Linear poly(alkyl ethylene imine) with varying side chain length: synthesis and physical properties
Hanneke M. L.
Lambermont-Thijs
a,
Lies
Bonami
b,
Filip E.
Du Prez
b and
Richard
Hoogenboom
*ac
aLaboratory of Macromolecular Chemistry and Nanoscience, Eindhoven University of Technology, P.O. Box 513, 5600 MB, Eindhoven, The Netherlands
bDepartment of Organic Chemistry, Polymer Chemistry Research Group, Ghent University, Krijgslaan 281 S4 bis, B-9000, Ghent, Belgium
cInstitute for Molecules and Materials, Radboud University Nijmegen, Heyendaalseweg 135, 6525 AJ, Nijmegen, The Netherlands. E-mail: r.hoogenboom@tue.nl
First published on 12th May 2010
Abstract
The synthesis of a series of poly(alkyl ethylene imine) (PalkylEI) with varying side chain lengths of 1, 2, 3 and 10 carbon atoms as well as a benzyl side group was performed. A series of polymers was synthesized from a direct reduction reaction of the corresponding poly(2-oxazoline) while another series was prepared by (reductive) alkylation of linear poly(ethylene imine) (PEI). The thermal and solubility properties of the resulting polymers were investigated and compared to the starting poly(2-oxazoline)s in order to study the effects of removing the carbonyl from the polymer structure. 1H NMR spectroscopy revealed quantitative formation of the PalkylEI, i.e. no residual signals of the poly(2-oxazoline) or linear PEI were observed. Thermal investigations revealed that the thermal transitions, melting and glass transition temperatures, significantly decreased after decarboxylation due to an increase in chain flexibility. Furthermore, solubility measurements indicated that the PalkylEIs are more hydrophobic compared to the corresponding poly(2-oxazoline)s, whereby poly(ethyl ethylene imine) revealed pH and temperature responsive solubility behavior in water.
Introduction
Poly(2-oxazoline)s are widely studied because of the ease and versatility to vary the side group. Methyl and ethyl substituents result in hydrophilic water soluble polymers while longer alkyl side chains as well as aromatic groups result in hydrophobic water insoluble polymers.1,2 Furthermore, the length of the alkyl side group has a significant influence on the thermal and surface properties.3 In literature, a large number of studies discuss structure–property relationships for poly(2-oxazoline)s, in some cases also using systematic variations in monomer composition.4–6 Moreover, the synthesis of poly(2-oxazolines) could be optimized by using single mode microwave reactors7,8 opening the gate to a broader applicability of poly(2-oxazoline)s. Commercially relevant applications of poly(2-oxazoline)s have been documented in numerous patents, including adhesives or coating formulations,9–11 pigment stabilizers in inks12 as well as personal care applications.13–18In contrast to poly(2-oxazoline)s, there are a limited number of studies reported on poly(alkyl ethylene imine)s (PalkylEIs). Because the PalkylEI and poly(2-oxazoline) only differ in molecular structure by the carbonyl group in the side chain, PalkylEIs are an interesting class of polymers for studying structure–property relationships and to evaluate the influence of this carbonyl group. It is not possible to synthesize linear alkylated PEI via the ring opening polymerization of N-alkyl ethylene imines because of chain transfer reactions causing branching. This branching occurs in a similar way as described for the cationic ring opening polymerization of ethylene imine.19,20 For the preparation of linear PalkylEI, two possible routes are known in literature. One route is the alkylation of linear PEI and a second route is the reduction of poly(2-oxazoline)s. Frech et al.21,22 synthesized linear poly(methyl ethylene imine) (PMEI) and linear poly(ethyl ethylene imine) (PEEI) via the alkylation of linear PEI and investigated their conductivities. Additionally, PMEI is also described in literature by Tanaka et al.23,24 who performed the Eschweiler–Clarke reductive N-methylation. The properties of PMEI were reported as viscous oil but when exposed to air it readily absorbed moisture and solidified by hydrate formation. Moreover, linear alkylated PEIs were also described in literature by Saegusa et al.25 who prepared linear PalkylEIs with various side chain lengths via the reduction of poly(2-oxazoline)s. Two possible routes were described utilizing LiAlH4 and AlH3, respectively, but a detailed study regarding the properties of the synthesized linear PalkylEI polymers was not reported in literature. Therefore, the effect of alkyl substitution on the properties of PalkylEI is not known and is expected to differ significantly from the corresponding poly(2-oxazoline)s due to the influence of the carbonyl group.
In this manuscript we report the synthesis of linear PEI with alkyl side chains of varying lengths. Three polymers were synthesized by reduction of poly(2-oxazoline)s, namely poly(2-ethyl-2-oxazoline) (PEtOx), poly(2-nonyl-2-oxazoline) (PNonOx) and poly(2-phenyl-2-oxazoline) (PPhOx). Furthermore, linear poly(methyl ethylene imine) (PMEI), poly(ethyl ethylene imine) (PEEI) and poly(benzyl ethylene imine) (PBnEI) were synthesized by (reductive) alkylation of linear PEI. The thermal and solubility properties of all resulting polymers were investigated and compared to the corresponding poly(2-oxazoline)s to reveal the structure–property relations and investigate the influence of the oxygen in the side chain of the poly(2-oxazoline).
Experimental section
Instrumentation
All chemicals were purchased from Aldrich and NonOx was kindly provided by Henkel. The 2-oxazoline monomers and methyl tosylate were distilled over barium oxide (BaO) and stored under argon. Acetonitrile (ACN, Aldrich) was dried over molecular sieves (3 Å). THF was dried over sodium. Polymerizations were carried out in the single-mode microwave reactor Emrys Liberator (Biotage) with capped reaction vials. These vials were heated to 105 °C, allowed to cool to room temperature, and filled with argon before use. All microwave polymerizations were performed under temperature control (IR sensor).1H NMR spectra were recorded on a Bruker AM500 spectrometer. All measurements were performed in deuterated chloroform. Chemical shifts are given in part per million with respect to tetramethylsilane or residual solvent signals. Size exclusion chromatography (SEC) was measured on a system equipped with a Waters 1515 Isocratic HPLC pump, a Waters 2414 refractive index detector (40 °C), a Waters 2707 autosampler, a PSS PFG guard column followed by 2 PFG-linear-XL (7 µm, 8 × 300 mm) columns in series at 40 °C. Hexafluoroisopropanol (HFIP, Apollo Scientific Limited) with potassium trifluoroacetate (3 g L−1) was used as eluent (flow rate of 0.8 mL min−1) since this eluent could suppress interactions between the PalkylEI and the column material. The molecular weights were calculated against poly(methyl methacrylate) standards (polymer laboratories, Mp = 580 Da up to Mp = 7.1 × 106 Da). Thermal transitions were determined on a DSC Q100 from TA Instruments. Sample weights were in the range from 3 to 10 mg. The samples were measured in closed aluminium cups with a heating and cooling rate of 20 K min−1 for both the glass transition temperatures and melting points (three measurements per sample after an initial first heating run that was not considered for the subsequent calculations).
The solubility measurements were performed by heating the polymer (5.0 mg mL−1) in a solvent mixture of ethanol (Biosolve) and deionized water (Laborpure, Behr Labor Technik) in a tightly closed vial allowing superheating of the solution. The temperature range was −20 °C to 105 °C with heating and cooling ramps of 1 °C min−1. During these controlled heating and cooling cycles (two cycles per sample), the transmission through the solutions was monitored with red light in a Crystal 16™ from Avantium Technologies.26 Four blocks of four parallel temperature controlled sample holders are connected to a Julabo FP40 cryostat allowing 16 simultaneous measurements. All vials were visually inspected after the heating program to facilitate the interpretation of the observed transmission profiles. The presented cloud point temperatures correspond to the dissolution temperature at 50% transmittance from the second heating run.
Synthesis of poly(2-oxazoline)s
The synthesis of the poly(2-oxazoline)s was performed as previously described.27 The polymerization mixtures for the microwave-assisted synthesis of poly(2-methyl-2-oxazoline) (PMeOx), poly(2-ethyl-2-oxazoline) (PEtOx), poly(2-phenyl-2-oxazoline) (PPhOx) and poly(2-nonyl-2-oxazoline) (PNonOx) homopolymers were prepared by weighing all reagents under an argon atmosphere. In each vial the corresponding 2-oxazoline monomer, methyl tosylate and acetonitrile were added, resulting in a total monomer concentration of 4 M and a monomer to initiator ratio of 100. The microwave vials were subsequently heated to 140 °C to reach full conversion. The polymerization mixtures were quenched after microwave heating by the automated addition of water. The resulting homopolymers were dried under reduced pressure at 40 °C to remove the solvent and residual monomer. All synthesized polymers were subsequently analyzed by SEC to reveal the molecular weight distribution and polydispersity index. Further characterization was performed by thermal and solubility measurements to determine selected polymer properties.Synthesis of PPEI, PBnEI and PDeEI via (reductive) alkylation
The synthesis of linear poly(propyl ethylene imine) (PPEI), linear poly(benzyl ethylene imine) (PBnEI) and linear poly(decyl ethylene imine) (PDeEI) was performed starting from the corresponding poly(2-oxazoline)s, namely PEtOx, PPhOx and PNonOx, respectively. The procedure is described in detail for the reduction of PEtOx to PPEI and is performed in a similar manner for PPhOx and PNonOx.The reduction of the amide was performed as described by Perner and Schulz.28 Two grams of PEtOx were dissolved in 60 mL dry tetrahydrofuran (THF) under nitrogen atmosphere, to which a solution of 25 mL of a 2 M borane/dimethylsulfide (BH3/DMS) solution in THF was slowly added. The resulting solution was allowed to reflux for 32 hours. After cooling to ambient temperature, THF and DMS were removed under reduced pressure. To destroy the excess borane and borane amine complex formed during the reduction, 20 mL of 6 N HCl were added and the mixture was refluxed for 2 hours. After cooling the reaction mixture to ambient temperature, NaOH was added until pH > 8 and the polymer was extracted with CH2Cl2. The resulting organic phase was dried with NaSO4, filtered and the remaining solvent was evaporated under reduced pressure (for a schematic representation of the synthesis route, see A in Fig. 1). The final products were characterized by 1H NMR spectroscopy and SEC.
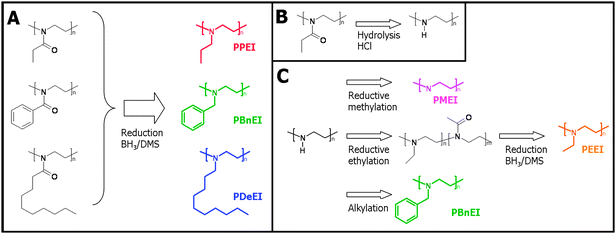 | ||
| Fig. 1 Schematic representation of the different synthesis routes to linear PalkylEI with varying side chain lengths. | ||
1H NMR PPEI (500 MHz, CDCl3): δ = 0.83 (-NCH2CH2CH3), δ = 1.43 (-NCH2CH2CH3), δ = 2.38 (-NCH2CH2CH3), δ = 2.51 (-NCH2CH2N-).
13C NMR PPEI (300 MHz, CDCl3): δ = 11.91 (-NCH2CH2CH3), δ = 20.39 (-NCH2CH2CH3), δ = 53.03 (-NCH2CH2CH3), δ = 57.30 (-NCH2CH2N-).
1H NMR PBnEI (500 MHz, CDCl3): δ = 2.37 (-NCH2CH2N-), δ = 3.38 (-NCH2C6H5), δ = 7.13–7.19 (-NCH2C6H5).
13C NMR PBnEI (300 MHz, CDCl3): δ = 52.44 (-NCH2Arom), δ = 59.30 (-NCH2CH2N-), δ = 126.69, 128.05, 128.69 and 139.75 (aromatic).
1H NMR PDeEI (500 MHz, CDCl3): δ = 0.87 (-NCH2(CH2)8CH3), δ = 1.25 (-NCH2(CH2)8CH3), δ = 2.42–2.80 (-NCH2CH2N- + -NCH2(CH2)8CH3).
13C NMR PDePEI (300 MHz, CDCl3): δ = 14.12 (-NCH2(CH2)8CH3), δ = 22.70, 27.23, 27.62, 29.39, 29.66, 29.72, 29.75, 31.94 (-NCH2(CH2)8CH3), δ = 53.01 (-NCH2CH2CH3), δ = 55.31 (-NCH2CH2N-).
Synthesis of linear PEI
Linear PEI was synthesized starting from 5.7 g poly(2-ethyl-2-oxazoline), which was subsequently hydrolyzed in 70 mL of concentrated (35%) HCl and 50 mL of water at 100 °C for 48 hours.29 The excess of HCl was removed under reduced pressure and the remaining solid was dissolved in 50 mL of water. The resulting mixture was made alkaline by adding aqueous NaOH and the formed white precipitate was filtrated and washed with water until neutral. The solid was recrystallized from water, isolated by filtration and finally lyophilized (for the schematic representation, see B in Fig. 1).1H NMR PEI (500 MHz, CDCl3): δ = 1.92 (-NH(CH2CH2)-), δ = 2.72 (-NH(CH2CH2)-).
13C NMR PEI (400 MHz, CDCl3): δ = 49.5 (-NCH2CH2N-).
Synthesis of PBnEI via the alkylation of PEI
Linear PEI was alkylated by nucleophilic substitution with benzyl bromide to obtain PBnEI. Therefore, 1 g of linear PEI was dissolved in 100 mL of ethanol. Subsequently, 4 g of benzyl bromide and 6.9 g of potassium carbonate (K2CO3) were added to the solution that was allowed to reflux for 16 hours. After cooling to room temperature, 100 mL of water were added and the ethanol was removed under reduced pressure. The product was finally extracted with CH2Cl2 from the water layer and dried with MgSO4. The remaining DCM was removed under reduced pressure. The characterization was performed with 1H NMR spectroscopy and SEC.1H NMR PBnEI (500 MHz, CDCl3): δ = 2.37 (-NCH2CH2N-), δ = 3.38 (-NCH2C6H5), δ = 7.13–7.19 (-NCH2C6H5).
13C NMR PBnEI (300 MHz, CDCl3): δ = 52.45 (-NCH2Arom), δ = 59.31 (-NCH2CH2N-), δ = 126.68, 128.05, 128.68 and 139.76 (aromatic).
Synthesis of PEEI
Linear poly(ethyl ethylene imine) (PEEI) was synthesized starting from linear PEI because it was found that PMeOx is not sufficiently soluble in any solvent suitable for the reduction reaction. The first step towards PEEI was performed by reductive alkylation as described by Gribble et al.,30 1.815 g of linear PEI was dissolved in 75 mL acetic acid and stirred at 55 °C under nitrogen atmosphere. During a period of 1 hour, 7.3 g sodium borohydride (NaBH4) were slowly added and the solution was allowed to react for 20 hours. After cooling to ambient temperature, the solution was diluted with 70 mL water and NaOH was added until pH > 8. The polymer was subsequently extracted with CH2Cl2 and the resulting organic phase was washed with saturated NaHCO3 solution and dried with Na2SO4. After filtration, CH2Cl2 was removed under reduced pressure and the resulting viscous product was further dried under reduced pressure. However, the obtained polymer, which is soluble in THF, still contained ∼50% amide units and, thus, further reduction was performed by the method described for PPEI (for the schematic representation see C in Fig. 1). It was found that during this method, the polymer was not easily extracted with CH2Cl2 from the alkaline solution, therefore the water phase was removed under reduced pressure and CHCl3 was added to extract the polymer from the remaining solid. The solution was filtered to remove the NaOH and dried over Na2SO4. To obtain the final polymer, CHCl3 was removed under reduced pressure. The presence of the N-ethyl group and absence of the acetamino-group were confirmed by 1H NMR, 13C NMR, COSY, HSQC and HMBC NMR spectroscopy.1H NMR PEEI (500 MHz, CDCl3): δ = 1.00 (-NCH2CH3), δ = 2.52 (-NCH2CH2N- + -NCH2CH3).
13C NMR PEEI (300 MHz, CDCl3): δ = 10.79 (-NCH2CH3), δ = 47.61 (-NCH2CH3), δ = 51.36 (-NCH2CH2N-).
Synthesis of PMEI
PMEI was prepared by reductive methylation of PEI as described by Tanaka et al.23 Therefore, 2.5 g of linear PEI were dissolved in 50 mL water at 70 °C to which 63 mL of formaldehyde and 90 mL of formic acid were added. The resulting solution was heated to 105 °C for 24 hours. After cooling down to ambient temperature, 100 mL of 12 M HCl solution were added. The excess of formic acid, formaldehyde and HCl was removed under reduced pressure and the remaining product was dissolved in water. Aqueous NaOH was added until pH > 8 and the product was extracted with CHCl3. The organic phase was evaporated and the product was dried under reduced pressure. The characterization was performed with 1H NMR spectroscopy and SEC.1H NMR PMEI (500 MHz, CDCl3): δ = 2.26 (-NCH3), δ = 2.52 (-NCH2CH2N-).
13C NMR PMEI (300 MHz, CDCl3): δ = 42.18 (-NCH3), δ = 54.02 (-NCH2CH2N-).
Results and discussion
The synthesis of a series of PalkylEI was performed to investigate structure–property relations as well as to study the influence of the carbonyl group on the properties of the polymers by comparison with the corresponding poly(2-oxazoline)s. Here we report the synthesis and properties of a series of PalkylEI polymers with varying side chain lengths of 1, 2, 3 and 10 carbon atoms as well as a benzyl side group. Three polymers were synthesized from a direct reduction reaction of the poly(2-oxazoline) while three polymers were synthesized via (reductive) alkylation of linear PEI. The thermal and solubility properties of the resulting polymers were investigated and compared to the corresponding poly(2-oxazoline)s to study the effects of the change in polymer structure.Synthesis of PalkylEI polymers
PalkylEI can be synthesized by alkylation of PEI via nucleophilic substitution of the corresponding alkylhalide. However, this reaction might lead to overalkylation resulting in a fraction of quaternary amine groups. Therefore, a series of PalkylEI polymers was synthesized by using one of the routes shown in Fig. 1. The first route involves direct reduction of poly(2-oxazoline)s resulting in linear PalkylEI (route A in Fig. 1). A second route was investigated starting from linear PEI, which was synthesized according to route B (Fig. 1) and subsequent (reductive) alkylation reactions (route C in Fig. 1).The direct reduction of PEtOx, PPhOx and PNonOx could be performed in THF with BH3/DMS. After 32 hours, the reduction was stopped and a purification step with HCl was performed to neutralize the excess of borane and destroy the borane amine complex to obtain the free tertiary amine. To prove complete reduction of the poly(2-oxazoline) to PalkylEI, 1H NMR spectroscopy was performed. Fig. 2 depicts the 1H NMR spectra of PEtOx and the corresponding synthesized PPEI. As can be observed from these two spectra, the signals corresponding to the PEtOx backbone, as well as the signals belonging to the CH2 and CH3 group of PEtOx side chain are shifting upfield and an additional peak of the CH2 group connected to the nitrogen is appearing at 2.4 ppm. The sharpening of the polymer signals upon reduction of the amide groups can be ascribed to the increased flexibility of the polymers and increased rotational freedom of the side chains resulting from the conversion of rigid amide groups into tertiary amines. For all three polymers, the 1H NMR spectra indicated a complete reduction, i.e. no residual signals of the poly(2-oxazoline)s were observed (not shown). SEC measurements were performed in hexafluoroisopropanol (HFIP) to suppress column interactions, except for PNonOx, which was not soluble in this particular solvent. The SEC chromatograms of the three polymers synthesized via route A are depicted in Fig. 3 (left) and the molecular weights as well as the polydispersity indices are listed in Table 1.
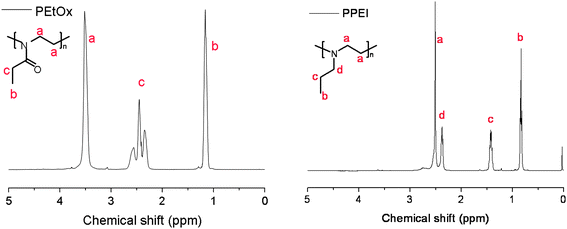 | ||
| Fig. 2 1H NMR spectra for PEtOx (left) and PPEI (right) indicating quantitative conversion after the reduction reaction. | ||
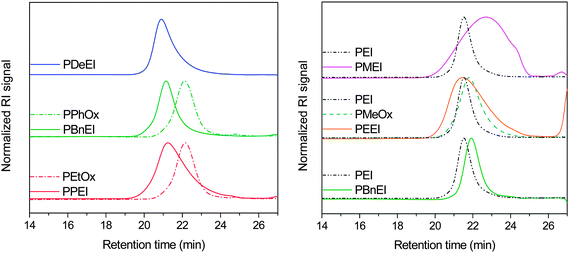 | ||
| Fig. 3 Left: SEC chromatograms for the poly(2-oxazoline)s and poly(alkyl ethylene imine)s from direct reduction (PNonOx was insoluble in the SEC eluent hexafluoroisopropanol). Right: SEC chromatograms for PEEI, PMEI and PBnEI prepared by alkylation compared to linear PEI and PMeOx. | ||
| M n,th/Da | M n,SEC/Da | PDI | T g/°C | ΔCp/J g−1 °C−1 | T m/°C | ΔH/J g−1 | |
|---|---|---|---|---|---|---|---|
| a M n and PDI determined by SEC with DMA/LiCl as eluent. b Highest peak in spectra is depicted as melting temperature. c PBnEI synthesized via route A. d PBnEI synthesized via route C. | |||||||
| Linear PEI | 4300 | 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
1.14 | 69 | 151.1 | ||
| PMEI | 5700 | 8100 | 1.70 | 29 | 0.39 | ||
| PMeOx | 8500 | 13800 |
1.27 | 87 | 0.45 | ||
| PEEI | 7100 | 12700 |
1.58 | −32 | |||
| PEtOx | 9900 | 11400 |
1.28 | 60 | 0.48 | ||
| PPEI | 8500 | 15500 |
1.53 | ||||
| PNonOx | 19700 |
15100a |
1.21a | 148 | 40.4 | ||
| PDeEI | 18300 |
21400 |
1.32 | −24b | |||
| PPhOx | 14700 |
13100 |
1.16 | 114 | 0.37 | ||
| PBnEIc | 13300 |
20600 |
1.31 | −18 | 0.46 | ||
| PBnEId | 13300 |
13600 |
1.17 | −14 | 0.44 |
In general, the experimental molecular weight increased after the reduction reaction. Since the removal of the carbonyl group decreases the molecular weight, these results indicate that the hydrodynamic volume of the poly(2-oxazoline) in HFIP is smaller compared to the PalkylEI, which might be related to the larger basicity of the tertiary amines compared to the amide groups resulting in stronger hydrogen bonding with HFIP, presumably resulting in a larger hydrodynamic volume. Furthermore, the shape of the SEC chromatograms for PPEI and PBnEI is similar to PEtOx and PPhOx, respectively, indicating that the backbone remains intact during the reduction. It should be mentioned, however, that the trace for PPEI is slightly broadened (tailing at higher elution times) compared to the starting PEtOx, which is probably caused by interactions between the tertiary amine groups with the column. The molecular weight (Mn) values from SEC, depicted in Table 1, are higher than the theoretical values since all synthesized polymers have a DP of 100, most likely due to the used calibration (PMMA standards) as was previously demonstrated for PEtOx by comparison with MALDI-TOF MS.31 MALDI-TOF MS of the synthesized PalkylEI derivatives only revealed low molar mass species (below 1000 g mol−1) indicating that ionization and desorption of the polymers were unsuccessful. Nonetheless, the expected mass spacing could be identified together with minor additional distributions separated by 14 g mol−1 resulting from incomplete reduction. However, no quantitative conclusions can be drawn from these results due to the partial ionization of the polymer samples.
The synthesis of PEEI was performed starting from linear PEI due to the insolubility of PMeOx in any suitable solvent for the reduction reaction (route C, Fig. 1). Linear PEI was prepared as described in literature by acidic hydrolysis of PEtOx (route B, Fig. 1).23,29 Subsequently, linear PEI was dissolved in acetic acid and NaBH4 was slowly added in a period of 1 hour. After the reaction, about half of the secondary amines were modified to ethylated monomer units, but some acetamido-groups were introduced as well. In a second step the reduction was completed utilizing BH3/DMS as reducing agent, facilitated by the solubility of the obtained polymer in THF. 1H NMR spectroscopy revealed a qualitative conversion to PEEI (1H NMR spectrum not shown). Further characterization was performed utilizing 13C NMR, COSY, and HMBC NMR spectroscopy, proving the absence of any acetamido-groups (results not shown). Moreover, to complete the series of PalkylEI, poly(methyl ethylene imine) (PMEI) was synthesized. A known method, namely the reductive methylation to linear PMEI with formaldehyde and formic acid23 was utilized with linear PEI as starting product. The polymer was purified by extraction with CH2Cl2 and characterization was performed with 1H NMR spectroscopy revealing complete conversion.
Finally, PBnEI was also synthesized starting from linear PEI by benzylation with benzyl bromide in ethanol. The purification of this polymer was simply performed by removal of the solvent and excess benzyl bromide under reduced pressure. Subsequent 1H NMR spectroscopy revealed the PBnEI structure and no remaining PEI signals. For the three polymers synthesized via route C (Fig. 1), SEC measurements were performed in HFIP as well (Fig. 3, right). The linear PEI has a narrower distribution compared to PMEI and PEEI, which is most likely due to interactions with the column material, which was also observed for PPEI. The shorter alkyl side chains seem to enhance the interactions with the column material even more due to a larger relative fraction of the interacting tertiary amine groups. Moreover, the molecular weight of PBnEI synthesized according to route C appears to have a lower molecular weight compared to the PBnEI synthesized according to route A. This might be due to the occurrence of unidentified side reactions that affect the hydrodynamic volume of the polymer, such as minor quaternization of PBnEI synthesized via route C due to the use of an excess of benzyl bromide. However, no signals indicative of side reactions were observed by 1H NMR spectroscopy obstructing a definite explanation for the different observed molar masses.
Thermal properties of the PalkylEI polymers
Selected thermal and solubility properties of the linear PalkylEI polymers were investigated and the results are compared to the corresponding poly(2-oxazoline)s. It is known from literature that only PNonOx is partially crystalline as a result of the crystallization of the long side chains, the reported melting temperature being 147 °C.31,32 The other three poly(2-oxazoline)s PMeOx, PEtOx and PPhOx are amorphous and exhibit glass transition temperatures of 83 °C,31,33 60 °C31,34 and 114 °C,35,31 respectively, which are close to literature values. Fig. 4 depicts the DSC traces of all polymers except PEtOx (Tg = 60 °C) and the corresponding PPEI for which no Tg or Tm could be detected. Tg and Tm values are listed in Table 1.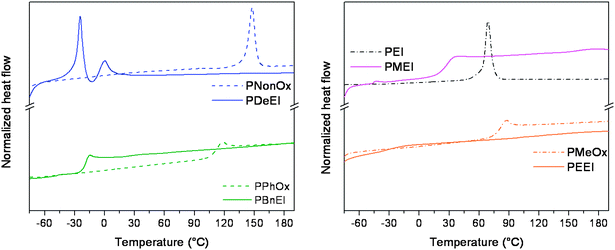 | ||
| Fig. 4 DSC heating curves obtained for the synthesized polymers. Left: PNonOx, PDeEI, PPhOx and PBnEI. Right: linear PEI, PMEI, PMeOx and PEEI (heating rate 20 °C min−1). | ||
As can be observed from Fig. 4 and Table 1, PNonOx exhibits a melting temperature of 148 °C, which is in agreement with earlier reported literature data.32,31 PDeEI also exhibits a melting temperature but this temperature decreased significantly to −24 °C. This tremendous decrease in melting temperature is due to the removal of the carbonyl group in the side chain causing an increase in the chain flexibility, i.e. the rigid amide groups are converted into freely rotating tertiary amines. In addition, retaining the crystallinity in PDeEI indirectly proves that the crystallinity of both PNonOx and PDeEI is side-chain crystallinity. Two melting transitions are observed for PDeEI, which is most likely caused by reorganization during melting. Furthermore, PPhOx and PBnEI both show a glass transition temperature as indicated in Fig. 4. As can be observed, the Tg is decreasing significantly from 113 °C to −18 °C for PBnEI and, as a result, the properties of the polymers are significantly different. PPhOx is a brittle solid polymer at room temperature while PBnEI is a viscous, sticky wax. Moreover, the Tg values of PBnEI prepared via routes A and C (Table 1) were in good agreement, indicating a similar polymer structure. The last PalkylEI, PPEI, which was synthesized according to route A, does not show a glass transition or melting temperature in DSC in the investigated temperature range from −80 to 200 °C. This might be caused by a transition that is too small to detect with DSC or by a glass transition temperature below −80 °C. In the second graph of Fig. 4 the thermal properties of PMEI and PEEI are depicted and compared to linear PEI and PMeOx. Linear PEI has a clear melting temperature of 69 °C (peak maximum), which is also known from literature,36 and no Tg could be detected. Due to the absence of any side groups, and the presence of both hydrogen bond donating and accepting groups, linear PEI has a strong tendency to crystallize. On the other hand, PMEI having a short alkyl side chain and no hydrogen bond donating groups was found to be amorphous with a Tg of 28.6 °C. PMEI with a DP of 2000 was reported in literature before and a Tg of −91 °C was determined.21,22 PMEI with a DP of 5000 was also reported by Tanaka et al. and they reported a Tg of −83 °C.23 Furthermore, PEEI with a DP of 2000 was also reported before with a Tg of −80 °C,21 while the linear PEEI with a DP of 100 synthesized within this report does not reveal a clear thermal transition in the measured temperature range. As can be observed from Fig. 4, a small step in the heat flow is observed at −32 °C but it does not show a sharp glass transition temperature as is observed for PMEI. The obtained differences in glass transition temperature between the literature and this work might be due to the possible formation of hydrates by the highly hygroscopic PMEI and PEEI.
From previous studies on linear poly(2-oxazoline)s with varying numbers of carbon atoms in the side chain, it is known that the glass transition temperature is decreasing linearly with increasing number of carbon atoms.3 This could be explained by the increased flexibility of the longer side chains. Also for PalkylEI, a decrease in Tg was expected in comparison to the poly(2-oxazoline) series. However, it was not anticipated that the removal of the carbonyl group would decrease the thermal transition temperatures with more than 100 °C for both amorphous and semi-crystalline polymers.
Solubility properties of the PalkylEI polymers
The solubility properties of the PalkylEIs were investigated in water and water/ethanol mixtures, which is an interesting low-toxicity solvent mixture. It is known that poly(2-oxazoline)s with a short side chain like methyl and ethyl are hydrophilic polymers while polymers with larger aliphatic or aromatic side chains show hydrophobic properties.1,2,37 Comparing these properties to the alkylated PEIs revealed a similar effect: PBnEI and PDeEI are not soluble in water or any water/ethanol mixture. Even though PPhOx is soluble in 80% EtOH solution38 due to hydration of the amide groups, PBnEI is not soluble in any water/ethanol mixture due to the increased hydrophobicity by removal of the carbonyl group in the side chain. A similar effect is observed for PDeEI and PNonOx. The latter is soluble in ethanol after heating to melt the polymer crystals37 while the increased hydrophobicity of PDeEI makes it insoluble in ethanol at room temperature and at elevated temperatures despite the lower melting temperature. The other polymers, namely PPEI, PEEI and PMEI, have a much shorter alkyl side chain and, therefore, exhibit different solubility properties in water or water/ethanol mixtures. The polymer with the shortest alkyl side chain, PMEI, is readily soluble in water and all water/ethanol mixtures at the investigated temperatures. Apparently, the amine groups are easily accessible to form hydrogen bonds with the water or ethanol resulting in dissolution of the polymer. The polymer with two carbon atoms in the side chain, PEEI, exhibits a LCST transition in water (Fig. 5).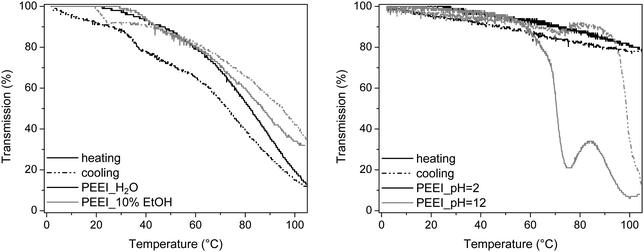 | ||
| Fig. 5 Transmittance versus temperature plots upon heating and cooling for PEEI in H2O, pH 2 and 12 and in 10 wt% EtOH solution at 5 mg mL−1. | ||
Below the cloud point, the polymer chains are soluble in water and exist in random coil formation because of the hydrogen bonding interactions between the polymer and water. However, when the temperature exceeds the cloud point (indicative for the LCST temperature), the interactions are broken and the polymer chains collapse and precipitate. From previous investigations it is known that PEtOx with a DP of 100 exhibits a cloud point temperature in water (5 mg mL−1) at 94 °C39 and by increasing the alkyl side chain to PPropOx, the cloud point at 5 mg mL−1 was decreased to 25 °C.39 Therefore, the hydrophobicity of the polymer has a large effect on the cloud point. This effect can also be observed for PPEI and PEEI, namely, PEEI exhibits a cloud point temperature in water at 81.7 °C and 87.0 °C in a 10 wt% EtOH solution while PPEI is insoluble in water and water with 10 wt% ethanol. The cloud points were determined at 5 mg mL−1 concentration from the transmission plots at 50% transmittance upon heating in the second heating run. Due to the increased hydrophobicity by removing the oxygen atom in the side chain, one side chain carbon atom less is sufficient to observe a cloud point when comparing the PalkylEI to the poly(2-oxazoline)s. Moreover, due to the improved solubility upon addition of ethanol, the cloud point temperature for PEEI is increasing in a 10 wt% ethanol in water solution. Further increase of the amount of ethanol results in complete solubility of PEEI over the measured temperature range. However, it should be noted that the LCST transition of PEEI is a broad transition compared to PEtOx or PPropOx. This can be understood by partial protonation of the PEEI, which significantly influences the solubility. In contrast, the amide groups in poly(2-oxazoline) are much less basic and will not be protonated. The LCST temperature of PEEI was therefore also investigated in acidic and basic conditions and the transitions are shown in Fig. 5, right.
From these results it can be observed that PEEI in acidic solution (pH 2) is soluble over the complete temperature range while PEEI in basic solution (pH 12) exhibits a much sharper transition compared to demineralized water (H2O in Fig. 5, left). Therefore, it can be concluded that the degree of protonation is of utmost importance for the solubility of PEEI. The transmission profile of the basic solution shows an unexpected increase in transmission at a temperature of 90 °C. By visual inspection it could be observed that the solution indeed becomes more clear and individual particles were visible, which might be due to the initial formation of large hydrated aggregates that further collapse upon increasing the temperature followed by the formation of larger aggregates. Furthermore, the solubility transition for heating and cooling of PEEI in basic medium shows a strong hysteresis and differs several tens of degrees in temperature, which might be related to hydrate formation.
Additional solubility measurements were performed with PPEI in water/ethanol mixtures (Fig. 6). In fact, PPEI is not soluble in water and only soluble in water/ethanol solutions with an ethanol content of at least 50 wt% EtOH. When increasing the temperature, a LCST transition was observed for the solutions containing 50, 60 and 70 wt% EtOH (Fig. 6). As can be observed, the LCST transition is sharp compared to PEEI, which is probably caused by a low protonation degree in ethanol/water. Furthermore, the transitions upon heating and cooling overlap within a few degrees indicating the absence of a hysteresis. The LCST temperature is increasing from 5.7 °C for the solution containing 50 wt% EtOH to 33.9 °C and 77.3 °C for the solutions containing 60 and 70 wt% EtOH respectively. The cloud point is increasing when adding ethanol to the aqueous solution due to a better solvation of the PPEI. When further increasing the amount of ethanol in the solution, the polymer becomes completely soluble in the range from −20 to 105 °C.
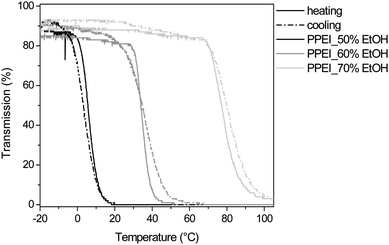 | ||
| Fig. 6 Transmittance versus temperature plots upon heating and cooling for PPEI in 50, 60 and 70 wt% EtOH solution at 5 mg mL−1. | ||
Conclusions
The synthesis of a series of PalkylEI was performed to investigate structure–property relations as well as to study the influence of the carbonyl group on the properties of the polymers. Such changes in the side chain between the poly(2-oxazoline) and the corresponding PalkylEI were not reported in detail before. The synthesis of a series of PalkylEI polymers with varying side chain lengths of 1, 2, 3 and 10 carbon atoms as well as a benzyl side group was performed via both direct reduction as well as (reductive) alkylation. From the four corresponding poly(2-oxazoline)s, only PNonOx is partially crystalline and exhibits a melting temperature at 150 °C. The corresponding PDeEI also exhibits a melting temperature but this temperature decreased significantly to −24 °C due to an increase in chain flexibility by the removal of the carbonyl group. A similar behavior was obtained for PBnEI and PMEI, i.e. the Tg of PPhOx was decreased from 113 °C to −18 °C for PBnEI. As such, removing the carbonyl group was found to lower the thermal transitions more than 100 °C.Solubility investigations revealed that PalkylEIs were more hydrophobic compared to the poly(2-oxazoline)s. For example, PBnEI and PDeEI are not soluble in water or any water/ethanol mixtures while the corresponding PPhOx and PNonOx are soluble in 80% EtOH or pure EtOH. Furthermore, PEEI exhibits a LCST transition in water and in 10 wt% EtOH while PPEI exhibits a LCST transition in water/ethanol solutions containing 50, 60 and 70 wt% EtOH. Interestingly, PEEI was found to be both pH and temperature responsive in water based on the basic tertiary amine groups while the solubility of the poly(2-oxazoline)s is not significantly changed by variation of the pH.
Acknowledgements
RH is grateful to NWO (Veni-grant) for financial support. Ing. Martin Fijten is acknowledged for the SEC measurements. F. E. Du Prez would like to thank the Belgian Program on Interuniversity Attraction Poles Initiated by the Belgian State, Prime Minister's office (Program P6/27) for the financial support.Notes and references
- S. Kobayashi, T. Igarashi, Y. Moriuchi and T. Saegusa, Macromolecules, 1986, 19, 535–541 CrossRef CAS.
- R. Jin, Adv. Mater., 2002, 14, 889–892 CrossRef CAS.
- R. Hoogenboom, M. W. M. Fijten, H. M. L. Thijs, B. M. van Lankvelt and U. S. Schubert, Des. Monomers Polym., 2005, 8, 659–671 Search PubMed.
- R. Hoogenboom, Macromol. Chem. Phys., 2007, 208, 18–25 CrossRef CAS.
- K. Aoi and M. Okada, Prog. Polym. Sci., 1996, 21, 151–208 CrossRef CAS.
- R. Hoogenboom, Angew. Chem., Int. Ed., 2009, 48, 7978–7994 CrossRef CAS.
- F. Wiesbrock, R. Hoogenboom, C. H. Abeln and U. S. Schubert, Macromol. Rapid Commun., 2004, 25, 1895–1899 CrossRef CAS.
- S. Sinnwell and H. Ritter, Macromol. Rapid Commun., 2005, 26, 160–163 CrossRef CAS.
-
J. F. Warchol and C. D. Walton, US Pat., 97–795911, 1998.
-
J. M. J. Frechet and K. Yui, US Pat., 96–660684, 1997.
-
R. H. G. Brinkhuis, WO Pat., 2002–EP13898, 2003.
-
S. H. Ma and J. N. Rodriguez-Parada, US Pat., 97–963839, 1998.
-
Y. Yoda, JP Pat., 04041600, 1992.
-
J. L. Herz, A. J. Levy, M. H. Litt and J. L. Zuckerman, US Pat., 3579630, 1969.
-
J. F. Grollier and C. Dubief, US Pat., 4867966, 1988.
-
J. Conradi, U. Eicken, W. Emmerling, A. Niemann and L. Schieferstein, WO Pat., 95/21602, 1995.
-
C. T. Morrissey, E. D. Smith, S. Midha and T. R. Nijakowski, US Pat., 6106820, 2000.
-
N. Hase and M. Ito, JP Pat., 10139650, 1998.
- S. Kobayashi, Prog. Polym. Sci., 1990, 15, 751–823 CrossRef CAS.
- D. A. Tomalia and G. R. Killat, Encycl. Polym. Sci. Eng., 1985, 1, 680–739 Search PubMed.
- R. Frech, G. A. Giffin, F. Y. Castillo, D. T. Glatzhofer and J. Eisenblätter, Electrochim. Acta, 2005, 50, 3963–3968 CrossRef CAS.
- R. A. Sanders, A. G. Snow, R. Frech and D. T. Glatzhofer, Electrochim. Acta, 2003, 48, 2247–2253 CrossRef CAS.
- R. Tanaka, I. Ueoka, Y. Takaki, K. Kataoka and S. Saito, Macromolecules, 1983, 16, 849–853 CrossRef CAS.
- R. Tanaka, M. Koike, T. Tsutsui and T. Tanaka, J. Polym. Sci., Polym. Lett. Ed., 1978, 16, 13–19 CrossRef CAS.
- T. Saegusa, A. Yamada, H. Taoda and S. Kobayashi, Macromolecules, 1978, 11, 435–436 CrossRef CAS.
- M. Birch, S. J. Fussel, P. D. Higginson, N. McDowall and I. Marziano, Org. Process Res. Dev., 2005, 9, 360–364 Search PubMed.
- F. Wiesbrock, R. Hoogenboom, M. A. M. Leenen, M. A. R. Meier and U. S. Schubert, Macromolecules, 2005, 38, 5025–5034 CrossRef CAS.
- T. Perner and R. C. Schulz, Br. Polym. J., 1987, 19, 181–188 CAS.
- H. M. L. Lambermont-Thijs, F. S. van der Woerdt, A. Baumgaertel, L. Bonami, F. E. Du Prez, U. S. Schubert and R. Hoogenboom, Macromolecules, 2010, 43, 927–933 CrossRef CAS.
- G. W. Gribble, J. M. Jasinski, J. T. Pellicone and J. A. Panetta, Synthesis, 1978, 766–768 CrossRef CAS.
- F. Wiesbrock, R. Hoogenboom, M. Leenen, S. F. G. M. van Nispen, M. van der Loop, C. H. Abeln, A. M. J. van den Berg and U. S. Schubert, Macromolecules, 2005, 38, 7957–7966 CrossRef CAS.
- M. Beck, P. Birnbrich, U. Eicken, H. Fischer, W. E. Fristad, B. Hase and H. J. Krause, Angew. Makromol. Chem., 1994, 223, 217–233 CrossRef CAS.
- R. H. Jin, J. Mater. Chem., 2003, 13, 672–675 RSC.
- J. Y. Kim, S. U. Hong, J. Won and Y. S. Kang, Macromolecules, 2000, 33, 3161–3165 CrossRef CAS.
- T. G. Bassiri, A. Levy and M. Litt, J. Polym. Sci., Part B: Polym. Lett., 1967, 5, 871–879 CrossRef.
- T. Saegusa, H. Ikeda and H. Fujii, Macromolecules, 1972, 5, 108 CrossRef CAS.
- H. M. L. Lambermont-Thijs, R. Hoogenboom, C. A. Fustin, C. Bomal-D'Haese, J. F. Gohy and U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 515–522 CrossRef CAS.
- R. Hoogenboom, H. M. L. Thijs, D. Wouters, S. Hoeppener and U. S. Schubert, Soft Matter, 2008, 4, 103–107 RSC.
- R. Hoogenboom, H. M. L. Thijs, M. J. H. C. Jochems, B. M. van Lankvelt, M. W. M. Fijten and U. S. Schubert, Chem. Commun., 2008, 5758–5760 RSC.
| This journal is © The Royal Society of Chemistry 2010 |