Cucurbit[7]uril host–guest complexes of cholines and phosphonium cholines in aqueous solution†
Ian W. Wyman and Donal H. Macartney*
Department of Chemistry, Queen's University, 90 Bader Lane, Kingston, ON K7L 3N6, Canada. E-mail: donal@chem.queensu.ca; Fax: +1 613 533 6669; Tel: +1 613 533 2617
First published on 24th November 2009
Abstract
The neutral host cucurbit[7]uril forms very stable complexes with a series of cationic cholines (R3NCH2CH2OR′+) and their phosphonium analogues (R3PCH2CH2OR′+) (R3 = Me3, Et3, or Me2Bz, or R3N = quinuclidinium, and R′ = H, COCH3, CO(CH2)2CH3, or PO3H), and (±)-carnitine, in aqueous solution. The complexation behaviour has been investigated using 1H and 31P NMR spectroscopies, and ESI mass spectrometry. The complexation-induced chemical shift changes of the guests clearly indicate the effects of replacing the N(CH3)3+ end group by P(CH3)3+, and changing the nature of R on the position of the guest with respect to the CB[7] cavity and its polar portal-lining carbonyl groups. This study demonstrates that molecular recognition of cholines in aqueous solution is achievable with a neutral host without the need for aromatic walls for cation–π interactions.
Introduction
The cucurbit[n]uril family (CB[n], n = 5–8, 10) of macrocyclic host molecules1 have been of increasing interest since the development of methods for improving the yields of the minor congeners (n = 5, 7, 8, and 10), in addition to the major CB[6] product, at the beginning of the millennium.2 With a hydrophobic cavity comparable in size to β-cyclodextrin or calix[4]arenes, and two restrictive portals lined with ureido carbonyl groups, the cucurbit[7]uril host (Scheme 1) has been shown to form remarkably stable complexes with a variety of guest molecules,3 particularly cationic organic3,4 and organometallic species,5 in aqueous solution. In addition to the hydrophobic effects of placing the guest within the cavity, the carbonyl groups are capable of stabilizing the host–guest complex through hydrogen bonding, ion–dipole, and dipole–dipole interactions with appropriate guests.1![Cucurbit[7]uril.](/image/article/2010/OB/b917610a/b917610a-s1.gif) | ||
| Scheme 1 Cucurbit[7]uril. | ||
There has been considerable recent interest in using cucurbiturils to aid in the delivery of molecules of biological and medicinal interest, through host–guest formation. Several potential antitumour platinum(II)6 and titanocene complexes7 have been shown to bind to CB[7] and CB[8]. Molecular recognition of amino acids and peptides by CB[7] and CB[8] has been reported by the groups of Urbach and Kim.8 The extremely stable complexation of ferrocenes with CB[7] (1010–1015 dm3 mol−1)5 has led to the development of systems which behave like the biotin–avidin system.5c We have recently reported studies of the pH-dependent host–guest interactions of CB[7] with the drug ranitidine9 and a series of local anaesthetics10 in aqueous solutions, with stability constants considerably greater than those reported for the analogous complexes with β-cyclodextrin. Nau and coworkers have likewise shown that the proton-pump inhibitors lansoprazole and omeprazole, and the fungicide thiabendazole form stable complexes with CB[7],11 improving activation and stabilization properties of the guests. We have observed that CB[7] binds the α-axial 5,6-dimethylbenzimidazole ligands of vitamin B12 and coenzyme B12 (stabilizing the ‘base-off’ form) and in the latter compound exhibits a subsequent binding of the β-axial 5′-deoxyadenosyl group.12 With these and other guests which bear nitrogen13 or carbon14 acid sites, the pKa values are observed to increase upon encapsulation of the guests, as the protonated forms bind more strongly through ion–dipole and hydrogen bonding interactions with the carbonyl groups on the CB[7] portals.
The choline family ((CH3)3NCH2CH2OR+) of important biological molecules have in common a trimethylammonium head group, which in the case of acetylcholine (R = COCH3), is used to bind the substrate to the active site of the acetylcholinesterase enzyme.15 The binding site consists of aromatic amino acid residues such as tryptophan, which employ cation–π interactions to hold the substrate in place.16 The majority of artificial macrocyclic host molecules which have previously exhibited binding of choline guests have contained aromatic walls and have been anionic in nature, thus employing electrostatic as well as cation–π interactions.17-30 These host molecules include acyclic receptors,17 cryptands,18 cyclophanes,19 molecular clefts,20p-tert-butylcalix[n]arene dianions (n = 4, 6),21p-sulfonated calix[n]arenes22 and other calix[n]arenes,23 tetracyanoresorcin[4]arene24 and other resorcin[n]arenes,25 pyrogallol[4]arenes,26 and other macrocycles.27 Kim and co-workers have reported that the water-soluble hexa(cyclohexyl)cucurbit[6]uril binds acetylcholine, with K*CB[6] = 1.3 × 103 dm3 mol−1, but does not include choline itself.28
Rebek and coworkers have designed deep, resorcarene-based cavitands for the binding of choline cations29 and have carried out catalytic formations of acetylcholine, and the aminolysis of the acetylcholine mimic p-nitrophenyl choline carbonate (PNPCC).30 Cuevas et al. had previously prepared ditopic, guanidinium-appended calix[6]arene receptors which act as an artificial acetylcholinesterase in the methanolysis of PNPCC.31
The phosphonium analogues of choline, acetylcholine and other related compounds have been known for some time and have been studied in terms of their relative activity to the corresponding nitrogen compounds.32 They have also been investigated as probes of choline and phospholipid metabolism employing in situ31P NMR experiments.32a
We have recently shown that while simple cations, such as protons and alkali metal and transition metal ions, as well as the positive sites on organic cations, bind at the polar portals of CB[7], more hydrophobic and charge-diffuse peralkylated onium cations, such as NR4+, PR4+ and SR3+, (R = methyl and ethyl) bind within the cavity of the host molecule in aqueous solution.4h With the larger NnPr4+ and NnBu4+ guests, energy-minimization calculations and the complexation-induced chemical shift changes in the two alkyl groups. In a related study we observed that the depolarizing muscle relaxing agent succinylcholine, as well as 1,10-bis(trimethylammonium)decane and its phosphonium analogue, can bind with CB[7] to form very stable [2]pseudorotaxanes at host![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :guest ratios up to 1:1.33 With additional CB[7], the migration of the host to encapsulate the ammonium or phosphonium end groups forms 2:1 host–guest complexes of varying stability.
:guest ratios up to 1:1.33 With additional CB[7], the migration of the host to encapsulate the ammonium or phosphonium end groups forms 2:1 host–guest complexes of varying stability.
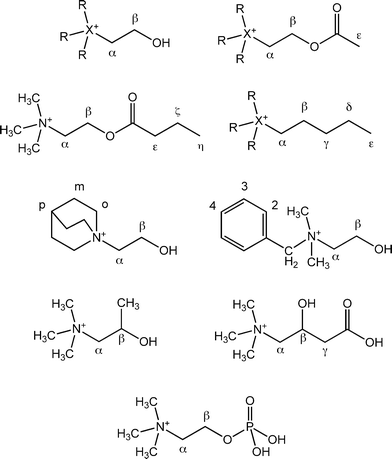
In conjunction with these studies, we have investigated the binding of quaternary ammonium and phosphonium guests including choline, acetylcholine, and their phosphonium analogues (Scheme 2), with CB[7] in aqueous solution. In addition, we have examined the binding of related guests, such as the zwitterionic (±)-carnitine and choline phosphate, and a series of R3X(CH2)4CH3+ (X = N or P and R = methyl or ethyl) cations, in which the pentyl chain has the same backbone length as the acetylcholine –CH2CH2OC(CO)CH3 group (Scheme 2). We have studied the host–guest interactions using 1H and 31P NMR spectroscopy to probe the nature of the complexes, and the stability constants, which have been determined by means of competitive 1H NMR spectral measurements, may be compared with reported values for the complexes with other artificial receptors.
Results and discussion
NMR and ESI-MS spectra of host–guest complexes
The formation of host–guest complexes between cucurbit[7]uril and the choline and phosphonium choline guests in aqueous solution have been established by ESI mass spectrometry34 and from 1H and 31P NMR spectroscopies. The titrations of the choline guests with CB[7] resulted in changes in the chemical shifts of the guest proton resonances, as illustrated in Fig. 1 for choline and its phosphonium analogues. The guests generally exhibit fast or intermediate exchange on the NMR timescale, with the guest resonances representing an average chemical shift of the resonances for the free and bound guest species, as seen with choline, or slow exchange in which there are resonances for the both free and bound guests, as in the case of the phosphonium choline, for example.![1H NMR spectra of choline (1.44 mmol dm−3) in (a) the absence of CB[7] and in the presence of (b) 0.25, (c) 0.65, and (d) 1.41 equivalents of CB[7], and (e) (2-hydroxyethyl)trimethylphosphonium bromide (1.50 mmol dm−3) in the absence of CB[7] and in the presence of (f) 0.29, (g) 0.74, and (h) 1.30 equivalents of CB[7], in D2O.](/image/article/2010/OB/b917610a/b917610a-f1.gif) | ||
| Fig. 1 1H NMR spectra of choline (1.44 mmol dm−3) in (a) the absence of CB[7] and in the presence of (b) 0.25, (c) 0.65, and (d) 1.41 equivalents of CB[7], and (e) (2-hydroxyethyl)trimethylphosphonium bromide (1.50 mmol dm−3) in the absence of CB[7] and in the presence of (f) 0.29, (g) 0.74, and (h) 1.30 equivalents of CB[7], in D2O. | ||
In the 1H NMR spectra of cucurbituril host–guest complexes, the complexation-induced shift changes (CIS = Δδlim = δbound−δfree) in the proton resonances of the guest molecule are very informative as to the average location of the guest with respect to the CB[7] cavity.1,3 Upfield shifts (Δδlim < 0) are observed for protons located within the hydrophobic cavity, while the deshielding of the polar carbonyl groups results in downfield shifts (Δδlim > 0) in the resonances of guest protons in the proximity of the carbonyl oxygens. The Δδlim values for the proton resonances of the choline guests are presented in Table 1.
| Guest | Δδlim, ppm | KCB[7], dm3 mol−1 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| CH3 | CH2 | P | Cα | Cβ | Cγ | Cδ | Cε | Cζ | Cη | ||
| a Data from reference 4h.b Using 1,4-phenylenediamine as the competitor.c Values of (7.0 ± 1.6) × 105 and (1.1 ± 0.3) × 106 dm3 mol−1 determined with PMe4+ and PEt4+, respectively, as the competitors.d Values of (5.7 ± 1.5) × 105 and (1.3 ± 0.4) × 106 dm3 mol−1 determined with PMe4+ and PEt4+, respectively, as the competitors.e Using TMSP as the competitor.f Methyl protons on Cβ.g Data from reference 35.h Δδ for the next methylene group is −0.23 ppm, while the remaining protons on the terminal end of the alkyl chain experience downfield shifts of 0.06 ppm or less upon complexation (ref. 35).i At pD 7.0.j In D2O with no added electrolyte.k At pD 2.0.l In D2O with 2.1 equiv of edta4− to complex Ca2+.m Methylene of benzyl group. Δδlim for phenyl resonances are −0.47 (p), −0.78 (m), and −1.07 ppm (o).n Methylene of benzyl group. Δδlim for phenyl resonances are −0.54 (p), −0.84 (m), and −1.04 (o).o Using NEt4+ as the competitor.p H3 protons.q Using p-xylylenediamine as a competitor.r H4 proton.s Value of (1.7 ± 0.2) × 106 dm3 mol−1 determined using 1,4-phenylenediamine as the competitor.t Value of (5.6 ± 1.3) × 105 dm3 mol−1 determined using TMSP as the competitor.u Values of (1.6 ± 0.4) × 106 and (3.0 ± 0.5) × 106 dm3 mol−1 determined using 1,4-phenylenediamine and NEt4+, respectively as the competitors. | |||||||||||
| N(CH3)4+a | −0.72 | (1.2 ± 0.4) × 105 | |||||||||
| (CH3)3N(CH2)2OH+ | −0.66 | −0.93 | −0.73 | (6.5 ± 1.2) × 105b,c | |||||||
| (CH3)3N(CH2)2O2CCH3+ | −0.28 | −0.65 | −0.85 | −0.68 | (7.0 ± 1.3) × 105b,d | ||||||
| (CH3)3NCH2CH(CH3)O2CCH3+ | −0.16 | −0.90 | −0.97 | −0.20 | (4.9 ± 0.9) × 106e | ||||||
| −0.65f | |||||||||||
| (CH3)3N(CH2)2O2C(CH2)2CH3+ | −0.10 | −0.36 | −0.62 | −0.82 | −0.65 | −0.76 | (1.7 ± 0.3) × 107b | ||||
| (2.2 ± 0.3) × 107e | |||||||||||
| (CH3)3N(CH2)11CH3g | −0.07 | −0.56 | −0.71 | −0.65 | −0.65 | −0.58 | −0.58 | −0.44 h | (5.8 ± 0.1) × 104 | ||
| (CH3)3N+CH2CH(OH)CH2CO2− | −0.78 | (3.8 ± 0.5) × 102i | |||||||||
| (CH3)3NCH2CH(OH)CH2CO2H+ | −0.70 | −0.91 | −0.65 | −0.14 | (8.0 ± 1.1) × 104j | ||||||
| −0.19 | (2.8 ± 0.4) × 103k | ||||||||||
| (CH3)3N+(CH2)2OP(O)(OH)O− | −0.82 | −0.43 | −0.04 | (1.2 ± 0.2) × 103l | |||||||
| (PhCH2)N(CH3)3+a | −0.25 | −0.70m | (2.5 ± 0.2) × 108 | ||||||||
| (PhCH2)(CH3)2N(CH2)2OH+ | −0.23 | −0.69n | −0.16 | −0.22 | (4.1 ± 1.2) × 108e | ||||||
| N(CH2CH3)4+a | −0.87 | −0.44 | (1.0 ± 0.2) × 106 | ||||||||
| (CH3CH2)3N(CH2)2OH+ | −0.46 | −0.83 | −0.87 | −0.45 | (1.8 ± 0.3) × 106e | ||||||
| (CH3CH2)3N(CH2)4CH3+ | −0.09 | −0.29 | −0.51 | −0.62 | −0.56 | −0.45 | −0.46 | (8.3 ± 1.9) × 105o | |||
| Quin(CH2)2OH+ | −0.76p | −0.90 | −0.39 | −0.07 | (1.3 ± 0.3) × 109q | ||||||
| −0.46r | |||||||||||
| P(CH3)4+a | −0.71 | −0.38 | (2.2 ± 0.4) × 106 | ||||||||
| (CH3)3P(CH2)4CH3+ | −0.04 | −0.02 | −0.74 | −0.69 | −0.64 | −0.55 | −0.64 | (1.4 ± 0.3) × 107b | |||
| (CH3)3P(CH2)2OH+ | −0.68 | −0.13 | −0.95 | −0.52 | (6.9 ± 1.8) × 106o,s | ||||||
| (CH3)3P(CH2)2O2CCH3+ | −0.67 | −0.56 | −0.90 | −0.40 | +0.02 | (8.9 ± 1.7) × 105b | |||||
| P(CH2CH3)4+a | −0.72 | −0.31 | −5.77 | (1.3 ± 0.3) × 105 | |||||||
| (CH3CH2)3P(CH2)4CH3+ | +0.18 | +0.04 | −1.65 | −0.54 | −0.74 | −0.76 | −0.68 | −0.73 | (1.6 ± 0.3) × 106e,t | ||
| (CH3CH2)3P(CH2)2OH+ | −0.32 | −0.72 | −4.75 | −0.69 | −0.18 | (1.3 ± 0.3) × 105o | |||||
| (CH3CH2)3P(CH2)2O2CCH3+ | −0.35 | −0.66 | −5.03 | −0.66 | −0.22 | −0.01 | (8.6 ± 1.6) × 105b,u | ||||
With choline and its phosphonium analogues, the Δδlim values are < 0 for all of the proton resonances, indicating that the entire guests are included in the cavity, including the trialkylonium head group. With acetylcholine, on the other hand, replacement of the N atom with P results in considerable changes in the Δδlim values (Table 1), indicating a change in the position of the guest relative to the CB[7] cavity. The Δδlim value for the onium methyl resonance is much smaller for acetylcholine (−0.28 ppm) than that of the phosphonium analogue (or choline itself), while the most upfield chemical shift change switches from the methylene group next to NMe3+ (Hα), to the proton next to the acetyl group (Hβ) with P. The acetyl methyl proton resonance exhibits a very slight downfield shift with X = P and a significant upfield shift with X = N. These differences clearly indicate that with the smaller N(CH3)3+ group, the acetyl group is included in the cavity, while the more hydrophobic P(CH3)3+ group is included in the case of the phosphonium acetylcholine analogue.
The complexation of acetylcholine is stabilized by ion–dipole and dipole–dipole interactions of the protonated and polar head groups of the guests with carbonyl laced portals. In addition, the Δδlim values of the Hα, Hβ, and acetyl methyl protons suggest that the carbonyl oxygen atom of the guests are located in the center of the CB[7] cavity. With the quadrupolar nature of the cucurbituril cavity, the oxygen may be involved in dipolar–quadrupolar interactions with the trimethylammonium group much closer to the carbonyls of the portal. We have recently shown that small polar molecules such as acetone and methyl acetate bind reasonably strongly to CB[7] as a result of contributions from dipole–quadrupole interactions, with the oxygen of the guest directed towards the center of the cavity wall.36
For the trimethylammonium compounds in general, the methyl protons all shift upfield, with the Δδlim values ranging from −0.07 to −0.82 ppm, indicating that on average the methyl protons are spending more time within the cavity. The least upfield shifts are for the most extended species, where the cavity is occupied by the long alkyl or ester chains, placing the cationic head group at or outside of the portal. Similar behaviour is observed for the trimethylphosphonium compounds (Table 1), with the exception of the aforementioned comparison between acetylcholine and the phosphonium analogue. The most upfield shifts are observed for choline and other species with β-hydroxyl groups and/or carboxylate or phosphate groups at the other end of the guest. The values are similar to the those observed for N(CH3)4+ (−0.72 ppm),4h which is fully encapsulated in the CB[7] cavity.
The 31P Δδlim values for the central P atom in the trimethyl- and triethylphosphonium analogues of the choline compounds were determined and are presented in Table 1. For the trimethylphosphonium compounds the Δδlim value is small and follows the trend in the magnitude of Δδlim for the methyl proton resonance. Notably, with the trimethylpentylphosphonium cation, very small upfield shifts are observed for the proton and phosphorus resonances of the trimethyl group, suggesting that it is located near the portal, rather than closer to the center of the cavity. For the triethylphosphonium guests, the 31P Δδlim values are considerably larger, as we observed previously for tetraethylphosphonium cation (Δδlim = −5.77 ppm),4h but follow a similar trend as observed with the trimethylphosphonium analogues. In the case of the phosphocholine, the phosphate P exhibits a small downfield shift, as the anionic groups are located outside the CB[7] cavity.
The effect of the acetyl group on the strength and position of the binding of acetylcholine to CB[7] was also probed by studying a series of R3X(CH2)4CH3+ (X = N or P and R = methyl or ethyl) cations, in which the pentyl chain has the same length as the acetylcholine –CH2CH2O2CCH3 group. In each case, the trimethyl- or triethylonium proton resonances display small upfield or downfield chemical shifts, while the pentyl methylene or methyl protons exhibit considerable upfield shifting upon CB[7] complexation (Table 1). With these guests, the more hydrophobic pentyl group is preferred for binding, while for the acetylcholine species, the trimethyl- or triethylonium centers are preferably bound in the CB[7] cavity.
Host–guest stability constants
The magnitude of the stability constants for the host–guest complexes formed between cucurbit[7]uril and the choline guests in this study would be expected to be dependent on a number of features of the guest structure including the nature of the quaternary center (central atom (N vs. P) and alkyl substituent(s)) and the size, charge, and hydrophobicity of the substituents on the hydroxyl/ester oxygen on the opposite side of the molecule, as well as the nature of any substituent on the ethylene linker between the quaternary center and the oxygen. While we have not examined the effects of the counter ion (Br−vs. Cl−) of these salts on the host–guest stability constants, it would be anticipated they would be minimal in the presence of an excess of acetate ions from the buffer employed. Biczók and co-workers have reported that there is not a significant change in the stability constants for CB[7] with the cationic berberine guest when the anion is varied from chloride to iodide to hydrogen phosphate.4jWith the zwitterionic (±)-carnitine and choline phosphate guests, the smallest stability constants were observed, and determined from least-squares fits to the 1:1 binding curves. The carboxylic acid group of the (±)-carnitine guest, (CH3)3NCH2CH(OH)CH2CO2H+, has a pKa of 3.78.37 The titrations of carnitine with CB[7] were performed at pD 2.0 and 7.0, using 0.010 mol dm−3 DCl–0.050 mol dm−3 NaCl and 0.050 NaO2C(CD3)2 solutions, respectively (Fig. 2). In the acidic solution, the stability constant was determined to be (2.8 ± 0.4) × 103 dm3 mol−1, with an upfield shift of the methyl protons of the NMe3+ group of 0.66 ppm, indicating that the trimethylammonium group was located in the cavity, while the carboxylic acid group was outside near the portal. In neutral solution, the deprotonation of the carboxylic acid group resulted in a significant decrease in the stability constant to (3.8 ± 0.5) × 102 dm3 mol−1, and an increase in the upfield shift of the methyl protons to 0.78 ppm. The repulsions between the carboxylate group and the polar portal-lining carbonyl groups result in a movement of the zwitterionic form of the guest, with respect to the cation form, with the NMe3+ head group now located further into the cavity.
![1H NMR chemical shift titrations of carnitine with CB[7] in D2O at (■) pD 2.0 (0.01 mol dm−3), (▲) pD 4.8 (0.050 mol dm−3 NaOAc/0.025 mol dm−3 DCl) and (●) pD 7.0 (no added electrolytes).](/image/article/2010/OB/b917610a/b917610a-f2.gif) | ||
| Fig. 2 1H NMR chemical shift titrations of carnitine with CB[7] in D2O at (■) pD 2.0 (0.01 mol dm−3), (▲) pD 4.8 (0.050 mol dm−3 NaOAc/0.025 mol dm−3 DCl) and (●) pD 7.0 (no added electrolytes). | ||
The choline phosphate guest was commercially available as its calcium salt. The CB[7] complexation of the zwitterionic form of the calcium salt of the choline phosphate, was examined by a 1H NMR chemical shift titration at pD 3.7. The result was a sigmoidal binding curve, due to competitive Ca2+ binding to the portals of the CB[7]. The additions of edta4− to the solution provided complexation of the Ca2+, allowing for uninhibited binding of the guest to CB[7]. With an excess of edta4− (2.1 equivalents) a normal 1:1 host–guest titration curve was obtained, with the stability constant of (1.2 ± 0.2) × 103 dm3 mol−1 being determined. At pD 7, where the phosphate group is dianionic (pKa of the zwitterion is 5.5),38 only small changes in chemical shifts of the guest were observed (Δδ = −0.07, −0.02, and 0.02 ppm for CH3, Hα and Hβ, respectively at [CB[7]/[guest] = 10), suggesting very weak binding of the NMe3+ head group.
The stability constants for the remainder of the CB[7] host–guest complexes are too large to calculate directly and were determined by 1H NMR competition experiments at pD 4.75 (acetate buffer) using a variety of competitor guests whose stability constants have been measured previously under these conditions.3,4h The values in Table 1 may be compared with the corresponding NR4+ and PR4+ cations determined previously.4h The magnitudes of the stability constants for the monocationic choline and acetylcholine guests, along with the NEt3+, PMe3+, and PEt3+ analogues may be related to the natures of the cationic head groups and the hydroxyl or acetyl end groups. For the choline series, the stability constants follow the same trend as observed for the tetraalkylonium cations, with PMe3+ > NEt3+ > PEt3+ > NMe3+ (choline). This trend is consistent with the observation from the Δδlim values that the trialkylonium head group is located within the cavity, along with the remainder of the ‘ethanol’ moiety of the cholines.
With the acetylcholine series, the stability constants are very similar, in the range of 7–9 × 105 dm3 mol−1. With this guest series, the location of the acetylcholines within the CB[7] is clearly different than that for the phosphonium species, where the Δδlim values (near zero) for the acetyl methyl protons indicated interior binding of the phosphonium head group, compared with the ammonium species, with a considerable upfield shift for the acetyl Me protons. The similarity of stability constants for the series is therefore likely coincidental. On comparing the phosphonium acetylcholine guests with the corresponding trialkylpentylphosphonium guests (same length backbone), the latter guests exhibit greater stability constants and form a complex with the CB[7] located over the pentyl group.
Within the choline/acetylcholine series (with NMe3+ head groups), the order of KCB[7] is butyrylcholine > β-methylacetylcholine > acetylcholine > choline > NMe4+. This trend is primarily related to the increased hydrophobicity of additional alkyl groups at the neutral end of the guest, or to the ethylene linker connecting the NMe3+ group to the hydroxyl or acetyl terminals. The trend in the stability constants is also paralleled by the trend in the absolute value of the N-methyl Δδlim parameter, with butyrylcholine (−0.10 ppm) < β-methylacetylcholine (−0.16) < acetylcholine (−0.28) < choline (−0.66) < NMe4+ (−0.72). By providing more hydrophobic aliphatic substituents, that are further from the quaternary ammonium center, the shielding cavity region of the CB[7] is drawn further away from the NMe3+ group.
By replacing the methyl or ethyl groups on the ammonium end of the choline guests with quinuclidinium or benzyl groups, the stability constants are increased to the 108–1010 dm3 mol−1 range. These end groups, whose protons experience large upfield shifts (Table 1), are located in the CB[7] cavity, with the Hα and Hβ protons being shifted far less upfield than the parent choline guest. The stability constants are similar in magnitude to those obtained for other cationic guests containing the quinuclidinium (KCB[7] = (5.8 ± 1.3) × 109 dm3 mol−1 for N-methylquinuclidinium)33 and benzyl (KCB[7] = (2.5 ± 0.6) × 108 dm3 mol−1 for (PhCH2)N(CH3)3+)4h substituents on the quaternary nitrogen. There are several other examples of guest molecules containing N(CH3)3+ head groups in which the strength of the binding to CB[7] is dictated by the hydrophobic portion of the molecule attached to the trimethylammonium group, including ferrocene,5c adamantane,3 and methyl viologen units.4i With these guests, as noted with quinuclidinium and benzyl substituents, the CB[7] host–guest stability constants are very similar in magnitude to those for other guests containing these hydrophobic centers.
The CB[7] host has a reasonable selectivity between butyrylcholine (2.0 × 107 mol dm−3) and acetylcholine (7.0 × 105 mol dm−3), the two natural substrates of butylcholinesterase and acetylcholinesterase, respectively. There is also good selectivity between acetylcholine (or choline) and (±)-carnitine (3.8 × 102 mol dm−3), which is also produced naturally through biosynthesis. The complexations of choline, acetylcholine, and related guests, such as carnitine, by a number of other artificial macrocyclic host molecules have been reported,17–31 although most of these have been measured in chloroform or other organic solvents. In the majority of these hosts, the repeating subunits contain aromatic rings and/or anionic substituents to provide for non-covalent cation–π and ion–dipole interactions, respectively, with the N(CH3)3+ head groups. There are several reports of the comparisons of stability constants for choline, acetylcholine, and carnitine with water-soluble macrocyclic host molecules. With p-sulfonated calix[4]arene, Nau and co-workers reported stability constants (pD 7.4) for these guests are 7 × 104, 1.0 × 105, and 1.7 × 103 dm3 mol−1, respectively, from a fluorescence displacement assay.22i Using a deep cavitand, Rebek and co-workers measured values of 2.6 × 104, 1.5 × 104, and 1.5 × 102 dm3 mol−1, respectively, utilizing isothermal calorimetry.29c Inoue and co-workers have determined stability constants of 3.4 × 103, 6.1 × 103, and 4.4 × 103 dm3 mol−1 (1.8 × 104 dm3 mol−1 for the zwitterion), respectively, for these guests with a pyrogallol[4]arene in ethanol.26
These values show similar trends to those obtained with CB[7], in that there is relatively little selectivity between choline and acetylcholine, and significantly lower values, with the exception of the pyrogallol[4]arene, for the zwitterionic carnitine. The comparisons of magnitudes of the stability constants for neutral CB[7] with the other host molecules indicate that the combination of ion–dipole interactions and the hydrophobic effect with the portals and cavity of the host, respectively, are sufficient to provide for significant molecular recognition of the choline and acetylcholine guests in aqueous solution.
Experimental
Materials
The cucurbit[7]uril was synthesized and characterized according to the method of Day and coworkers.2b The choline, acetylcholine, trimethylphosphine, triethylphosphine, trimethylamine, triethylamine, 2-bromoethanol, 1-bromopentane, 2-bromoethylacetate, and butyrylcholine (Sigma-Aldrich), and benzyldimethylaminocholine bromide (Fluka) were used as received. The yields of the novel compounds have not been optimized.Triethylpentylammonium bromide
A solution of 7.82 g (51.8 mmol) 1-bromopentane and 5.79 g (57.2 mmol) triethylamine in 8 mL CH3CN was refluxed for 48 h. Removal of the solvent gave a hygroscopic reddish-orange solid. Yield 4.76 g (36%). mp. 155–159 °C (lit. 145–147 °C,39 147 °C40). 1H NMR (400 MHz, D2O) δ 3.24 (q, 6H, J = 7.2 Hz, CH2), 3.12 (t, 2H, J = 7.8 Hz, Hα), 1.54 (qn, 2H, J = 7.8 Hz, Hβ), 1.32 (m, 4H, Hγ and Hδ), 1.23 (tt, 9H, J = 7.2 Hz, 1.9 Hz, CH3), 0.87 (t, 3H, J = 7.0 Hz, Hε) ppm. 13C NMR (100 MHz, D2O) δ 55.82 (t, J = 2.7 Hz, Cα), 51.96 (t, J = 2.7 Hz, N+-CH2CH3), 26.88 (Cδ), 20.66 (Cγ), 19.71 (Cβ), 12.19 (Cε), 5.76 (N+-CH2CH3) ppm. HR-ESI-MS: calcd. for C11H26N (M-Br+) m/z = 172.2060; found 172.2059.(2-Hydroxyethyl)triethylammonium bromide
A mixture of 0.724 g (5.79 mmol) of 2-bromoethanol and 0.665 g (6.57 mmol) of triethylamine in 3 mL of ethanol was stirred at room temperature for 4 days. The precipitate was washed with ether and dried. Yield 0.224 (17%). mp. 250–253 °C (lit.41 256–257 °C). 1H NMR (400 MHz, D2O) δ 3.92 (br t, 2H, Hβ), 3.32 (q, 6H, J = 7.2 Hz, CH2), 3.31 (t, 2H, Hα), 1.22 (t, 9H, J = 7.2 Hz, CH3) ppm. 13C NMR (100 MHz, D2O) δ 57.43 (Cα), 54.44 (Cβ), 53.38 (N+-CH2CH3), 6.69 (N+-CH2CH3) ppm. HR-ESI-MS: calcd. for C8H20O2N (M − Br+) m/z = 146.1534; found 146.1534.(2-Hydroxyethyl)quinuclidinium bromide
The quinuclidine was prepared by placing 1.989 g (13.5 mmol) of quinuclidine hydrochloride in 270 ml of 0.10 M NaOH (27 mmol). The neutralized quinuclidine was extracted with chloroform and, upon removing the solvent under reduced pressure, 0.981 g (8.82 mmol, yield 65%) was obtained. A solution of 0.517 g (4.14 mmol) of 2-bromoethanol and 0.391 g (3.52 mmol) of quinuclidine in 5 mL of THF was heated under argon at 50 °C for 4 h. The product was washed with ether and dried. Yield 0.476 g (57%). mp 231–233 °C. 1H NMR (400 MHz, D2O) δ 3.95 (br t, 2H, Hβ), 3.44 (t, 6H, H2, J = 8.0 Hz), 3.24 (t, 2H, Hα, J = 5.2 Hz), 2.12 (hp, 1H, H4, J = 3.2 Hz), 1.93 (m, 6H, H3) ppm. 13C NMR (100 MHz, D2O) δ 65.31 (Cα), 55.50 (C2), 54.80 (Cβ), 23.61 (C3), 19.01 (C4) ppm. HR-ESI-MS: calcd. for C9H18NO (M − Br+) m/z = 156.1382; found 156.1382.Trimethylpentylphosphonium bromide
A solution of 0.755 g (5.00 mmol) of 1-bromopentane and 5.00 mL of 1.0 M trimethylphosphine (5.00 mmol) in THF was heated under argon at 70 °C for 12 h. The precipitate was washed with ether and dried. Yield 0.0754 g (7%). mp. 174–176 °C. 1H NMR (400 MHz, D2O) δ 2.18 (m, 2H, Hα), 1.84 (d, 9H, CH3, JP–H = 15.6 Hz), 1.61 (m, 2H, Hβ, J = 7.2 Hz), 1.44 (qn, 2H, Hγ, J = 7.2 Hz), 1.35 (sx, 2H, Hδ, J = 7.2 Hz), 0.89 (td, 3H, Hε, J = 7.2 Hz, JP–H = 1.6 Hz) ppm. 13C NMR (100 MHz, D2O) δ 32.14 (d, J = 15.5 Hz, Cγ), 22.95 (d, J = 52.3 Hz, Cα), 21.55 (s, Cδ), 20.66 (d, J = 4.2 Hz, Cβ), 13.26 (s, Cε), 7.53 (d, J = 54.9 Hz, CH3) ppm. 31P NMR (133 MHz, D2O) δ 25.93 ppm. HR-ESI-MS: calcd. for C8H20P (M − Br)+m/z = 147.1298; found 147.1298.Triethylpentylphosphonium bromide
A solution of 0.801 g (5.30 mmol) of 1-bromopentane and 6.00 mL of 1.0 M triethylphosphine (6.00 mmol) in THF were stirred under argon in a pressure tube for 12 h, followed by a further 6 h at 60 °C. The product was washed with ether and dried. Yield 0.244 g (17%). mp. 176–178 °C. 1H NMR (400 MHz, D2O) δ 2.20 (m, 2H, Hα), 2.19 (dq, 6H, CH2CH3, JP–H = 18.0 Hz, J = 7.8 Hz), 1.59 (m, 2H, Hβ, JP–H = 23.2 Hz, J = 7.2 Hz), 1.44 (q, 2H, Hγ, J = 7.2 Hz), 1.36 (sx, 2H, Hδ, J = 7.2 Hz), 1.22 (dt, 9H, CH3, JP–H = 18.0 Hz, J = 7.8 Hz), 0.89 (t, 3H, Hε, J = 7.2 Hz) ppm. 13C NMR (100 MHz, D2O) δ 32.39 (d, Cγ, JP–C = 15 Hz), 21.54 (s, Cδ), 20.47 (d, Cβ, JP–C = 6 Hz), 16.90 (d, Cα, JP–C = 48 Hz), 13.28 (s, Cε), 11.24 (d, P+-CH2CH3, JP–C = 50 Hz), 4.94 (d, P+-CH2CH3, JP–C = 5 Hz) ppm. 31P NMR (133 MHz, D2O) δ 38.56 ppm. HR-ESI-MS: calcd. for C11H26P (M-Br)+m/z = 189.1766; found 189.1766.(2-Hydroxyethyl)trimethylphosphonium bromide
A solution of 0.618 g (5.95 mmol) of 2-bromoethanol and 5.00 mL of 1.0 M trimethylphosphine (5.00 mmol) in THF was heated at 70 °C under argon in a pressure tube for 12 h. The precipitate was washed with ether and dried. Yield 0.483 g (48%). mp. 290–292 °C. 1H NMR (400 MHz, D2O) δ 3.97 (dt, 2H, Hβ, JP–H = 20.0 Hz, J = 6.2 Hz), 2.46 (dt, 2H, Hα, JP–H = 13.2 Hz, J = 6.2 Hz), 1.87 (d, 9H, CH3, J = 14.8 Hz) ppm. 13C NMR (100 MHz, D2O) δ 54.88 (d, Cβ, JP–C = 5 Hz), 26.20 (d, Cα, JP–C = 49 Hz), 8.15 (d, P+-CH3, JP–C = 49 Hz), 5.00 (d, CH3JP–C = 5 Hz) ppm. 31P NMR (133 MHz, D2O) δ 26.33 ppm. HR-ESI-MS: calcd. for C5H14OP (M − Br)+m/z = 121.0776; found 121.0772.(2-Hydroxyethyl)triethylphosphonium bromide
A solution of 0.800 g (6.40 mmol) of 2-bromoethanol and 7.00 mL of 1.0 M triethylphosphine (7.00 mmol) in THF was heated at 70 °C under argon in a pressure tube for 24 h. The product was washed with ether and dried. Yield 0.158 g (10%). mp. 221–225 °C (lit.42 223 °C). 1H NMR (400 MHz, D2O) δ 3.91 (dt, 2H, Hβ, 3J = 6.2 Hz, JP–H = 18 Hz), 2.42 (dt, 2H, Hα, 3J = 6.2 Hz, JP–H = 12 Hz), 2.22 (dq, 6H, CH2CH3, 3J = 7.8 Hz, JP–H = 13 Hz), 1.19 (dt, 9H, CH3CH2, 3J = 7.8 Hz, JP–H = 18 Hz) ppm. 13C NMR (100 MHz, D2O) δ 64.60 (d, J = 5.6 Hz, Cβ), 20.48 (d, J = 48.9 Hz, Cα), 11.60 (d, J = 49.4 Hz, P+-CH2CH3), 4.62 (d, J = 5.4 Hz, P+-CH2CH3) ppm. 31P NMR (133 MHz, D2O) δ 37.93 ppm. HR-ESI-MS: calcd. for C8H20OP (M − Br)+m/z = 163.1246; found 163.1246.(2-Acetoxyethyl)trimethylphosphonium bromide
A solution of 0.707 g (4.23 mmol) of 2-bromoethyl acetate and 5.10 mL of 1.0 M trimethylphosphine (5.10 mmol) in THF was heated at 70 °C under argon for 72 h. The hygroscopic product was washed with ether and dried. Yield 0.348 g (34%). mp. 59–62 °C. 1H NMR (400 MHz, D2O) δ 4.42 (dt, 2H, CH2-O, JP–H = 19.9 Hz, J = 6.2 Hz), 2.64 (dt, 2H, CH2-P+, JP–H = 13.2 Hz, J = 6.2 Hz), 2.09 (s, 3H, CH3C(O)), 1.90 (d, 9H, CH3, JP–H = 14.4 Hz) ppm. 13C NMR (100 MHz, D2O) δ 173.32 (s, CO), 57.93 (d, Cβ, JP–C = 6 Hz), 23.04 (d, Cα, JP–C = 54 Hz), 20.19 (s, CH3), 7.89 (d, P+-CH3, JP–C = 55 Hz) ppm. 31P NMR (133 MHz, D2O) δ 25.84 ppm. HR-ESI-MS: calcd. for C7H16O2P (M − Br)+m/z = 163.0882; found 163.0888.(2-Acetoxyethyl)triethylphosphonium bromide
A solution of 0.749 g (4.48 mmol) of 2-bromoethylacetate and 5.00 mL of 1.0 M triethylphosphine (5.00 mmol) in THF was heated at 60 °C under argon in a pressure tube for 6 h, followed by a further 12 h at room temperature. The hygroscopic product was washed with ether and dried. Yield ∼2%. mp. 58–64 °C (lit.42 74.5 °C). 1H NMR (400 MHz, D2O) δ 4.41 (dt, 2H, Hβ, JP–H = 17.5 Hz, J = 6.5 Hz), 2.65 (dt, 2H, CH2-P+, JP–H = 12.5 Hz, J = 6.5 Hz), 2.27 (dq, 6H, CH2CH3, JP–H = 13.0 Hz, J = 7.6 Hz), 2.10 (s, 3H, CH3C(O)), 1.23 (dt, 9H, CH3CH2, JP–H = 18.5 Hz) ppm. 13C NMR (100 MHz, D2O) δ 173.30 (s, CO), 57.75 (d, J = 5.4 Hz, Cβ), 20.17 (s, COCH3), 17.26 (d, J = 49.3 Hz, Cα), 11.51 (d, J = 48.8 Hz, P+-CH2CH3), 4.62 (d, J = 5.4 Hz, P+-CH2CH3) ppm. 31P NMR (133 MHz, D2O) δ 38.34 ppm. HR-ESI-MS: calcd. for C10H22O2P (M − Br)+m/z = 205.1351; found 205.1352.Methods
The 1H, 13C, and 31P NMR spectra were recorded on Bruker Avance 400 or 500 spectrometers in D2O. The high-resolution electrospray ionization time-of-flight mass spectra were recorded on a QStar XL QqTOF instrument with an ESI source. The host–guest stability constants for the cucurbit[7]uril complexes with the cationic guests (KCB[7]) were determined by competitive 1H NMR binding studies at 298 K as described by Isaacs and co-workers.3 The solutions were prepared in D2O containing acetate buffer (0.050 mol dm−3 NaOAc-d3–0.025 mol dm−3 DCl) at pD 4.75 with 3-trimethylsilylpropionic-2,2,3,3-d4 acid (KCB[7] = (1.82 ± 0.22) × 107 dm3 mol−1),3 tetraethylammonium bromide ((1.0 ± 0.2) × 106 dm3 mol−1),4h tetramethylphosphonium bromide ((2.2 ± 0.4) × 106 dm3 mol−1),4h tetraethylphosphonium chloride ((1.3 ± 0.3) × 105 dm3 mol−1),4hp-xylylenediamine ((1.84 ± 0.34) × 109 dm3 mol−1),3 or 1,2-phenylenediamine ((8.04 ± 1.28) × 104 dm3 mol−1)3 (Sigma-Aldrich, used as received) as the competing guests.Conclusions
The cucurbit[7]uril host molecule forms very stable complexes with a variety of choline cations and their phosphonium analogues in aqueous solution. The stability of the host–guest complexes depends on the nature of the cationic head group as well as the size and charge on the substituents on the opposite end of the guest and on the ethylene linker. The neutral CB[7] host is able to recognize the cholines in water without the use of aromatic or anionic components in macrocycle subunits. We are currently investigating the use of CB[7] in assisting the metal-catalyzed hydrolyses of choline phosphate esters, such as 4-nitrophenylphosphocholine, by binding the choline ester and helping to stabilize transition states involving binding of the metal ion, such as zinc, at the carbonyl-lined portal(s) of the cucurbit[7]uril host.Acknowledgements
The financial support of this work by the Natural Sciences and Engineering Research Council of Canada is gratefully acknowledged (D.H.M.). The Ontario Government and the Walter C Sumner Foundation are thanked for scholarship support (I.W.W.).Notes and references
- (a) W. L. Mock, Top. Curr. Chem., 1995, 175, 1 CAS; (b) K. Kim, Chem. Soc. Rev., 2002, 31, 96 RSC; (c) O. A. Gerasko, D. G. Samsonenko and V. P. Fedin, Russ. Chem. Rev., 2002, 71, 741 RSC; (d) J. W. Lee, S. Samal, N. Selvapalam, H.-J. Kim and K. Kim, Acc. Chem. Res., 2003, 36, 621 CrossRef CAS; (e) J. Lagona, P. Mukhopadhyay, S. Chakrabarti and L. Isaacs, Angew. Chem., Int. Ed., 2005, 44, 4844 CrossRef CAS; (f) N. J. Wheate, Aust. J. Chem., 2006, 59, 354 CrossRef CAS; (g) K. Kim, N. Selvapalam, Y. H. Ko, K. M. Park, D. Kim and J. Kim, Chem. Soc. Rev., 2007, 36, 267 RSC; (h) L. Isaacs, Chem. Commun., 2009, 619 RSC.
- (a) J. Kim, I.-S. Jung, S.-Y. Kim, E. Lee, J.-K. Kang, S. Sakamoto, K. Yamaguchi and K. Kim, J. Am. Chem. Soc., 2000, 122, 540 CrossRef CAS; (b) A. Day, A. P. Arnold, R. J. Blanch and B. Snushall, J. Org. Chem., 2001, 66, 8094 CrossRef CAS.
- S. Liu, C. Ruspic, P. Mukhopadhyay, S. Chakrabarti, P. Y. Zavalij and L. Isaacs, J. Am. Chem. Soc., 2005, 127, 15959 CrossRef CAS.
- See for example: (a) W. Ong, M. Gomez-Kaifer and A. E. Kaifer, Org. Lett., 2002, 4, 1791 CrossRef CAS; (b) H.-J. Kim, W. S. Keon, Y. H. Ko and K. Kim, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 507 CrossRef; (c) Choi, S. H. Park, A. Y. Ziganshina, Y. H. Ko, J. W. Lee and K. Kim, Chem. Commun., 2003, 2176 RSC; (d) K. Moon and A. E. Kaifer, Org. Lett., 2004, 6, 185 CrossRef CAS; (e) V. Sindelar, M. A. Cejas, F. M. Raymo and A. E. Kaifer, New J. Chem., 2005, 29, 280 RSC; (f) D. S. N. Hettiarachchi and D. H. Macartney, Can. J. Chem., 2006, 84, 905 CrossRef CAS; (g) R. Wang, L. Yuan, H. Ihmels and D. H. Macartney, Chem.–Eur. J., 2007, 13, 6468 CrossRef CAS; (h) A. D. St-Jacques, I. W. Wyman and D. H. Macartney, Chem. Commun., 2008, 4936 RSC; (i) G. A. Vincil and A. R. Urbach, Supramol. Chem., 2008, 20, 681 CrossRef CAS; (j) M. Megyesi, L. Biczók and I. Jablonkai, J. Phys. Chem. C, 2008, 112, 3410 CrossRef CAS.
- (a) W. S. Jeon, K. Moon, S. H. Park, H. Chun, Y. K. Ho, J. Y. Lee, S. E. Lee, S. Samal, N. Selvapalam, M. V. Rekharsky, V. Sindelar, D. Sobransingh, Y. Inoue, A. E. Kaifer and K. Kim, J. Am. Chem. Soc., 2005, 127, 12984 CrossRef CAS; (b) R. Wang, L. Yuan and D. H. Macartney, Organometallics, 2006, 25, 1820 CrossRef CAS; (c) M. V. Rekharsky, T. Mori, C. Yang, Y. H. Ko, N. Selvapalam, H. Kim, D. Sobransingh, A. E. Kaifer, S. Liu, L. Isaacs, W. Chen, S. Moghaddam, M. K. Gilson, K. Kim and Y. Inoue, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 20737 CrossRef CAS.
- (a) N. J. Wheate, A. I. Day, R. J. Blanch, A. P. Arnold, C. Cullinane and J. G. Collins, Chem. Commun., 2004, 1424 RSC; (b) Y. J. Jeon, S. Y. Kim, Y. H. Ko, S. Sakamoto, K. Yamaguchi and K. Kim, Org. Biomol. Chem., 2005, 3, 2122 RSC; (c) N. J. Wheate, D. P. Buck, A. I. Day and J. G. Collins, Dalton Trans., 2006, 451 RSC; (d) S. Kemp, N. J. Wheate, S. Wang, J. G. Collins, S. F. Ralph, A. I. Day, V. J. Higgins and J. P. Aldrich-Wright, J. Biol. Inorg. Chem., 2007, 12, 969 CrossRef CAS.
- D. P. Buck, P. M. Abeysinghe, C. Cullinane, A. I. Day, J. C. Collins and M. M. Harding, Dalton Trans., 2008, 2328 RSC.
- (a) M. E. Bush, N. D. Bouley and A. R. Urbach, J. Am. Chem. Soc., 2005, 127, 14511 CrossRef CAS; (b) M. L. Heitmann, A. B. Taylor, P. J. Hart and A. R. Urbach, J. Am. Chem. Soc., 2006, 128, 12574 CrossRef CAS; (c) A. Henning, H. Bakirci and W. M. Nau, Nat. Methods, 2007, 4, 629 CrossRef CAS; (d) A. Hennig, G. Ghale and W. M. Nau, Chem. Commun., 2007, 1614 RSC; (e) M. V. Rekharsky, H. Yamahura, Y. H. Ko, N. Selvapalam, K. Kim and Y. Inoue, Chem. Commun., 2008, 2236 RSC.
- R. Wang and D. H. Macartney, Org. Biomol. Chem., 2008, 6, 1955 RSC.
- I. W. Wyman and D. H. Macartney, Org. Biomol. Chem., accepted for publication. 10.1039/b915694a.
- N. Saleh, A. L. Koner and W. M. Nau, Angew. Chem., Int. Ed., 2008, 47, 5398 CrossRef CAS.
- R. Wang, B. C. MacGillivray and D. H. Macartney, Dalton Trans., 2009, 3584 RSC.
- (a) R. Wang, L. Yuan and D. H. Macartney, Chem. Commun., 2005, 5867 RSC; (b) R. Wang, L. Yuan and D. H. Macartney, J. Org. Chem., 2006, 71, 1237 CrossRef CAS; (c) J. Mohanty, A. C. Bhasikuttan, W. M. Nau and H. Pa, J. Phys. Chem. B, 2006, 110, 5132 CrossRef CAS; (d) M. Shaikh, J. Mohanty, P. K. Singh, W. M. Nau and H. Pal, Photochem. Photobiol. Sci., 2008, 7, 408 RSC.
- R. Wang, L. Yuan and D. H. Macartney, Chem. Commun., 2006, 2908 RSC.
- R. Silman and J. L. Sussman, Curr. Opin. Pharmacol., 2005, 5, 293 CrossRef CAS.
- (a) D. A. Dougherty and D. A. Stauffer, Science, 1990, 250, 1558 CrossRef CAS; (b) C. A. Ma and D. A. Dougherty, Chem. Rev., 1997, 97, 1303 CrossRef CAS; (c) A. Zacharias and D. A. Dougherty, Trends Pharmacol. Sci., 2002, 23, 281 CrossRef CAS.
- (a) M. J. Minch, J. P. Sevenair and C. Henling, J. Org. Chem., 1979, 44, 3247 CrossRef CAS; (b) K. Ito, K. Nagase, N. Morohashi and Y. Ohba, Chem. Pharm. Bull., 2005, 53, 90 CrossRef CAS.
- (a) M. Dhaenens, L. Lacombe, J.-M. Lehn and J.-P. Vigneron, J. Chem. Soc., Chem. Commun., 1984, 1097 RSC; (b) T. H. Webb, H. Suh and C. S. Wilcox, J. Am. Chem. Soc., 1991, 113, 8554 CrossRef CAS; (c) R. Meric, J. P. Vigneron and J.-M. Lehn, J. Chem. Soc., Chem. Commun., 1993, 129 RSC; (d) S. M. Ngola and D. A. Dougherty, J. Org. Chem., 1998, 63, 4566 CrossRef CAS; (e) S. M. Ngola, P. C. Kearney, S. Mecozzi, K. Russell and D. A. Dougherty, J. Am. Chem. Soc., 1999, 121, 1192 CrossRef CAS.
- (a) P. C. Kearney, L. S. Mizoue, R. A. Kumpf, J. E. Forman, A. Forman and D. A. Dougherty, J. Am. Chem. Soc., 1993, 115, 9907 CrossRef CAS; (b) R. Meric, J.-M. Lehn and J.-P. Vigneron, Bull. Soc. Chem. Fr., 1994, 131, 579; (c) S. Bartoli and S. Roelens, J. Am. Chem. Soc., 2002, 124, 8307 CrossRef CAS; (d) P. Sarri, F. Venturi, F. Cuda and S. Roelens, J. Org. Chem., 2004, 69, 3654 CrossRef CAS; (e) P. T. Corbett, J. K. M. Sanders and S. Otto, Chem.–Eur. J., 2008, 14, 2153 CrossRef CAS.
- S. Tomàs, R. Prohens, M. Vega, M. C. Rotger, P. M. Deyà, P. Ballester and A. Costa, J. Org. Chem., 1996, 61, 9394 CrossRef CAS.
- J. M. Harrowfield, W. R. Richmond and A. M. Sobolev, J. Inclusion Phenom. Mol. Recognit. Chem., 1994, 19, 257 CrossRef CAS.
- (a) J.-M. Lehn, R. Meri, J.-P. Vigneron, M. Cesario, J. Guilhem, C. Pascard, Z. Asfari and J. Vicens, Supramol. Chem., 1995, 5, 97 CrossRef CAS; (b) K. N. Koh, K. Araki, A. Ikeda, H. Otsuka and S. Shinkai, J. Am. Chem. Soc., 1996, 118, 755 CrossRef CAS; (c) J. L. Atwood, L. J. Barbour, P. C. Junk and G. W. Orr, Supramol. Chem., 1995, 5, 105 CrossRef CAS; (d) G. Arena, A. Casnati, L. Mirone, D. Sciotto and Ungaro, Tetrahedron Lett., 1997, 38, 1999 CrossRef CAS; (e) G. Arena, A. Casnati, A. Contino, G. G. Lombardo, D. Sciotto and R. Ungaro, Chem.–Eur. J., 1999, 5, 738 CrossRef CAS; (f) G. Arena, A. Casnati, A. Contino, G. F. Gulino, D. Sciotto and R. Ungaro, J. Chem. Soc., Perkin Trans. 2, 2000, 419 RSC; (g) T. Jin, J. Inclusion Phenom. Macrocyclic Chem., 2003, 45, 195 CrossRef CAS; (h) A. Specht, F. Ziarelli, P. Bernard, M. Goeldner and L. Ping, Helv. Chim. Acta, 2005, 88, 2641 CrossRef CAS; (i) H. Bakirci and W. M. Nau, Adv. Funct. Mater., 2006, 16, 237 CrossRef CAS; (j) L.-H. Wang, D.-S. Guo, Y. Chen and Y. Liu, Thermochim. Acta, 2006, 443, 132 CrossRef CAS; (k) N. Norbakov, P. Timmerman, N. Lidich, B. Urbach, A. Sa'ar and S. Yitzchaik, Langmuir, 2008, 24, 2580 CrossRef CAS.
- (a) V. Böhmer, A. Dalla Cort and L. Mandolini, J. Org. Chem., 2001, 66, 1900 CrossRef CAS; (b) B. Masci, S. L. Mortela, D. Persiani and P. Thuéry, J. Org. Chem., 2006, 71, 504 CrossRef CAS; (c) B. Masci, D. Persiani and P. Thuéry, J. Org. Chem., 2006, 71, 9784 CrossRef CAS; (d) T. Haino, H. Fukuoka, M. Katayama and Y. Fukazawa, Chem. Lett., 2007, 36, 1054 CrossRef CAS; (e) M. Fehlinger and W. Abraham, J. Inclusion Phenom. Macrocyclic Chem., 2007, 58, 263 CrossRef CAS.
- (a) S.-D. Tan, W.-H. Chen, A. Satake, B. Wang, Z.-L. Zu and Y. Kobuke, Org. Biomol. Chem., 2004, 2, 2719 RSC; (b) W.-H. Chen, Y. Wei, S.-D. Tan, B. Wang and Z.-L. Xu, Supramol. Chem., 2005, 17, 469 CrossRef CAS.
- (a) H.-J. Schneider, D. Guttes and U. Schneider, Angew. Chem., Int. Ed. Engl., 1986, 25, 647 CrossRef; (b) H.-J. Schneider and U. Schneider, J. Org. Chem., 1987, 52, 1613 CrossRef CAS; (c) H.-J. Schneider, D. Guttes and U. Schneider, J. Am. Chem. Soc., 1988, 110, 6449 CrossRef CAS; (d) H.-J. Schneider and U. Schneider, Chem. Ber., 1994, 127, 2455 CAS; (e) H.-J. Schneider and U. Schneider, J. Inclusion Phenom. Mol. Recognit. Chem., 1994, 19, 67 CrossRef CAS; (f) M. Inouye, K. Hashimoto and K. Isagawa, J. Am. Chem. Soc., 1994, 116, 5517 CrossRef CAS; (g) S. J. Park and J.-I. Hong, Tetrahedron Lett., 2000, 41, 8311 CrossRef CAS; (h) K. Salorinne, T.-R. Tero, K. Riikonen and M. Nissinen, Org. Biomol. Chem., 2009, 7, 4211 RSC.
- B. Schnatwinkel, M. V. Rehkarsky, V. V. Borovkov, Y. Inoue and J. Mattay, Tetrahedron Lett., 2009, 50, 1374 CrossRef CAS.
- (a) P. S. Bates, R. Kataky and D. Parker, J. Chem. Soc., Chem. Commun., 1993, 691 RSC; (b) D. Parker, R. Kataky, P. M. Kelly and S. Palmer, Pure Appl. Chem., 1996, 68, 1219 CAS; (c) R. L. E. Furlan, Y.-F. Ng, G. R. L. Cousins, J. E. Redman and J. K. M. Sanders, Tetrahedron, 2002, 58, 771 CrossRef CAS; (d) M.-L. Dumartin, C. Givelet, P. Meyrand, B. Bibal and I. Gosse, Org. Biomol. Chem., 2009, 7, 2725 RSC.
- J. Zhao, H.-J. Kim, J. Oh, S.-Y. Kim, J. W. Lee, S. Hakamoto, K. Yamaguchi and K. Kim, Angew. Chem., Int. Ed., 2001, 40, 4233 CrossRef CAS.
- (a) P. Ballester, A. Shivanyuk, A. Rafir Far and J. Rebek, Jr., J. Am. Chem. Soc., 2002, 124, 14014 CrossRef CAS; (b) F. Hof, L. Trembleau, E. C. Ullrich and J. Rebek, Jr., Angew. Chem., Int. Ed., 2003, 42, 3150 CrossRef; (c) S. M. Biros, E. C. Ullrich, F. Hof, L. Trembleau and J. Rebek, Jr., J. Am. Chem. Soc., 2004, 126, 2870 CrossRef CAS; (d) A. Lledó, R. J. Hooley and J. Rebek, Jr., Org. Lett., 2008, 10, 3669 CrossRef CAS.
- (a) A. Gissot and J. Rebek Jr., J. Am. Chem. Soc., 2004, 126, 7424 CrossRef CAS; (b) R. Sebastien and J. Rebek, Jr., J. Am. Chem. Soc., 2004, 126, 16280 CrossRef CAS; (c) F. H. Zelder and J. Rebek, Jr., Chem. Commun., 2006, 753 RSC.
- F. Cuevas, S. Di Stefano, J. O. Magrans, P. Pridox, L. Mandolini and J. de Mendoza, Chem.–Eur. J., 2000, 6, 3228 CrossRef CAS.
- (a) B. S. Szwergold, F. Kappler, M. Molder, C. Shaller and T. R. Brown, NMR Biomed., 1994, 7, 121 CrossRef CAS; (b) J. V. Jimenez, T. L. Richards, A. C. Heide, J. R. Grierson and E. G. Shankland, Magn. Res. Med., 1995, 33, 285 CrossRef CAS; (c) J. C. Street, B. X. Szwergold, C. Matei, F. Kappler, U. Mahmood, T. R. Brown and J. A. Koutcher, Magn. Res. Med., 1997, 38, 769 CrossRef CAS.
- I. W. Wyman and D. H. Macartney, J. Org. Chem., 2009, 74, 8031 CrossRef CAS.
- See Electronic Supplementary Information (ESI)†.
- Y. H. Ko, H. Kim, Y. Kim and K. Kim, Angew. Chem., Int. Ed., 2008, 47, 4106 CrossRef CAS.
- I. W. Wyman and D. H. Macartney, Org. Biomol. Chem., 2008, 6, 1796 RSC.
- P. De Maria, A. Fontana, S. Frascari, G. Gargaro, D. Spinelli and M. O. Tinti, J. Pharm. Sci., 1994, 83, 742 CrossRef CAS.
- P. Gettins, M. Potter, R. J. Leatherbarrow and R. A. Dwek, Biochemistry, 1982, 21, 4927 CrossRef CAS.
- J. C. Kellett and W. C. Doggett, J. Pharm. Sci., 1966, 55, 414 CrossRef CAS.
- J. M. Orosz and J. G. Wetmur, Biopolymers, 1977, 16, 1183 CrossRef CAS.
- M. F. Shostakovskii, Izv. Akad. Nauk SSSR, Ser. Khim., 1958, 204 Search PubMed.
- R. R. Renshaw and R. A. Bishop, J. Am. Chem. Soc., 1938, 60, 946 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details of guest syntheses and characterizations, ESI-MS spectra, and 1H NMR spectra of competitive binding experiments. See DOI: 10.1039/b917610a |
| This journal is © The Royal Society of Chemistry 2010 |