Protein thin film machines
Stefania
Federici
a,
Giulio
Oliviero
a,
Kimberly
Hamad-Schifferli
*bc and
Paolo
Bergese
*ab
aChemistry for Technologies Laboratory and INSTM, University of Brescia, Via Branze, 38, 25123, Brescia, Italy
bDepartment of Mechanical Engineering, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139, USA. E-mail: paolo.bergese@ing.unibs.it
cDepartment of Biological Engineering, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139, USA. E-mail: schiffer@mit.edu
First published on 8th October 2010
Abstract
We report the first example of microcantilever beams that are reversibly driven by protein thin film machines fuelled by cycling the salt concentration of the surrounding solution. We also show that upon the same salinity stimulus the drive can be completely reversed in its direction by introducing a surface coating ligand. Experimental results are throughout discussed within a general yet simple thermodynamic model.
Natural molecular machines drive essential biological processes and have generated a great deal of interest, being even attempted as an argument to confute Darwin's evolution theory.1 More recently, the ability to induce molecular motion upon external stimulation has been mimicked by synthetic molecules and eventually is beginning to be harnessed to generate nano- and even microscale mechanical work.2 Flexible microcantilever beams can not only serve for this task but also as tools to study the inter- and intra-molecular interactions set in action by the molecular machines themselves. However, to date, molecular machine driven microcantilevers with reversible and multi-stage performances have been only reported for synthetic molecules, such as rotaxanes,3 polymer brushes,4 aptamers,5 or short polypeptides.6
Proteins, on the other hand, are naturally engineered to undergo complex and large scale conformational changes7 that can be tuned by environmental stimuli such as salt concentration and pH, but exploiting them for synthetic molecular devices presents significant challenges. Any useful machine requires an interface for inputs and outputs,2 but inorganic interfaces are especially problematic for proteins, causing denaturation or non-specific adsorption, thus obscuring desired effects.8 Furthermore, inorganic surfaces for biomolecules are very often not bare, but have surface coating ligands, and these molecules dominate the nature of the biological–inorganic interface.9 Unfortunately, interactions with the ligand are difficult to measure directly, so much about the behavior of the interface remains unknown.10 Clearly, if hybrid molecular devices are to exploit the unique properties of proteins, an understanding of the effect of the surface coating ligand is also essential.
Here, we report the first example of a microcantilever (MC) that is reversibly driven by a protein thin film machine which is fuelled by cycling the salinity of the surrounding solution (Fig. 1) and explore the effect of surface coating ligand. We find that the ligand can significantly affect the MC movement, completely reversing its direction. These results show that choice of ligand plays an important role in tuning machine behavior and action. We discuss results using a simple thermodynamic framework,11 and find that the molecular forces that build up to drive the MC are hydration forces,12 consistent in magnitude with unfolding forces measured in single-molecule experiments.13
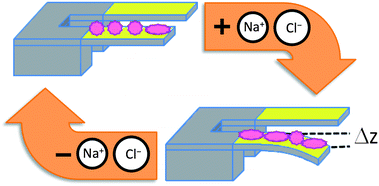 | ||
| Fig. 1 Scheme of a protein driven microcantilever (MC) which is fuelled by cycling the salt concentration of the surrounding solution. A self-assembled monolayer of protein is deposited on the top face of a MC beam (for our experiments we use Au coated Si MC beams). At the starting stage the system is in chemomechanical equilibrium with the surrounding saline buffer solution. Modulation of the salt concentration shifts the conformational equilibrium of the proteins and in turn unbalances the in-plane intermolecular forces within the film. The resultant forces cumulate and trigger a macroscopic surface work (i.e. induce a change of the surface pressure) that the MC counterbalances by bending till chemomechanical equilibrium is restored. Note: to rule out “unspecific” effects, the MC bending, Δz, is referred to an unmodified MC (reference MC). | ||
The equilibrium between the conformational states of a protein can be represented by two states, A and B. In dilute electrolyte solutions, equilibrium can be significantly shifted toward one of the states by tuning the salt concentration, which changes the number of ions bound by the protein and the Debye–Hückel screening of the charge interactions on the protein.14 These changes can either expose peptide groups that are buried in A to the solution in B, denaturing the protein, or drive the opposite phenomenon, causing the protein to fold. Thus, A and B can be regarded as two opposite states of a molecular switch (one of the two basic types of molecular machines2) that can be cyclically populated by varying the salt concentration.
By adopting a formalism analogous to the one used to include counterion binding in DNA hybridization,15 we can express the equilibrium between states A and B as follows:
| A + jC ⇄ B | (1) |
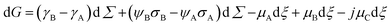 | (2) |
From eqn (2) the overall change in the molar Gibbs free energy, ΔrG, is:
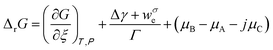 | (3) |
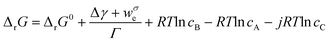 | (4) |
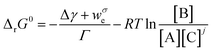 | (5) |
It is of note that typical equations describing thermodynamics of protein folding are particular cases of eqn (5). In bulk solution the surface terms disappear and eqn (5) reduces to the classic definition of conformational stability free energy.25,26 Instead, when only mechanical work exists (that is weσ = 0), eqn (5) matches the equation introduced by Bustamante to account for the effect of force on the free energy of a single-molecule transformation.13
Cytochrome c (cyt-c) was selected as a model protein because its structure and folding have been extensively characterized experimentally and computationally in both free solution and at the Au–solution interface.27–32 In particular, it was found that when conjugated to gold NPs, cyt-c changes its α-helical structure. Unfolding varies with salt concentration9 and the choice of labeling site for the NP can have a profound influence on structure and folding ability.23
First, we probed the mechanical work performed by thin films of cytochrome c from Saccharomyces cerevisiae (YCC), which possesses a unique exposed cysteine at position 102 (C102) that can covalently bind to Au. YCC was bound to the Au coated face of Si MCs with (YCC–BPS–Au MCs) and without (YCC–Au MCs) the ligand bis-(p-sulfonatophenyl)phenylphosphine, BPS. BPS caps the Au surface, resulting in a negatively charged surface (see Fig. 3).
Arrays of eight Si MCs (500 × 100 × 1 µm3) with the top faces coated by 20 nm of Au (Concentris, Basel) were modified (Fig. 1). As-received arrays were all cleaned by ozone-UV before modification. Arrays whose modification included a pre-treatment by BPS Au capping were exposed overnight to a 1 mM water solution of BPS (Strem Chemicals). On each MC array (BPS pre-treated and bare) YCC thin films were deposited onto four of the eight MCs by overnight incubation in a 10 mM PBS solution of YCC 20 µM using a capillary delivery system that could incubate each MC separately. The other four MCs were left unmodified and served as references. After YCC immobilization all arrays were carefully rinsed with PBS.
MC arrays were actuated by a Cantisens Research platform (Concentris, Basel) equipped with a microfluidic system to handle liquid delivery to the MCs and multiple lasers for simultaneous measurement of the deflection of the individual MCs. All buffer solutions were flowed through the microfluidic chamber at a rate of 0.83 µL s−1.
Fig. 2 shows the mean absolute deflection of the four MCs modified with YCC (YCC–Au, black solid line) and the four reference MCs (dashed blue line) under cycling of the salt concentration between 50 and 10 mM PBS. Error bars are reported at significant points and represent the standard deviation of the mean of homologous MCs. There is a significant separation between the YCC–Au and the Au deflection signals, indicating that the ionic cycle drives a specific surface work of the YCC thin film. The absolute deflection of the reference MCs is non-zero because of the surface work due to ion adsorption at the Au–solution interface,33 and the bending of the laser beam used for the deflection readout after the change of the solution refraction index upon salinity variation.34 This indicates the importance of using reference MCs to isolate unspecific contributions to the surface work. The differential signal relative to the Ref-MCs also allows minimization of instrumental artifacts such as fluctuations induced by solution flow, which result in peaks upon switching between the 50 mM PBS and 10 mM PBS solutions.
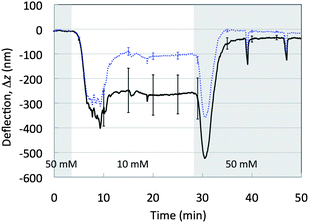 | ||
| Fig. 2 Mean absolute deflection signals of the four MCs supporting the YCC thin film (YCC–Au), black solid line, and of the four reference MCs, dashed blue line, when the solution is cycled from 50 mM PBS (grey area) to 10 mM PBS (white area). Error bars are reported at significant points and represent the standard deviation of the mean of homologous MCs. | ||
The differential deflection, Δz, for the YCC–Au MCs is reported in the left panel of Fig. 3 (black solid line). Upon a decrease of the salt concentration from 50 mM to 10 mM the YCC–Au MCs undergo a downward mean differential deflection of Δz = −160 nm, thus balancing a compressive (repulsive) surface work of the YCC thin film onto the Au MC face. The figure also shows that the work is performed in a reversible and sharp fashion, comparable to the behavior of thin film switches made of synthetic molecules.3–6 Differential deflection can be analyzed using eqn (5) to separate surface work into the mechanical and electrical contributions. We can thus assume that the MC balances by bending the surface pressure, −Δγ, which comprises the mechanical contribution of the surface work. To a first approximation, surface pressure can be related to Δz by Stoney's Equation,11 which yields for Δz = −160 nm a surface pressure of −34 mN m−1.35 Supposing a typical monolayer of YCC (∼1012 molecules cm−2) we can infer that each YCC molecule contributes to change the surface pressure of about −60 pN m−1. Remarkably, the repulsive forces generating this pressure are of the same order of magnitude needed to mechanically unfold biomolecules, as measured in single-molecule experiments, ranging from tens to hundreds of pN.13
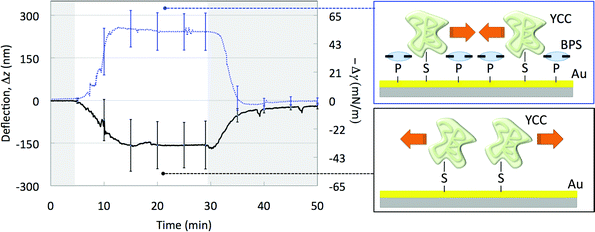 | ||
| Fig. 3 (Left panel) Mean differential deflection signals (left ordinate axis) and mean differential surface pressure (right ordinate axis) of the YCC–Au MCs (black solid line) and of the YCC–BPS–Au MCs (blue dashed line) when the solution is cycled from 50 mM PBS (grey area) to 10 mM PBS (white area). (Right top panel) Scheme of the top face of a YCC–BPS–Au MC and of the in-plane inter-protein attractive forces that are triggered by the salt concentration decrease and cumulate to deflect the MC upward. (Right bottom panel) Scheme of the top face of a YCC–Au MC and of the in-plane inter-protein repulsive forces that are triggered by the salt concentration decrease and cumulate to deflect the MC downward. | ||
However, under the same salinity changes, the Au coated MCs previously capped with the BPS ligand, YCC–BPS–Au MCs, undergo a mean differential upward deflection of Δz = +240 nm (Fig. 3, blue dashed line), corresponding to a tensile (attractive) surface pressure of +52 mN m−1.35 This shows that the surface coating ligand can significantly affect the behavior of the protein thin film switch.
The thermodynamic framework introduced before can explain these results. Eqn (5) suggests that the intermolecular force that builds up molecule by molecule to a macroscopic mechanical effect is not electrical in nature, but that its extent and sign as well as the work it performs (Δγ/Γ) are determined by the work necessary to modify the interfacial electric double layer (weσ/Γ) and by the chemical work (last term at the right hand side of the equation), in agreement with previous empirical findings.3–6 Accordingly, since they have different electric potentials ψ, YCC–Au and YCC–BPS–Au films under the same salinity conditions perform different electrical and chemical work that must be balanced by different mechanical work, resulting in different MC bending.
From a molecular perspective both the sign and modulus of the surface pressures directly recall molecular hydration forces,12 which become significant in confined geometries with nanoscale surface separations (typically below 10 molecular water diameters). These hydration forces oscillate with distance, varying between attraction and repulsion with a periodicity equal to the water diameter. Since the inter-protein separation in the YCC–MC and YCC–BPS–MC films differs due to BPS intercalation23 (Fig. 3) the intermolecular forces that are responsible for the mechanical work may be these hydration forces. This result is similar to recent studies of self-assembled monolayers (SAMs) of single strand DNA36 which point at the key role played by the structural forces in water in the mechanisms that allow harnessing molecular transformations of surface confined biopolymers to perform a larger scale mechanical motion.
In order to get further insight into the role played by the ligand, we probed the behavior of horse cytochrome c (HCC) on Au coated MCs capped with BPS. HCC has nearly identical structure to YCC but lacks C102 and so it cannot covalently bind to the Au MC. HCC electrostatically adsorbs onto BPS capped Au surfaces without intercalating BPS and in a way that its structure is maintained9,37 (Fig. 4). The used MC arrays, the functionalization scheme and procedures and the actuation modes were the same adopted for the YCC–BPS arrays. The left panel of Fig. 4 shows the mean differential deflection signal of HCC on BPS coated MCs under three salinity cycles. Upon a decrease of salt concentration from 50 mM to 10 mM the HCC–BPS–Au MCs undergo a downward mean differential bending of −105 nm, balancing a compressive (repulsive) surface pressure of −23 mN m−1. Albeit performing less mechanical work the HCC–BPS–Au MCs behave similarly to the YCC–Au MCs. This suggests that BPS switches the protein–protein interaction from repulsive to attractive only when proteins are intercalated in it. It remains open if BPS is involved in the molecular mechanism only as a “spacer” that changes protein–protein interactions, or if it plays a more significant role in these interactions. In turn, it is an equally fundamental matter for future development to investigate the threshold density of protein coverage to perform reversible mechanical work.
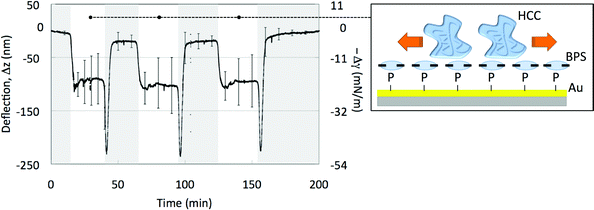 | ||
| Fig. 4 (Left panel) Mean differential deflection signals (left ordinate axis) and mean differential surface pressure (right ordinate axis) of the HCC–BPS–Au MCs when the solution is cycled from 50 mM PBS (grey area) to 10 mM PBS (white area). The downward spikes appearing at the switching between the 10 mM PBS solution and the 50 mM PBS solution are due to the different responses of the HCC–BPS–Au MCs and of the reference MCs to the change. (Right panel) Scheme of the top face of a HCC–BPS–Au MC and of the repulsive in-plane inter-protein forces that are triggered by the salt concentration decrease and cumulate to deflect the MC downward. | ||
Fig. 4 finally evidences that the HCC film switch could perform several fully reversible motion cycles, giving a design hint for future more application-oriented experiments. Very likely, the fact that HCC seats onto BPS by retaining its folded structure contributes to enhance the machine robustness over more cycles.
Before concluding, it is worth noticing that the presented experiments as well as their thermodynamic analysis are of primary interest also for interface biological systems other than thin film switches. Consider eqn (5), it suggests that the surface work required to confine a protein at a solid–solution interface can be compensated by a shift of the folding/unfolding equilibrium. This is fully consistent with previous observations showing that YCC covalently tethered by C102 onto charged Au NPs presents a decrease of α-helicity,9 but adds the surface work parameter to the picture. This aspect is of basic importance, since surface work means significant protein–protein interactions and charge screening that very likely concur with folding degree and binding sites accessibility to affect the protein surface functionality. Remarkably, as evidenced in Fig. 3, similar folding behaviors such as the increase of denaturation of YCC at charged NPs induced by lowering the salt concentration,23 can be accompanied by both attractive or repulsive intense forces, that probably differently impact YCC surface functions.
In summary, we are able to create hybrid molecular driven devices based on MCs and protein thin films with motion fuelled by salinity cycles. Upon the same salinity stimulus, the film mechanical work and the MC direction of motion are tunable over a range of approximately 90 mN m−1 and 400 nm, respectively, and depend on the nature of the attachment of the protein to the MC and also the surface coating ligand at the protein–MC interface. These results aid in progress towards hybrid biomolecular devices, and show that the nature of the interface between proteins and inorganic surfaces can have a profound effect on biomolecular machines. How the nanoscale rearrangements of the protein can cumulate to create microscale mechanical work can be described in thermodynamic terms; that with the experimental evidences point to building up of inter-protein hydration forces as the primary mechanism. The proposed thermodynamic description is amenable to be extended to other breakthrough yet few understood interfaces in biology, including NP–protein conjugates10 and stimuli-responsive surfaces realized with biomolecules.38
Acknowledgements
This work was developed and written in the framework of the UniBS-MIT-MechE faculty exchange Program co-sponsored by the CARIPLO Foundation, Italy, under the Grant 2008-2290.Notes and references
- (a) W. Chiu, M. Baker and S. Almo, Trends Cell Biol., 2006, 16, 144–150 CrossRef CAS; (b) A. Clements, D. Bursac, X. Gatsos, A. Perry, S. Civciristov, N. Celik, V. Likic, S. Poggio, C. Jacobs-Wagner, R. Strugnell and T. Lithgow, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 15791–15795 CrossRef CAS.
- (a) E. R. Kay, D. A. Leigh and F. Zerbetto, Angew. Chem., Int. Ed., 2007, 46, 72–191 CrossRef CAS; (b) V. Balzani, A. Credi and M. Venturi, ChemPhysChem, 2008, 9, 202–220 CrossRef.
- B. K. Juluri, A. S. Kumar, Y. Liu, T. Ye, Y.-W. Yang, A. H. Flood, L. Fang, J. F. Stoddart, P. S. Weiss and T. J. Huang, ACS Nano, 2009, 3, 291–300 CrossRef CAS.
- F. Zhou, W. Shu, M. Welland and W. Huck, J. Am. Chem. Soc., 2006, 128, 5326–5327 CrossRef CAS.
- W. M. Shu, D. S. Liu, M. Watari, C. K. Riener, T. Strunz, M. E. Welland, S. Balasubramanian and R. A. McKendry, J. Am. Chem. Soc., 2005, 127, 17054–17060 CrossRef CAS.
- A. Valiev, N. I. Abu-Lail, D. W. Lim, A. Chilkoti and S. Zausher, Langmuir, 2007, 23, 339–344 CrossRef CAS.
- K. Wang, J. G. Forbes and A. J. Jin, Prog. Biophys. Mol. Biol., 2001, 77, 1 CrossRef CAS.
- A. E. Nel, L. Madler, D. Velegol, T. Xia, E. M. V. Hoek, P. Somasundaran, F. Klaessig, V. Castranova and M. Thompson, Nat. Mater., 2009, 8, 543–557 CrossRef CAS.
- M.-E. Aubin-Tam and K. Hamad-Schifferli, Langmuir, 2005, 21, 12080–12084 CrossRef CAS.
- (a) C. You, A. Chompoosor and V. Rotello, Nano Today, 2007, 2, 34–43 CrossRef; (b) M.-E. Aubin-Tam and K. Hamad-Schifferli, Biomed. Mater., 2008, 3, 034001 Search PubMed (17p); (c) S. Park and K. Hamad-Schifferli, Curr. Opin. Chem. Biol., in press, DOI:10.1016/j.cbpa.2010.06.186.
- P. Bergese, G. Oliviero, I. Alessandri and L. E. Depero, J. Colloid Interface Sci., 2007, 316, 1017–1022 CrossRef CAS.
- For a throughout treatment of intermolecular forces in biological molecules see: D. Leckband and J. Israelachvili, Q. Rev. Biophys., 2001, 34, 105–267 Search PubMed.
- C. Bustamante, Annu. Rev. Biochem., 2004, 73, 705–748 CrossRef CAS.
- T. M. Record, C. F. Anderson and T. M. Lohman, Q. Rev. Biophys., 1978, 2, 103–178 CrossRef.
- See: P. Gong and R. Levicky, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 5301–5306 Search PubMed , and references cited therein.
- More generally, j represents the difference between the number of charged ligands C bound to A and the number of the ones bound to B. This or more sophisticated formalisms are unessential for the present discussion; the interested reader is redirected to ref. 14.
- W. Norde, Colloids and Interfaces in Life Sciences, Marcel Dekker, New York, 2003, ch. 9 Search PubMed.
- Eqn (2) is strictly true only for plane and spherical surfaces. For other geometries the surface curvature must be taken into account. More details can be found in: W. A. Adamson and A. P. Gast, Physica Chemistry of Surfaces, Wiley, New York, 6th edn, 2000, pp. 48–100 Search PubMed.
- More details on the concept underpinning eqn (2) can be found in: (a) Ref. 11 ; (b) P. Bergese, G. Oliviero, I. Colombo and L. E. Depero, Langmuir, 2009, 25, 4271–4273 CrossRef CAS; (c) G. Oliviero, S. Federici, P. Colombi and P. Bergese, J. Mol. Recognit., DOI:10.1002/jmr.1019, published in early view.
- A. M. Moulin, S. J. O'Shea, R. A. Badley, P. Doyle and M. E. Welland, Langmuir, 1999, 15, 8776–8779 CrossRef CAS.
- P. Roach, D. Farrar and C. C. Perry, J. Am. Chem. Soc., 2005, 127, 8168–8173 CrossRef CAS.
- M. Friedel, A. Baumketner and J. Shea, J. Chem. Phys., 2007, 126, 095101 CrossRef.
- M.-E. Aubin-Tam, W. Hwang and K. Hamad-Schifferli, Proc. Natl. Acad. Sci. U. S. A., 2009, 11, 4095–4100 CrossRef.
- S. Lacerda, J. Park, C. Meuse, D. Pristinski, M. Becker, A. Karim and J. Douglas, ACS Nano, 2009, 4, 365–379.
- C. N. Pace, S. Trevino, E. Prabhakaran and J. M. Scholts, Philos. Trans. R. Soc. London, Ser. B, 2004, 359, 1225–1235 CrossRef CAS.
- D. T. Haynie, Biological Thermodynamics, Cambridge University Press, Cambridge, 2001, pp. 165–171 Search PubMed.
- G. V. Louie, W. L. B. Hutcheon and G. D. Brayer, J. Mol. Biol., 1988, 199, 205–314.
- G. V. Louie and G. D. Brayer, J. Mol. Biol., 1990, 214, 527–555 CAS.
- H. Maity, M. Maity and S. W. Englander, J. Mol. Biol., 2004, 343, 223–233 CrossRef CAS.
- D. R. Hostetter, G. T. Weatherly, J. R. Beasley, K. Bortone, D. S. Cohen, S. A. Finger, P. Hardwidge, D. S. Kakouras, A. J. Saundersm, S. K. Trojak, J. C. Waldner and G. J. Pielak, J. Mol. Biol., 1999, 289, 639–644 CrossRef CAS.
- D. Roccatano, I. Daidone, M. A. Ceruso, C. Bossa and A. Di Nola, Biophys. J., 2003, 84, 1876–1883 CrossRef CAS.
- A. E. García and G. Hummer, Proteins: Struct., Funct., Genet., 1999, 36, 175–191 CrossRef CAS.
- S. Cherian and T. Thundat, Appl. Phys. Lett., 2002, 80, 2219–2221 CrossRef CAS.
- Y. Huang, H. Liu, K. Li, Y. Chen, Q. Zhang and X. Wu, Sens. Actuators, A, 2008, 148, 329–334 CrossRef.
- We used the following form of the Stoney's equation: −Δγ = (ΔzEt2)/[4L2(1 − ν)], where Δz is the cantilever deflection (with the z axis individuated by the unitary vector normal to the top surface of the cantilever base, i.e. Δz < 0 for a downward cantilever bending), E is the cantilever Young's modulus, t is the cantilever thickness, L is the cantilever length and ν is the cantilever Poisson's ratio. For the used Si MCs: E = 168.5 GPa, t = 1 µm, L = 100 µm and ν = 0.25. For further details see ref. 11 and references cited therein.
- J. Mertens, C. Rogero, M. Calleja, D. Ramos, J. A. Martin-Gago, C. Briones and J. Tamayo, Nat. Nanotechnol., 2008, 3, 301–307 CrossRef CAS.
- S. Srivastava, A. Verma, B. L. Frankamp and V. M. Rotello, Adv. Mater., 2005, 17, 617–621 CrossRef CAS.
- (a) P. M. Mendes, Chem. Soc. Rev., 2008, 37, 2361–2580 RSC; (b) H. Nandivada, A. M. Ross and J. Lahann, Prog. Polym. Sci., 2010, 35, 141–154 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |