather than in aqueous conditions and which exhibit properties that range from elastic to extremely rigid. Among all the different silks, dragline silk and flagelliform silk are the most studied and exhibit completely different mechanical properties. The dragline silk has a very high tensile strength and extends only 30%, and is used as the frame of the orb web. Flagelliform silk forms the core fibers of the capture spiral of the web and, like elastin and resilin, meets the criteria as an elastomeric protein with low stiffness and high extensibility (>200%), similar to lightly cross-linked rubbers.51However, the rubber-like behavior of flagelliform silk fibres are unique because these fibers function in air while other elastomeric proteins function in aqueous media. Previous studies reported that molar concentrations of low molecular weight organic compounds in flagelliform silk network maintain its molecular mobility by drawing water out of air and plasticizing the silk protein.52,53 Like all elastic proteins, the flagelliform silk also consists of repetitive amino acid sequences, based on pentapeptide repeats of GPGGX, in addition to two other domains; GGX and a spacer (Fig. 3) that disrupts the glycine-rich regions and may contribute to the alignment of monomers into fibers.54 The exact mechanism of crosslinking in flagelliform silk protein is unknown, however interactions between the spacer domains are thought to be involved.55
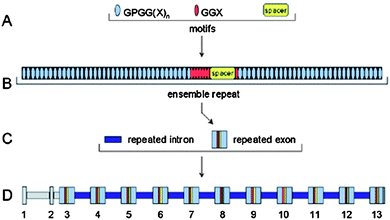 | ||
| Fig. 3 The flagelliform gene contains hierarchical sets of components.56,57 (A) The repetitive coding region is composed of codons for three different amino acid sequence motifs. (B) Iterations of the three motifs are organized into complex ensemble repeats of about 440 amino acids. (C) Each ensemble repeat is encoded by a single exon. These repeated exons are separated by repeated introns. (D) The flagelliform silk gene spans about 30 kb. Exons and introns are numbered, and regions of nonrepetitive sequence are shaded gray. Reproduced with permission from ref. 56, © 2000 Science. | ||
Conticello and co-workers have synthesized a recombinant polypeptide which mimics the repeating sequence GPGGX of the N. Clavipes flagelliform silk protein in E. coli. Characterization of the conformational properties of the recombinant polypeptide in aqueous solution by CD, FTIR and NMR spectroscopy indicated the presence of type-II β-turns, and an increase in the β-turn content was observed in solid films.58 Since these turn conformers are sparsely populated under ambient solution conditions, they are probably in rapid exchange with open and/or random coil conformation. This finding is consistent with the hypothesis of an entropic mechanism of elasticity, resembling that proposed for the other β-turn-forming polypeptides, including elastin. The combination of the semi-crystalline structure, which imparts mechanical strength and toughness, with the protein’s interaction with water, which allows a stable protein solution to dry into a fiber in benign environments, provides multiple opportunities in materials design.
Resilin
Resilin, the elastomeric protein mainly found in the specialized regions of the cuticle in most insects, was first discovered five decades ago by Weis-Fogh during a study of flight systems in locusts.59 Almost all types of insect cuticle contain resilin and the presence of this proteinaceous matrix has a prominent effect on the mechanical properties of the cuticle. Resilin is the most efficient elastomeric protein known and is present in most, if not all insects, where it has been adapted for flight,59,60 jumping61,62 and sensory mechanisms. It confers long-range elasticity to the cuticle and functions both as an energy store13,63 as well as a damper of vibrations in sound-producing organs of some insects such as cicadas64 and moths,65 where it withstands frequencies of vibration up to 4 kHz. Resilin obtained from elastic tendons of dragonfly and locust ligament shows extraordinary elasticity with very little loss and an elastic modulus of 0.588–0.883 MPa.19,66,67 Naturally occurring resilin is crosslinked; di- and tri-tyrosine crosslinks are formed by peroxide oxidation of tyrosine residues to connect the resilin polypeptides in a three-dimensional network.Ardell and Andersen identified a gene from Drosophila melanogaster (CG15290) as the likely precursor for Drosophila resilin.68 The N-terminal region of CG15290 is dominated by 18 slightly varying copies of a 15-residue motif: GGRPSDSYGAPGGGN. The overall resilin sequence has a very high content of glycine (35–40%) and proline (7–10%).69 Andersen and Andrell suggested that these repeats may form β-spirals but with larger and more irregular loops than in other elastomeric proteins. Recent circular dichroic spectroscopy investigations on short resilin peptides, by Bochicchio et al., revealed the coexistence of polyproline-II conformation with type II β-turns.23,70 These results are in agreement with the predictions made by Andersen and the conformations are similar to those found in other elastomeric proteins. Weis-Fogh initially suggested that resilin behaves as an entropic elastomer consisting of unordered chains linked through stable crosslinks and that its restoring elastic force arises due to the loss in conformational entropy upon stretching.67 However, the presence of β-turns may indicate the formation of sliding β-turns as suggested for other elastomeric proteins.23,71 Unlike elastin, the resilin sequence is dominated by hydrophilic residues, suggesting that the hydrophobic interactions are minimal and that there could be an energetic component, in addition to an entropic component, responsible for the elastic recoil. Further spectroscopic studies on RLPs will help unveil the details of the mechanism of elasticity of resilin.
Genetically engineered elastomeric polypeptides
Recombinant methods have afforded a powerful tool to enable the synthesis of polypeptides with desirable sequences in a host organism such as E. coli. Owing to the control over sequence, molecular weight, polydispersity and functional group placement afforded by these methods, it is straightforward to produce modular polypeptides equipped with both structural and bioactive domains. Polypeptides with distinctive architectures have been designed; original examples include polypeptides rich in β-sheet structures in which the lamellar thickness can be controlled by the sequence periodicity,72 smectic liquid crystal phase of stiff helical rods with uniform molecular weight73 and pH- and temperature-sensitive hydrogels.74,75 In addition, certain structures in natural proteins have been recapitulated in multimeric consensus peptide repeats such as the VPGXG sequence from elastin and AG-rich repeats from silk,76,77 although it has remained difficult to reproduce the mechanical properties of the natural proteins. In addition to engineering mechanical and biological properties of the recombinant polypeptides, the chemical versatility of the polypeptides may also be engineered with techniques to incorporate non-natural amino acids, which has expanded options to include multiple functionalities in a polypeptide chain. These advances have facilitated the creation of biomaterials with desirable mechanical and biological properties, targeted for use in either rigid, permanent bone-like implants or soft, degradable hydrogels for wound healing. A variety of biologically-active domains, i.e. heparin binding domains,78–82 growth factors,83–86 protease degradation sites37,78 and cell binding domains such as RGD,37,87 REDV88 and E-cadherin89 have been incorporated into biosynthetic polypeptides to enhance cell-biomaterial interactions. Although there have been many repetitive polypeptides produced, we focus here on more recent work on elastin- and resilin-like polypeptides.Elastin-like polypeptides (ELPs)
Interest in emulating the highly elastic behavior and excellent mechanical properties of the natural elastins has motivated the enormous growth of research on these polypeptides over the past few decades. As discussed above, the amino acid sequence of naturally occurring elastin mainly comprises a pentameric repeating unit, VPGVG. Initial studies, carried out by Urry on chemically synthesized poly(VPGVG) that was cross-linked by γ-irradiation,90 indicated that the polypeptide exhibited entropic elasticity and also showed a supramolecular organization very similar to that of elastin. More recently, with the standardization of molecular biological protocols, ELPs of more complex yet well-defined structures have been produced.The thermally responsive behavior of elastin-like polypeptides (ELP) has been a strong motivation behind their extensive study and application. ELPs exhibit an inverse transition temperature similar to a lower critical solution temperature, in which the polypeptide undergoes phase separation from solution with increasing temperature, upon its collapse into a β-spiral structure and simultaneous release of water molecules associated with the polypeptide chain. This thermally responsive behavior is conserved for all protein polymers of the general sequence (VPGXG)n, where X can be any amino acid except proline, as determined by Urry and coworkers.91–93 The temperature at which the protein polymer undergoes its inverse transition, Tt, can be controlled via changes in the identity of X, which offers many promising opportunities for various applications including drug delivery, surface modification, nanoparticle formation, actuation, and purification. The thermally responsive behavior of ELPs is completely reversible, and the polypeptide returns to its soluble state when the temperature is lowered below Tt. It has been shown that one repeat unit of VPGVG is sufficient to allow the transition from random conformation into an ordered β-turn,94 although higher molecular weight polymers are required for useful material properties.
Tirrell and coworkers have produced ELPs with lower Tts for applications as vascular graft replacement materials. The lower transition temperatures (22–28 °C) of these VPGIG-based ELPs facilitate the use of these polypeptides under physiological conditions,95 and the inclusion of the cell-binding domain of CS5 has been useful for promoting endothelial cell attachment to these materials. Chilkoti and coworkers have used varying ratios of valine, glycine, or alanine residues for the production of ELPs with physiologically relevant Tts for drug delivery applications.96–98
A variety of crosslinking techniques have been employed to obtain crosslinks in ELP, including gamma-irradiation,99 physical100 and chemical crosslinking.101 The pentapeptide sequence of VPGVG has been modified by the insertion of amino acids with functionalized side-chains (lysine and glutamic acid), that can be crosslinked with appropriate bifunctional reagents. Selective crosslinking of amino groups of ornithine and lysine residues in the polypeptides has been accomplished via the use of chemicals such as glutaraldehyde,101 hexamethyl isocyanate,102 bis(sulfosuccinimidyl) suberate,103 hydroxymethylphosphines104 and many others. The Tirrell group has demonstrated that elastic hydrogels formed via chemical crosslinking of ELPs containing cell-binding domains could support the growth and spreading of endothelial cells. These ELPs, consisting of a repeating structure composed of alternating lysine-containing ELP domains and integrin-binding fibronectin-derived domains, were crosslinked with glutaraldehyde,101 bis(sulfosuccinimidyl) suberate (BS3),103 and hexamethylene diisocyanate,105 and it was found that the moduli of the resulting hydrogels were very close to the modulus of native elastin, 0.3–0.6 MPa, with extension ratios of 200–400%. In addition, the modulus can be tuned by altering the protein molecular weight, distance between lysine crosslinking sites, positioning of lysine crosslinking sites, molar ratio of crosslinker to primary amine, the type of crosslinker used and the solvent conditions. Endothelial cells cultured on films of these ELPs successfully proliferated and formed monolayers upon attachment to the integrin-binding domains. The cell growth and spreading was shown to be largely dependent on the density of RGD or CS5 cell-binding domains as well as on the location of the crosslinkable lysine residues.87,88,106 The tunable, elastin-like mechanical properties of the ELP hydrogels and their ability to mediate cellular adhesion and proliferation make them suitable candidates for use in small diameter vascular grafts.
The Chilkoti group has reported the use of an aqueous, biocompatible Mannich-type condensation reaction to crosslink lysine-containing ELPs over a wide range in pH (2–13), with the advantage that this reaction produces only water as a by-product.108 In this reaction, an organophosphorous crosslinker, β-[tris(hydroxymethyl)phosphino]propionic acid (THPP), reacts with the amines of the lysine residues in the ELP to create trifunctional intra- or intermolecular crosslinks. The ELPs undergo gelation within minutes under physiological conditions, indicating that they are suitable for use as injectable biomaterials. The shear modulus of the crosslinked hydrogels is comparable to or higher than that of some connective tissues, such as nucleus pulposus or meniscus, which can further be modulated by adjusting the lysine density or arrangement. The hydrogels were also tested for biocompatibility, and it was found that they are non-cytotoxic.104,108 These results illustrate that THPP-crosslinked ELPs provide a biocompatible and injectable biomaterial that may support tissue regeneration in a load-bearing environment.
Very recently, Straley and Heilshorn employed these principles in the design of a scaffold composed of crosslinked, multi-component, engineered ELPs that allow for independent tuning of elastic modulus, degradation rate, and cell adhesion.37 The scaffold primarily comprises an elastin-like structural sequence to impart mechanical properties to the scaffold, an RGD sequence to promote cell attachment, as well as a urokinase plasminogen activator sequence to control matrix degradation rate. This multi-component engineered protein was shown to support cell attachment, neuronal differentiation and neurite outgrowth of a PC-12 neuronal cell line. These studies highlight the versatility of the genetic engineering approach in the design of modular proteins with a combination of optimal material and biological properties.
ELPs containing non-natural amino acids have also been produced to expand the range of crosslinking methods, to provide opportunities for photopatterning of ELPs, and to elucidate some of the conformational features of the polypeptide that contribute to the inverse temperature transition. Tirrell and coworkers synthesized an ELP with the general sequence [(VPGVG)2(VPGFG)(VPGVG)2]5VPGC, substituting the phenylalanine residue with a photosensitive amino acid, p-azidophenylalanine, as a crosslinking site. The photosensitivity of the non-natural amino acid permitted photopatterning of ELP-films onto surfaces for immobilization of target proteins.109
Conticello and coworkers have conducted elegant studies in which proline analogues have been quantitatively incorporated into the polypeptide [(VPGVG)4VPGIG]16 to study the role of polypeptide conformation on the inverse temperature transition. The stereoelectronic properties of the proline analogues 4-fluoroproline and 3-fluoroproline cause the epimers of these analogues to either promote or inhibit β-turn formation, and these studies have thus substantiated the importance of the β-turn structure in the coacervation and potential elastomeric properties of elastin.110,111 Incorporation of the 4R-substituted prolines, which are known to stabilize the β-turn structure, promote coacervation and a lower Tt; the 4S epimers show the opposite effect. Similar effects were observed for 3-hydroxyprolines, showing the generality of this approach. Studies to correlate extent of β-turn formation with tensile properties have yet to be reported, but would provide further insight into the potential origin of elasticity in these polypeptides.
The variations in coacervation behavior of hydrophobic and hydrophilic ELPs have been widely used as a strategy for producing crosslinked networks and nanoparticles. Conticello and coworkers have reported systematic rheological studies of genetically synthesized self-assembling ELP copolymers.100,112–114 For example, BAB triblock copolymers have been produced in which the B block, [(IPAVG)4(VPAVG)]n, is hydrophobic and plastic, and the A block, [(VPGVG)4(VPGEG)]m, is hydrophilic and elastic. Variations in processing conditions yield elastomeric films that can show strain-to-break values up to approximately 1200%; the greatest extension-to-break values are observed for films produced under stronger phase-separation conditions. They found that changing the length or hydrophilicity of the middle block largely affected the viscoelastic and mechanical behavior of the copolymers when the same hydrophobic and plastic domain was used.112 In addition, nanoparticles can be formed from triblock copolymers with similarly designed blocks, as well as by diblock ELPs with blocks of differing hydrophilicity.115 The assembly process can be manipulated via sequence variations that allow triggered assembly (into nanoparticles or networks) on the basis of changes in pH, temperature, and ionic strength.
Dreher et al. reported a series of ELP diblock copolymers, of various molecular weights and different hydrophilic-to-hydrophobic ratios, that carried an N-terminal peptide ligand; these polymers are capable of self-assembling into spherical micelles when heated slightly above body temperature (Fig. 4). The critical micelle temperature is controlled by the hydrophobic block, and the size of the micelle is determined by both the total ELP length and hydrophilic-to-hydrophobic block ratio. These studies have demonstrated the capability of ELP's bearing terminal ligands to form multivalent spherical micelles in the clinically relevant temperature range 37–42 °C and to target cancer cells.35
![Temperature triggered self-assembly of an ELPBC to form multivalent spherical micelles. An N-terminal ELP[V1A8G7-n] gene (hydrophilic, high Tt) and C-terminal ELP[V5-n] gene (hydrophobic, low Tt) are seamlessly fused together to create a gene that encodes an ELPBC. When the size and ratio of the blocks are correctly selected, the ELPBC self-assembles into a spherical micelle at ∼40 °C. In the cartoon shown, upon self-assembly the spherical micelles present multiple copies of an affinity targeting moiety (green triangle) and sequester a drug or imaging agent (lightning bolt) within the core of the micelle. Reproduced with permission from ref. 35, © 2008 Journal of the American Chemistry Society.](/image/article/2010/PY/b9py00346k/b9py00346k-f4.gif) | ||
| Fig. 4 Temperature triggered self-assembly of an ELPBC to form multivalent spherical micelles. An N-terminal ELP[V1A8G7-n] gene (hydrophilic, high Tt) and C-terminal ELP[V5-n] gene (hydrophobic, low Tt) are seamlessly fused together to create a gene that encodes an ELPBC. When the size and ratio of the blocks are correctly selected, the ELPBC self-assembles into a spherical micelle at ∼40 °C. In the cartoon shown, upon self-assembly the spherical micelles present multiple copies of an affinity targeting moiety (green triangle) and sequester a drug or imaging agent (lightning bolt) within the core of the micelle. Reproduced with permission from ref. 35, © 2008 Journal of the American Chemistry Society. | ||
Employing the introduction of orthogonal functionalities via biosynthetic strategies, Teeuwen et al. produced clickable ELPs bearing non-natural reactive groups—azidohmoalanine or homopropargylglycine—via expression from auxotrophic expression hosts.116 These clickable ELPs were used as versatile modular building blocks by applying Cu-catalyzed azide-alkyne cycloaddition (CuAAC) to conjugate three different moieties, namely fluorescent probes, a polymer, and the enzyme CalB, These approaches enable the introduction of the characteristic LCST behavior of ELPs to biohybrid structures for applications such as protein separations, catalytically active coacervates, microfluidics, and targeting of tumor tissue.
Taken together, these studies highlight that ELPs can be used for a wide variety of different applications such as protein purification, drug delivery vehicles, surface engineering, biosensing and tissue engineering.
Resilin-like polypeptides (RLPs)
Although the outstanding materials properties of resilin were discovered five decades ago, only very recently have useful quantities of recombinant resilin been obtained from E. coli cultures. Elvin and coworkers successfully cloned the cDNA of N-terminal region of Drosophila CG15290 gene and expressed the protein in E. coli in its soluble form with yields up to 15 mg L−1.117 After successful optimization of the expression conditions, RLP has been expressed in E. coli at 300 mg L−1via lactose-induced fermentation procedures.118,119 The recombinant resilin can be crosslinked via Ru(II)-mediated photochemical methods to obtain di-tyrosine linkages. The resilience of the crosslinked hydrated RLP, as suggested by scanning probe microscopy and tensile testing measurements, was reported to be 97% with an elastic modulus of 2.5 kPa; with only 3% of the stored energy lost as heat.Recently, Elvin and co-workers continued and expanded the structural and mechanical measurements to three different resilin-based recombinant proteins: Rec1-resilin (the first exon of the Drosophila CG15920 gene), Dros16 ((GGRPSDSYGAPGGGN)16) and An16 ((AQTPSSQYGAP)16). As indicated by CD data, Rec1-resilin and An16 share similar spectra that display mainly unordered conformations with minor contributions from β-sheet, β-turns, and PPII conformations while surprisingly, Dros16 contains a relatively high level of β-sheet and forms a more folded structure. Although the secondary structure of Dros16 is significantly different from Rec1-resilin and An16, the proteins exhibit similar mechanical features such as high resilience, elastomeric stretch, and low moduli, as indicated via SPM measurements and tensile testing.120 In order to understand the functional and structural properties of native resilin proteins more completely, Qin et al. expressed the three exons of the Drosophila CG15920 gene, which encode the full native resilin protein with chitin binding domain.121 This full length recombinant resilin was compared with previously reported resilins and found to be comparable in both structure and function, with the additional possiblities for formation of resilin-chitin composites due to the introduction of the chitin-binding domain. There have not been many investigations on the applications for RLPs, but opportunities are plentiful, given that naturally occuring resilin is expected to maintain high resilience even at relatively high frequencies.13,122 Furthermore, the RLPs provide attractive alternatives for developing materials in the regeneration of mechanically active tissue where high frequency responsiveness and superior fatigue resistance are important.
These unique mechanical properties of RLPs have motivated our modular design of the first-reported resilin sequences that combine resilin's excellent mechanical properties with biological domains.123 The sequence of a resilin-like polypeptide with 12 repeats (RLP12) is shown below in Fig. 5. The same 15 amino acid repeating unit identified from the D. melanogaster resilin gene was used as a structural domain to impart mechanical strength.The capacity to support the attachment of mammalian cells to the scaffold was provided by the incorporation of the cell-binding ligand RGDSP, which is derived from fibronectin as it has been demonstrated as a positive tool to promote integrin mediated cell adhesion.124,125 A matrix metalloproteinase (MMP) sensitive sequence (GPQGIWGQ), derived from human α1(I) collagen chain, is also introduced to enhance the control of proteolytic degradation.126,127 The heparin-binding domain, CKAAKRPKAAKDKQTK, which effectively promotes the noncovalent sequestration of both heparin and growth factors (VEGF and bFGF), is also included in the sequence.80,82,128–131
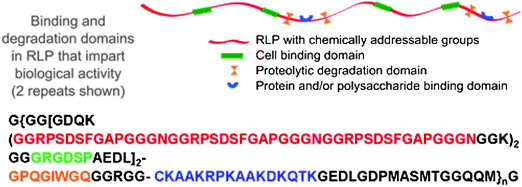 | ||
| Fig. 5 A schematic and amino acid sequence of a resilin-like polypeptide (RLP). This sequence contains 12 repeats of the resilin consensus sequence. Reproduced with permission from ref. 123, © 2009 The Royal Society of Chemistry. | ||
Successful expression with higher yields was achieved by employing Studier auto-induction as an alternative method to IPTG induction.132 The expression of a pure RLP12 of the desired sequence was confirmed by SDS-PAGE, MALDI-MS and amino acid analysis. Consistent with previous results and the anticipated conformational behavior of the naturally occurring resilin, the overall conformation of this RLP12 was suggested by CD and FTIR studies to be highly unordered with a small contribution from type-II β-turn. These results also indicate that modification of the resilin sequence with biological domains does not appreciably alter the expected conformational behavior of the resilin-like polypeptide.
The desired mechanical properties of this polypeptide were measured by both oscillatory rheology and tensile testing. The cross-linked hydrated RLP12 hydrogel exhibits a Young's modulus of approximately 50 kPa with an average extension-to-break ratio of 180% (Fig. 6), consistent with the mechanical properties reported for other recombinant resilin-like polypeptides.
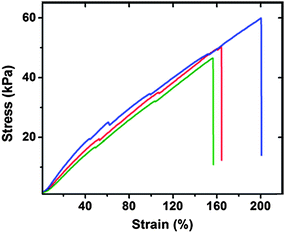 | ||
| Fig. 6 Tensile testing data for hydrated RLP12 films. Stress-strain data was recorded at a strain rate of 10% gauge length per minute at 25 °C. Reproduced with permission from ref. 123, © 2009 The Royal Society of Chemistry. | ||
Given the presence of fibroblasts in mechanically active vocal fold tissue, preliminary studies of the adhesion of NIH-3T3 fibroblasts on the surface of the RLP12 films were undertaken; the data indicated good cytocompatibility and that the material supports the adhesion and proliferation of the cells (Fig. 7). These reported properties suggest the potential of modular RLPs as matrices in vocal fold regeneration and cardiovascular applications, and optimization of the materials for support of primary cell adhesion and proliferation is under way.123
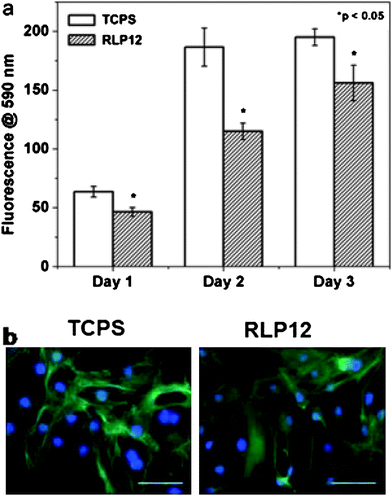 | ||
| Fig. 7 Proliferation data (days 1–3) and fluorescence microscopy images (day 1) of NIH-3T3 cells on control TCPS and on films formed from RLP12. The scale bar represents 100 μm. Reproduced with permission from ref. 123, © 2009 The Royal Society of Chemistry. | ||
Conclusions
The increasing amount of knowledge acquired from recent studies on elastomeric proteins has inspired the design and synthesis of biopolymers with interesting mechanical and biological properties. Results obtained suggest an optimistic vision for the production of efficient elastomeric biomaterials. A substantial amount of work on recombinant versions of silk, elastin and collagen has afforded materials with useful mechanical and biological properties, as well as with a multitude of applications including drug delivery, tissue engineering, and tissue substitutes. Although most studies have been performed on elastomeric proteins such flagelliform silk and elastin, the recent and efficient production of resilin-like polypeptides in E.coli has offered access to a class of hydrophilic elastomers with excellent resilience and high frequency responsiveness. These materials may be useful for encapsulating hydrophilic drugs such as proteins, and for providing extracellular matrix-mimetic, cell-adhesive, and enzymatically degradable substrates for tissue engineering applications. However, a lack of detailed understanding of the molecular folding mechanisms of these elastomeric proteins limits the structure-based manipulation of related mechanical properties. Increasing the expression yield by employing large scale fermentation, or using alternative approaches such as expression in higher eukaryotic organisms to permit elaboration of polypeptides would be of significant value. Potential long-term issues for the clinical use of these materials, such as their inflammatory response, endotoxin contamination, and immune response, must be assessed prior to their translation. Nevertheless, continued advances in the characterization of the conformational properties of these materials, their expression, and their processing into biologically-friendly matrices, promises to expand their use in biomaterials applications.Acknowledgements
Related work in the authors' laboratories has been supported by the University of Delaware Research Foundation, the National Science Foundation (DMR 0239744), and the National Center for Research Resources (NCRR), a component of the National Institutes of Health (P20-RR017716, and P20-RR015588 for instrument resources). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of NCRR or NIH.Notes and References
- H. C. Bennetclark and E. C. A. Lucey, J. Exp. Biol., 1967, 47, 59 Search PubMed.
- M. Burrows, S. R. Shaw and G. P. Sutton, BMC Biol., 2008, 6, 41 CrossRef.
- H. C. Bennetclark, J. Exp. Biol., 1975, 63, 53–83 Search PubMed.
- M. E. G. Evans, J. Zool., 1973, 169, 181–194 Search PubMed.
- D. G. Furth, W. Traub and I. Harpaz, J. Exp. Zool., 1983, 227, 43–47 CrossRef.
- J. Norman and T. Desai, Ann. Biomed. Eng., 2006, 34, 89–101 CrossRef.
- W. Ryu, S. W. Min, K. E. Hammerick, M. Vyakarnam, R. S. Greco, F. B. Prinz and R. J. Fasching, Biomaterials, 2007, 28, 1174–1184 CrossRef CAS.
- N. E. Fedorovich, J. Alblas, J. R. de Wijn, W. E. Hennink, A. J. Verbout and W. J. A. Dhert, Tissue Eng., 2007, 13, 1905–1925 CrossRef CAS.
- H. Park, C. Cannizzaro, G. Vunjak-Novakovic, R. Langer, C. A. Vacanti and O. C. Farokhzad, Tissue Eng., 2007, 13, 1867–1877 CrossRef CAS.
- L. A. Smith, X. H. Liu and P. X. Ma, Soft Matter, 2008, 4, 2144–2149 RSC.
- A. Subramanian, U. M. Krishnan and S. Sethuraman, J. Biomed. Sci., 2009, 16, 108 CrossRef.
- J. H. Jang, O. Castano and H. W. Kim, Adv. Drug Delivery Rev., 2009, 61, 1065–1083 CrossRef CAS.
- J. Gosline, M. Lillie, E. Carrington, P. Guerette, C. Ortlepp and K. Savage, Philos. Trans. R. Soc. London, Ser. B, 2002, 357, 121–132 CrossRef CAS.
- D. W. Urry, T. Hugel, M. Seitz, H. E. Gaub, L. Sheiba, J. Dea, J. Xu and T. Parker, Philos. Trans. R. Soc. London, Ser. B, 2002, 357, 169–184 CrossRef CAS.
- Z. Indik, H. Yeh, N. Ornsteingoldstein, P. Sheppard, N. Anderson, J. C. Rosenbloom, L. Peltonen and J. Rosenbloom, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 5680–5684 CrossRef CAS.
- D. W. Urry, J. Protein Chem., 1988, 7, 1–34 CAS.
- D. W. Urry, J. Protein Chem., 1988, 7, 81–114 CrossRef CAS.
- B. Vrhovski and A. S. Weiss, Eur. J. Biochem., 1998, 258, 1–18 CrossRef CAS.
- D. W. Urry, T. Hugel, M. Seitz, H. E. Gaub, L. Sheiba, J. Dea, J. Xu and T. Parker, Philos. Trans. R. Soc. London, Ser. B, 2002, 357, 169–184 CrossRef CAS.
- C. M. Venkatachalam and D. W. Urry, Macromolecules, 1981, 14, 1225–1229 CrossRef CAS.
- A. M. Tamburro, V. Guantieri, L. Pandolfo and A. Scopa, Biopolymers, 1990, 29, 855–870 CrossRef CAS.
- F. Lelj, A. M. Tamburro, V. Villani, P. Grimaldi and V. Guantieri, Biopolymers, 1992, 32, 161–172 CAS.
- B. Bochicchio, A. Pepe and A. M. Tamburro, Chirality, 2008, 20, 985–994 CrossRef CAS.
- M. Kajitani, Y. Wadia, H. Xie, M. T. Hinds, S. W. Shalaby, K. R. Swartz and K. W. Gregory, ASAIO J., 2000, 46, 409–414 CrossRef CAS.
- F. J. Schoen and R. J. Levy, J. Biomed. Mater. Res., 1999, 47, 439–465 CrossRef CAS.
- J. D. Kakisis, C. D. Liapis, C. Breuer and B. E. Sumpio, J. Vasc. Surg., 2005, 41, 349–354 CrossRef.
- G. E. Amiel, M. Komura, O. Shapira, J. J. Yoo, S. Yazdani, J. Berry, S. Kaushal, J. Bischoff, A. Atala and S. Soker, Tissue Eng., 2006, 12, 2355–2365 CrossRef CAS.
- M. T. Hinds, D. W. Courtman, T. Goodell, M. Kwong, H. Brant-Zawadzki, A. Burke, B. A. Fox and K. W. Gregory, J. Biomed. Mater. Res., 2004, 69a, 55–64 Search PubMed.
- W. J. Paule, S. Bernick, B. Strates and M. E. Nimni, J. Biomed. Mater. Res., 1992, 26, 1169–1177 CrossRef CAS.
- M. E. Nimni, D. Myers, D. Ertl and B. Han, J. Biomed. Mater. Res., 1997, 35, 531–537 CrossRef CAS.
- N. Vyavahare, M. Ogle, F. J. Schoen and R. J. Levy, Am. J. Pathol., 1999, 155, 973–982 Search PubMed.
- A. Singla and C. H. Lee, J. Biomed. Mater. Res., 2003, 64a, 706–713 Search PubMed.
- D. T. Simionescu, J. J. Lovekamp and N. R. Vyavahare, J. Heart Valve Dis., 2003, 12, 217–225 Search PubMed.
- A. Kurane, D. T. Simionescu and N. R. Vyavahare, Biomaterials, 2007, 28, 2830–2838 CrossRef CAS.
- M. R. Dreher, A. J. Simnick, K. Fischer, R. J. Smith, A. Patel, M. Schmidt and A. Chilkoti, J. Am. Chem. Soc., 2008, 130, 687–694 CrossRef CAS.
- S. Dublin, Y. Zimenkov and V. P. Conticello, 2009.
- K. S. Straley and S. C. Heilshorn, Soft Matter, 2009, 5, 114–124 RSC.
- W. Kim and V. P. Conticello, Polym. Rev., 2007, 47, 93–119 Search PubMed.
- W. F. Daamen, J. H. Veerkamp, J. C. M. van Hest and T. H. van Kuppevelt, Biomaterials, 2007, 28, 4378–4398 CrossRef CAS.
- D. Chow, M. L. Nunalee, D. W. Lim, A. J. Simnick and A. Chilkoti, Mater. Sci. Eng., R, 2008, 62, 125–155 CrossRef.
- A. J. Simnick, D. W. Lim, D. Chow and A. Chilkoti, Polym. Rev., 2007, 47, 121–154 Search PubMed.
- S. Kyle, A. Aggeli, E. Ingham and M. J. McPherson, Trends Biotechnol., 2009, 27, 423–433 CrossRef CAS.
- A. S. Tatham, L. Hayes, P. R. Shewry and D. W. Urry, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 2001, 1548, 187–193 Search PubMed.
- A. S. Tatham, A. F. Drake and P. R. Shewry, J. Cereal Sci., 1990, 11, 189–200 CrossRef CAS.
- S. M. Gilbert, N. Wellner, P. S. Belton, J. A. Greenfield, G. Siligardi, P. R. Shewry and A. S. Tatham, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 2000, 1479, 135–146 Search PubMed.
- M. A. Bowie, J. D. Layes and M. E. Demont, J. Exp. Biol., 1993, 175, 311–315 Search PubMed.
- Q. P. Cao, Y. J. Wang and H. Bayley, Curr. Biol., 1997, 7, R677–R678 CrossRef CAS.
- Y. Kikuchi and N. Tamiya, J. Biochem., 1981, 89, 1975–1976 CAS.
- A. S. Tatham and P. R. Shewry, Philos. Trans. R. Soc. London, Ser. B, 2002, 357, 229–234 CrossRef CAS.
- B. Bochicchio, F. Jimenez-Oronoz, A. Pepe, M. Blanco, L. B. Sandberg and A. M. Tamburro, Macromol. Biosci., 2005, 5, 502–511 CrossRef CAS.
- J. M. Gosline, M. E. Demont and M. W. Denny, Endeavour, 1986, 10, 37–43 CrossRef.
- F. Vollrath and D. T. Edmonds, Nature, 1989, 340, 305–307 CrossRef.
- M. A. Townley, D. T. Bernstein, K. S. Gallagher and E. K. Tillinghast, J. Exp. Zool., 1991, 259, 154–165 CrossRef.
- C. Y. Hayashi and R. V. Lewis, J. Mol. Biol., 1998, 275, 773–784 CrossRef CAS.
- C. Y. Hayashi, N. H. Shipley and R. V. Lewis, 1999.
- C. Y. Hayashi and R. V. Lewis, Science, 2000, 287, 1477–1479 CrossRef CAS.
- J. E. Garb, T. DiMauro, V. Vo and C. Y. Hayashi, Science, 2006, 312, 1762–1762 CrossRef CAS.
- Y. T. Zhou, S. X. Wu and V. P. Conticello, Biomacromolecules, 2001, 2, 111–125 CrossRef CAS.
- T. Weis-Fogh, J. Exp. Biol., 1960, 37, 889 Search PubMed.
- S. N. Gorb, Naturwissenschaften, 1999, 86, 552–555 CrossRef CAS.
- M. Burrows, Nature, 2003, 424, 509–509 CrossRef CAS.
- M. Rothschild, J. Schlein, K. Parker, C. Neville and S. Sternberg, Philos. Trans. R. Soc. London, Ser. B, 1975, 271, 499 CrossRef CAS.
- S. O. Andersen., in Elastomeric Proteins, ed. T. Shewry P. R., A. S.; Bailey, A. J., ed., Cambridge University Press, Cambridge, Editon edn., 2003, pp. pp259-278 Search PubMed.
- D. Young and H. C. Bennetclark, J. Exp. Biol., 1995, 198, 1001–1019 Search PubMed.
- N. Skals and A. Surlykke, J. Exp. Biol., 1999, 202, 2937–2949 Search PubMed.
- T. Weisfogh, J. Mol. Biol., 1961, 3, 520 CrossRef CAS.
- T. Weisfogh, J. Mol. Biol., 1961, 3, 648 CrossRef CAS.
- D. H. Ardell and S. O. Andersen, Insect Biochem. Mol. Biol., 2001, 31, 965–970 CrossRef CAS.
- K. Bailey and T. Weisfogh, Biochim. Biophys. Acta, 1961, 48, 452 CAS.
- S. P. Antonio, M. Tamburro, Valentina Santopietro, Angelo Bracalello, Brigida Bochicchio and Antonietta Pepe, ChemBioChem, 2009, 11, 83–93.
- K. M. Nairn, R. E. Lyons, R. J. Mulder, S. T. Mudie, D. J. Cookson, E. Lesieur, M. Kim, D. Lau, F. H. Scholes and C. M. Elvin, Biophys. J., 2008, 95, 3358–3365 CrossRef CAS.
- M. T. Krejchi, E. D. T. Atkins, A. J. Waddon, M. J. Fournier, T. L. Mason and D. A. Tirrell, Science, 1994, 265, 1427–1432 CrossRef CAS.
- S. J. M. Yu, V. P. Conticello, G. H. Zhang, C. Kayser, M. J. Fournier, T. L. Mason and D. A. Tirrell, Nature, 1997, 389, 167–170 CrossRef CAS.
- W. A. Petka, J. L. Harden, K. P. McGrath, D. Wirtz and D. A. Tirrell, Science, 1998, 281, 389–392 CrossRef CAS.
- W. Shen, J. A. Kornfield and D. A. Tirrell, Soft Matter, 2007, 3, 99–107 RSC.
- J. P. O'Brien, S. R. Fahnestock, Y. Termonia and K. C. H. Gardner, Adv. Mater., 1998, 10, 1185 CrossRef CAS.
- J. T. Prince, K. P. McGrath, C. M. Digirolamo and D. L. Kaplan, Biochemistry, 1995, 34, 10879–10885 CrossRef CAS.
- S. Halstenberg, A. Panitch, S. Rizzi, H. Hall and J. A. Hubbell, Biomacromolecules, 2002, 3, 710–723 CrossRef CAS.
- L. Zhang, E. M. Furst and K. L. Kiick, J. Controlled Release, 2006, 114, 130–142 CrossRef CAS.
- N. Yamaguchi, L. Zhang, B. S. Chae, C. S. Palla, E. M. Furst and K. L. Kiick, J. Am. Chem. Soc., 2007, 129, 3040 CrossRef CAS.
- K. L. Kiick, Soft Matter, 2008, 4, 29–37 RSC.
- T. Nie, R. E. Akins and K. L. Kiick, Acta Biomater., 2009, 5, 865–875 CrossRef CAS.
- K. Ogiwara, M. Nagaoka, C. S. Cho and T. Akaike, Biotechnol. Lett., 2005, 27, 1633–1637 CrossRef CAS.
- I. Elloumi, R. Kobayashi, H. Funabashi, M. Mie and E. Kobatake, Biomaterials, 2006, 27, 3451–3458 CrossRef CAS.
- D. H. Kim, J. T. Smith, A. Chilkoti and W. M. Reichert, Biomaterials, 2007, 28, 3369–3377 CrossRef CAS.
- J. G. Doheny, E. J. Jervis, M. M. Guarna, R. K. Humphries, R. A. J. Warren and D. G. Kilburn, Biochem. J., 1999, 339, 429–434 CrossRef CAS.
- J. C. Liu, S. C. Heilshorn and D. A. Tirrell, Biomacromolecules, 2004, 5, 497–504 CrossRef CAS.
- S. C. Heilshorn, K. A. DiZio, E. R. Welsh and D. A. Tirrell, Biomaterials, 2003, 24, 4245–4252 CrossRef.
- M. Nagaoka, H. Ise and T. Akaike, Biotechnol. Lett., 2002, 24, 1857–1862 CrossRef CAS.
- D. W. Urry, Methods Enzymol., 1982, 82, 673–716 Search PubMed.
- D. T. McPherson, C. Morrow, D. S. Minehan, J. G. Wu, E. Hunter and D. W. Urry, Biotechnol. Prog., 1992, 8, 347–352 CrossRef CAS.
- D. W. Urry, D. C. Gowda, T. M. Parker, C. H. Luan, M. C. Reid, C. M. Harris, A. Pattanaik and R. D. Harris, Biopolymers, 1992, 32, 1243–1250 CrossRef CAS.
- D. W. Urry, C. H. Luan, T. M. Parker, D. C. Gowda, K. U. Prasad, M. C. Reid and A. Safavy, J. Am. Chem. Soc., 1991, 113, 4346–4348 CrossRef CAS.
- H. Reiersen, A. R. Clarke and A. R. Rees, J. Mol. Biol., 1998, 283, 255–264 CrossRef CAS.
- T. Yamaoka, T. Tamura, Y. Seto, T. Tada, S. Kunugi and D. A. Tirrell, Biomacromolecules, 2003, 4, 1680–1685 CrossRef CAS.
- J. A. Mackay and A. Chilkoti, Int. J. Hyperthermia, 2008, 24, 483–495 CrossRef.
- M. F. Shamji, H. Betre, V. B. Kraus, J. Chen, A. Chilkoti, R. Pichika, K. Masuda and L. A. Setton, Arthritis Rheum., 2007, 56, 3650–3661 CrossRef CAS.
- A. Chilkoti, M. R. Dreher, D. E. Meyer and D. Raucher, Adv. Drug Delivery Rev., 2002, 54, 613–630 CrossRef CAS.
- J. Lee, C. W. Macosko and D. W. Urry, Macromolecules, 2001, 34, 4114–4123 CrossRef CAS.
- K. Nagapudi, W. T. Brinkman, J. Leisen, B. S. Thomas, E. R. Wright, C. Haller, X. Y. Wu, R. P. Apkarian, V. P. Conticello and E. L. Chaikof, Macromolecules, 2005, 38, 345–354 CrossRef CAS.
- E. R. Welsh and D. A. Tirrell, Biomacromolecules, 2000, 1, 23–30 CrossRef CAS.
- P. J. Nowatzki and D. A. Tirrell, Biomaterials, 2004, 25, 1261–1267 CrossRef CAS.
- K. Di Zio and D. A. Tirrell, Macromolecules, 2003, 36, 1553–1558 CrossRef.
- D. W. Lim, D. L. Nettles, L. A. Setton and A. Chilkoti, Biomacromolecules, 2008, 9, 222–230 CrossRef CAS.
- J. F. V. Vincent and U. G. K. Wegst, Arthropod Struct. Dev., 2004, 33, 187–199 CrossRef.
- S. C. Heilshorn, J. C. Liu and D. A. Tirrell, Biomacromolecules, 2005, 6, 318–323 CrossRef CAS.
- D. E. Berning, K. V. Katti, C. L. Barnes and W. A. Volkert, J. Am. Chem. Soc., 1999, 121, 1658–1664 CrossRef CAS.
- D. W. Lim, D. L. Nettles, L. A. Setton and A. Chilkoti, Biomacromolecules, 2007, 8, 1463–1470 CrossRef CAS.
- K. C. Zhang, M. R. Diehl and D. A. Tirrell, J. Am. Chem. Soc., 2005, 127, 10136–10137 CrossRef CAS.
- W. Kim, R. A. McMillan, J. P. Snyder and V. P. Conticello, J. Am. Chem. Soc., 2005, 127, 18121–18132 CrossRef CAS.
- W. Kim, K. I. Hardcastle and V. P. Conticello, Angew. Chem., Int. Ed., 2006, 45, 8141–8145 CrossRef CAS.
- K. Nagapudi, W. T. Brinkman, B. S. Thomas, J. O. Park, M. Srinivasarao, E. Wright, V. P. Conticello and E. L. Chaikof, Biomaterials, 2005, 26, 4695–4706 CrossRef CAS.
- E. R. Wright and V. P. Conticello, Adv. Drug Delivery Rev., 2002, 54, 1057–1073 CrossRef CAS.
- E. R. Wright, V. P. Conticello and R. P. Apkarian, Microsc. Microanal., 2003, 9, 171–182 CrossRef CAS.
- E. R. Wright, R. A. McMillan, A. Cooper, R. P. Apkarian and V. P. Conticello, Adv. Funct. Mater., 2002, 12, 149–154 CrossRef CAS.
- R. L. M. Teeuwen, S. S. van Berkel, T. H. H. van Dulmen, S. Schoffelen, S. A. Meeuwissen, H. Zuilhof, F. A. de Wolf and J. C. M. van Hest, Chem. Commun., 2009, 4022–4024 RSC.
- C. M. Elvin, A. G. Carr, M. G. Huson, J. M. Maxwell, R. D. Pearson, T. Vuocolo, N. E. Liyou, D. C. C. Wong, D. J. Merritt and N. E. Dixon, Nature, 2005, 437, 999–1002 CrossRef CAS.
- M. Kim, C. Elvin, A. Brownlee and R. Lyons, Protein Expression Purif., 2007, 52, 230–236 CrossRef CAS.
- R. E. Lyons, E. Lesieur, M. Kim, D. C. C. Wong, M. G. Huson, K. M. Nairn, A. G. Brownlee, R. D. Pearson and C. M. Elvin, Protein Eng., Des. Sel., 2007, 20, 25–32 CrossRef CAS.
- R. E. Lyons, K. M. Nairn, M. G. Huson, M. Kim, G. Dumsday and C. M. Elvin, Biomacromolecules, 2009, 10, 3009–3014 CrossRef CAS.
- G. K. Qin, S. Lapidot, K. Numata, X. Hu, S. Meirovitch, M. Dekel, I. Podoler, O. Shoseyov and D. L. Kaplan, Biomacromolecules, 2009, 10, 3227–3234 CrossRef CAS.
- W.-F. T. Jensen and M., Philos. Trans. R. Soc. London, Ser. B, 1962, 245, 137–169 CrossRef.
- M. B. Charati, J. L. Ifkovits, J. A. Burdick, J. G. Linhardt and K. L. Kiick, Soft Matter, 2009, 5, 3412–3416 RSC.
- S. C. Rizzi, M. Ehrbar, S. Halstenberg, G. P. Raeber, H. G. Schmoekel, H. Hagenmuller, R. Muller, F. E. Weber and J. A. Hubbell, Biomacromolecules, 2006, 7, 3019–3029 CrossRef CAS.
- U. Hersel, C. Dahmen and H. Kessler, Biomaterials, 2003, 24, 4385–4415 CrossRef CAS.
- H. Nagase and G. B. Fields, Biopolymers, 1996, 40, 399–416 CrossRef CAS.
- M. R. Lutolf, F. E. Weber, H. G. Schmoekel, J. C. Schense, T. Kohler, R. Muller and J. A. Hubbell, Nat. Biotechnol., 2003, 21, 513–518 CrossRef CAS.
- S. H. Kim and K. L. Kiick, Peptides, 2007, 28, 2125–2136 CrossRef CAS.
- T. Nie, A. Baldwin, N. Yamaguchi and K. L. Kiick, J. Controlled Release, 2007, 122, 287–296 CrossRef CAS.
- S. C. Liu, F. Y. Zhou, M. Hook and D. D. Carson, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 1739–1744 CrossRef CAS.
- S. C. Liu, J. Julian and D. D. Carson, J. Biol. Chem., 1998, 273, 9718–9726 CrossRef CAS.
- F. W. Studier, Protein Expression Purif., 2005, 41, 207–234 CrossRef CAS.
Footnote |
| † These authors are equal contributors. |
| This journal is © The Royal Society of Chemistry 2010 |