Small molecule induced control in duplex and triplex DNA-directed chemical reactions†
Mikkel F. Jacobsen, Jens B. Ravnsbæk and Kurt V. Gothelf*
Danish Research Foundation: Centre for DNA Nanotechnology at the Interdisciplinary Nanoscience Center and the Department of Chemistry, AarhusUniversity, Langelandsgade 140, 8000 Aarhus C, Denmark. E-mail: kvg@chem.au.dk; Fax: 86196199
First published on 9th November 2009
Abstract
Triplex DNA binders can effectively control copper-catalysed alkyne–azide click reactions in DNA architecture, such that either duplex or triplex DNA directed reactions of terminally attached azides and alkynes occur, in the absence or presence of triplex DNA binder, respectively.
Nature utilizes a multitude of macromolecular recognition elements to control chemical reactivity such as nucleic acids and proteins.1 For example, it is well-known that the reactivity of allosteric enzymes can be modulated by the recognition of small molecule allosteric regulators through changes in conformation.2 A plethora of different small molecules bind to various nucleic acid architectures inducing stabilization.3 This includes higher order structures such as G-quadruplex and triplex DNA. Along these lines, triplex DNA binders are small and usually cationic molecules which stabilize triplex DNA and often possess highly selective binding to triplex over duplex DNA.4,5
It has been demonstrated by Mao et al. how DNA-triplex structures containing CGC triplets can be used in a pH regulated triplex formation to control the formation of an amide bond.6 Here we have used triplex DNA as a model system to demonstrate how the non-covalent interaction of a small molecule regulator, in this case a triplex DNA binder, can be used to control the covalent chemical reactions of conjugated functional groups. The recent surge in the use of DNA-directed chemistry,7e.g. in drug discovery,8 may benefit from the development of reactions based on novel and controllable higher-order DNA architectures.
The Huisgen–Sharpless–Meldal copper-catalyzed 1,3-dipolar cycloaddition (CuAAC) has been used extensively for the modification of nucleosides (e.g. conjugation of an fluorophor/biomolecule) prior to or following their incorporation into oligonucleotides.9,10 Contrarily, only very few examples of DNA-directed CuAAC reactions have been reported, in which an alkyne–DNA conjugate and an azide–DNA conjugate, in low concentrations are brought in close proximity either by direct hybridization or by hybridization of the two conjugates to a template strand.7 In two papers Liu et al. performed DNA-directed CuAAC reactions, and in both cases moderate yields were obtained in part due to DNA degradation in the presence of Cu(I).11 More recently Brown et al. described a chemical ligation type DNA-directed CuAAC reaction which proceeded almost quantitatively,12,13 and notably DNA degradation was avoided by including a ligand for copper.
To demonstrate the small-molecule-induced control of the CuAAC reaction we have applied a dialkyne–DNA conjugate, an azide–DNA conjugate and a triplex forming oligonucleotide (TFO) azide conjugate (Scheme 1). The depicted triplex DNA is unstable at room temperature in the absence of a triplex DNA binder and duplex control prevails leading to only one click reaction.
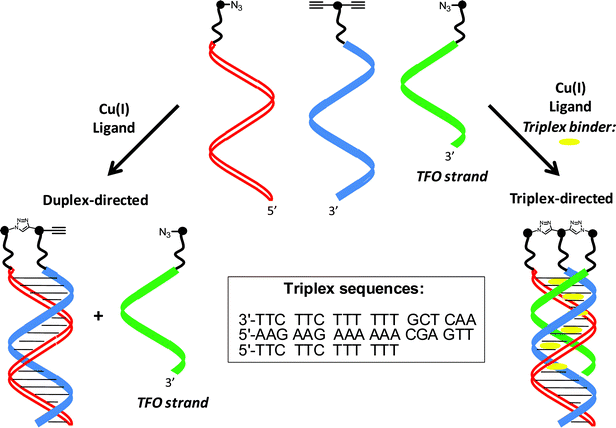 | ||
| Scheme 1 Controlling nucleic acid-directed chemical reactions with triplex DNA binders. | ||
In the presence of a triplex DNA binder triplex control takes over forming the parallel pyrimidine–purine–pyrimidine triplex with Hoogsteen base pairing to the triplex-forming oligonucleotide (TFO), leading to two click reactions. Thereby, the central dialkyne-modified oligonucleotide becomes ligated to the two azide-modified oligonucleotides in one step via 1,2,3-triazole head groups formed in the double click reaction resulting in non-symmetrical three-way branched oligonucleotides.
A selection of azide and dialkyne-modified oligonucleotides was synthesized (Fig. 1, Supporting Information). Both azide (X1, X2) and dialkyne modifications (conjugated and non-conjugated dialkyne, Y1, Y2 and Z1, Z2, respectively) were investigated with two different lengths of linkers (C6 or C6+C12) (O1–O8). Since the degradation of DNA in the presence of Cu(I) and oxygen can seriously affect the outcome of any CuAAC reaction,14 ligands for Cu(I) were employed to protect it from oxidation and partly shield the DNA. Two different ligands, BPDS (1)15 and THTA (2)16 were tested. For binding and stabilization of triplex DNA two triplex DNA binders, coralyne chloride (3)4c and the more potent triplex DNA binder 44d were employed.
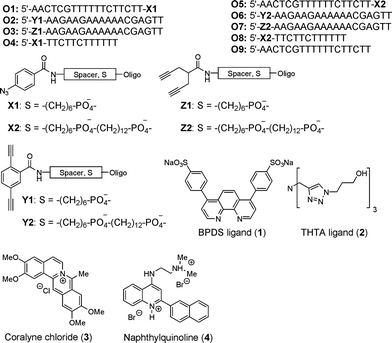 | ||
| Fig. 1 Oligonucleotides, ligands, and triplex DNA binders prepared and/or applied in this study. | ||
Triplex formation is a kinetically slow process compared to duplex formation. Therefore, preformed duplexes were subjected to annealing for an extended period (overnight) at 5 °C with the TFO strand (and any triplex DNA binder), and subsequently warmed slowly to 22 °C before adding the copper-catalyst mixture and allowing the conjugates to react for 2 h. Longer reaction times were not beneficial for the yield of the reactions, due to degradation of DNA.
The triplex and duplex-directed reactions were analyzed by denaturing PAGE (Fig. 2). Most prominent is the complete change in chemical reactivity in the presence (lane 1 and 2) and absence (lane 3) of triplex DNA binder 4, and the high selectivity of the reactions. On the other hand, the weaker triplex DNA binder coralyne (3) led to a less clean reaction with the contaminant formation of both duplex product and the product which corresponds to the ligation of the TFO strand and one of the duplex strands (Hoogsteen product, lane 4). Thus, the strength of stabilization with the triplex DNA binders is reflected in the reaction outcome.
 | ||
Fig. 2 Analysis of the CuAAC reactions by denaturing PAGE (reaction conditions, unless otherwise noted: 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 strand ratio; triplex DNA binder, ligand, CuSO4 and sodium ascorbate in PBS buffer. T1–T3: intramolecular triplexes). :1:1 strand ratio; triplex DNA binder, ligand, CuSO4 and sodium ascorbate in PBS buffer. T1–T3: intramolecular triplexes). | ||
High reaction yields for the formation of the triplex products from the three strands were obtained in the reaction of both non-conjugated and conjugated dialkyne-ODNs by employing the ligand 2 and triplex DNA binder 4 (lane 1 and 2) as indicated by denaturing PAGE and RP-HPLC. Yields of 90% and 80% for the duplex (lane 3) and triplex directed reaction (lane 1), respectively, were obtained as estimated by image analysis (GeneTools software) and corroborated by RP-HPLC analysis.16,17 In the case of dialkyne-oligonucleotide O6 weak higher bands besides the triplex product were observed which we assign to oligomerization reactions (homocouplings) of the dialkyne catalyzed by Cu(I) (lane 2). Similar faint bands were not observed with the non-conjugated dialkyne-oligonucleotides O3 and O7.18
As controls the exclusive formation of duplex product from O5 and O7 was carried out (lane 5), as was the ligation of the TFO strand O8 with O7 by employing the non-azide-modified duplex strand O9 (lane 6). As expected the absence of copper catalyst resulted in no reaction (lane 11).
Optimization of the reaction conditions revealed that subtle differences in the efficiency of C6 linkers compared to C6+C12 linkers existed, which demonstrates the distance dependence of the reaction.19 Minor amounts of both Hoogsteen and duplex product were seen in the case of short linkers (Fig. 2, lane 7), while the combination of short linkers on the duplex strands and a long linker on the TFO strand also gave a clean reaction (lane 8). The use of BPDS ligand (1) led to considerably reduced yields due to degradation of DNA (lane 9), as indicated by the faint band and confirmed by RP-HPLC, while the absence of ligand barely provided any product (lane 10).
The identity of the formed intramolecular triplexes was confirmed by MALDI-TOF MS, and they could readily be isolated by RP-HPLC, and characterized by thermal denaturation experiments. The intramolecular triplexes T1–T4 exhibit similar melting behavior characterized by a weak Hoogsteen transition at 53 °C, much higher than their intermolecular counterparts (in the absence of triplex binder), and a duplex/random coil transition at 64 °C (Fig. 3 and Table 1). Notably, no hysteresis was observed due to fast hybridization kinetics.
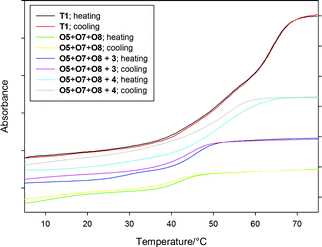 | ||
| Fig. 3 Thermal denaturation profiles of inter and intramolecular triplexes at 260 nm (different absorbance scale for T1 and O5+O7+O8 triplexes). | ||
These results are supported by the thermal denaturation profiles of the triplex O5+O7+O8 (Fig. 3 and Table 1). Hence, the Tm3 → 2 value for the weak Hoogsteen transition (triplex/duplex transition) in the absence of triplex binder is 11.0 °C, well below room temperature, and only the duplex-directed reaction occurs. On the other hand, in the presence of 4 it raises to 54.0 °C, where stabilization is so pronounced that the Hoogsteen and duplex/random coil transitions coincide,4d the triplex-directed reaction is highly favored. Coralyne (3) provides less stabilization (29.6 °C) with a broad Hoogsteen transition closer to room temperature, and a less selective reaction ensues. Considerable hysteresis of the melting curves was observed due to the slow kinetics involved in the formation of intermolecular triplexes.4e
In summary, we have demonstrated the use of triplex DNA binders as external stimuli for controlling chemical reactivity in higher-order DNA-directed reactions. Thus, the efficient double click reactions reported here could be controlled by simply adding a strong triplex DNA binder such as naphthylquinoline (4), while selectivity decreased with the weaker binder coralyne (3). Given the huge number of small molecules available for binding and stabilization of almost any DNA architecture, the methodology may be expanded considerably in scope, which is desirable in other complex DNA-based reaction systems where precise control of reactivity is required. Furthermore, the method provides a new high-yielding method for the formation three-way branched nonsymmetrical DNA sequences that each in principle may be extended in a unique sequence, and could be useful in the construction of DNA nanostructures.
Acknowledgements
Financial support for this work by the Danish National Research Foundation, and the Carlsberg Foundation is gratefully acknowledged.References
- (a) Nucleic Acids in Chemistry and Biology, 3rd Ed. (Eds: G. M. Blackburn, M. J. Gait, D. Loakes, D. M. Williams), Royal Society of Chemistry, Cambridge, 2006 Search PubMed; (b) Principles of Nucleic Acid Structure, S. Neidle, Elsevier, 2008 Search PubMed; (c) Proteins: Structure and Function, D. Whitford, Wiley, Chichester, 2005 Search PubMed.
- Allosteric Regulatory Enzymes, T. Traut, Springer, New York, 2008 Search PubMed.
- M. J. Hannon, Chem. Soc. Rev., 2007, 36, 280–295 RSC.
- (a) J. B. Chaires, J. Ren, M. Henary, O. Zegrocka, G. R. Bishop and L. Strekowski, J. Am. Chem. Soc., 2003, 125, 7272–7283 CrossRef; (b) C. Escudé, C. H. Nguyen, S. Kukreti, Y. Janin, J.-S. Sun, E. Bisagni, T. Garestier and C. Hélène, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 3591–3596 CrossRef CAS; (c) J. S. Lee, L. J. P. Latimer and K. J. Hampel, Biochemistry, 1993, 32, 5591–5597 CrossRef CAS; (d) W. D. Wilson, F. A. Tanious, S. Mizan, S. Yao, A. S. Kiselyov, G. Zon and L. Strekowski, Biochemistry, 1993, 32, 10614–10621 CrossRef CAS; (e) J. L. Mergny, G. Duval-Valentin, C. H. Nguyen, L. Perrouault, B. Faucon, M. Rougée, T. Montenay-Garestier, E. Bisagni and C. Hélène, Science, 1992, 256, 1681–1684 CrossRef CAS.
- B. P. Belotserkovskii, E. De Silva, S. Tornaletti, G. Wang, K. M. Vasquez and P. C. Hanawalt, J. Biol. Chem., 2007, 282, 32433–32441 CrossRef CAS.
- Y. Chen and C. Mao, J. Am. Chem. Soc., 2004, 126, 13240 CrossRef CAS.
- D. R. Liu and X. Li, Angew. Chem., Int. Ed., 2004, 43, 4848–4870 CrossRef CAS.
- (a) B. N. Tse, T. M. Snyder, Y. Shen and D. R. Liu, J. Am. Chem. Soc., 2008, 130, 15611–15626 CrossRef CAS; (b) E. M. Driggers, S. P. Hale, J. Lee and N. K. Terrett, Nat. Rev. Drug Discovery, 2008, 7, 608–624 CrossRef CAS; (c) L. A. Haff, C. G. M. Wilson, Y. Huang, B. K. Benton, J. F. Bond, A. M. Stern, R. F. Begley and J. M. Coull, Clin. Chem., 2006, 52, 2147–2148 CrossRef CAS.
- J. E. Moses and A. D. Moorhouse, Chem. Soc. Rev., 2007, 36, 1249–1262 RSC.
- (a) F. Amblard, J. H. Cho and R. F. Schinazi, Chem. Rev., 2009, 109, 4207–4220 CrossRef CAS; (b) P. M. E. Gramlich, C. T. Wirges, A. Manetto and T. Carell, Angew. Chem., Int. Ed., 2008, 47, 8350–8358 CrossRef CAS.
- (a) Z. J. Gartner, R. Grubina, C. T. Calderone and D. R. Liu, Angew. Chem., Int. Ed., 2003, 42, 1370–1375 CrossRef; (b) M. W. Kanan, M. M. Rozenman, K. Sakurai, T. M. Snyder and D. R. Liu, Nature, 2004, 431, 545–549 CrossRef CAS.
- R. Kumar, A. El-Sagheer, J. Tumpane, P. Lincoln, L. M. Wilhelmsson and T. Brown, J. Am. Chem. Soc., 2007, 129, 6859–6864 CrossRef CAS.
- A. H. El-Sagheer and T. Brown, J. Am. Chem. Soc., 2009, 131, 3958–3964 CrossRef CAS.
- (a) R. L. Weller and S. R. Rajski, Org. Lett., 2005, 7, 2141–2144 CrossRef CAS; (b) C. J. Burrows and J. G. Muller, Chem. Rev., 1998, 98, 1109–1152 CrossRef CAS.
- W. G. Lewis, F. G. Magallon, V. V. Fokin and M. G. Finn, J. Am. Chem. Soc., 2004, 126, 9152–9153 CrossRef CAS.
- T. R. Chan, R. Hilgraf, K. B. Sharpless and V. V. Fokin, Org. Lett., 2004, 6, 2853–2855 CrossRef CAS.
- RP-HPLC analysis indicated that the observed non-quantitative conversion is due to partial degradation of the oligonucleotides.
- The higher propensity for oligomerisation of oligonucleotides carrying Y1, Y2 groups compared to Z1, Z2 may be explained by the higher acidity of the terminal alkynes.
- (a) Z. J. Gartner, M. W. Kanan and D. R. Liu, Angew. Chem., Int. Ed., 2002, 41, 1796 CrossRef CAS; (b) Z. J. Gartner and D. R. Liu, J. Am. Chem. Soc., 2001, 123, 6961 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, MALDI-TOF and RP-HPLC data, and spectral data for all new compounds. See DOI: 10.1039/b919387a |
| This journal is © The Royal Society of Chemistry 2010 |