DNA-mediated site-specific deposition of gold nanoparticles on silicon wafers†
Katharina J. C.
Heimann
a and
Clemens
Richert
*ab
aInstitute of Organic Chemistry and Center for Functional Nanostructures (CFN), Karlsruhe Institute of Technology (KIT), 76131, Karlsruhe, Germany. E-mail: katharina@rrg.uka.de; Fax: +49 721 608 4825; Tel: +49 721 608 7212
bInstitut für Organische Chemie, Universität Stuttgart, 70569, Stuttgart, Germany. E-mail: lehrstuhl-2@oc.uni-stuttgart.de; Fax: +49 711 685 64321; Tel: +49 711 685 64311
First published on 21st October 2010
Abstract
We report a method for the site-specific deposition of gold nanoparticles, as programmed by DNA sequences immobilized on the surface of silicon oxide-coated silicon wafers. After optimization of surface chemistries, selectivities of between 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 118:1 were achieved for the DNA-based sorting of populations of gold nanoparticle of 15 nm and 60 nm diameter from a common suspension via oligonucleotide duplex formation.
:1 and 118:1 were achieved for the DNA-based sorting of populations of gold nanoparticle of 15 nm and 60 nm diameter from a common suspension via oligonucleotide duplex formation.
Deoxyribonucleic acid (DNA) is emerging as one of the most attractive materials for nanostructuring via the bottom-up approach.1 Synthetic oligodeoxynucleotides form antiparallel duplexes of predictable strength and dimensions, and are readily available via automated DNA synthesis. Modified oligonucleotides may self-assemble to form functional nucleic acid nanoarchitectures.2 Tethering nanoscale cargo to one oligonucleotide and covalently linking a complementary strand to a surface gives a predictable attraction between the two and thus a means for the site-specific deposition of the cargo. In principle, this includes the deposition of metallic nanoparticles that may act as components of molecular electronic circuits or electro-optic devices.3 But, while the design principle is simple, the practical implementation can be challenging.
Gold nanoparticles coated with oligonucleotides have been shown to assemble in sequence-specific fashion.4–8 The assembly process can be used to detect target strands, leading to powerful diagnostic devices.9,10 Harnessing the forces of multivalent binding between DNA-coated nanoparticles and planar surfaces is far from trivial. Unspecific adsorption can be very strong, and nanoparticles of different diameters bring very different contact areas to the surfaces, so that large particles with much larger contact areas stick more strongly than smaller ones. After pioneering work of Mirkin and coworkers,11 we were recently able to demonstrate DNA-based ‘sorting’ of gold nanoparticles of different diameters onto DNA-coated areas of planar gold surfaces.12 Inducing selective hybridization, and thus deposition, from a mixture required careful optimization of the surface coating of nanoparticles and surfaces.
While useful as a model system, depositing gold nanoparticles onto gold surfaces is unlikely to lead to novel applications in molecular electronics. Here we demonstrate DNA-based sorting of nanoparticles of 15 and 60 nm diameter onto different surface areas of a silicon wafer. For this, an immobilization strategy for the capture strands on the surface was developed, together with an improved synthesis of the thiol DNA coating the nanoparticles.
Fig. 1 shows molecular details of the surface chemistries employed. The synthesis of coating strands 1 and 2 started from hexaethylene glycol, which was converted to phosphoramidite 3 in just three steps. This streamlined synthesis is four steps shorter than the route to the corresponding, disulfide-based building block reported earlier.12 With 3 and 5′-phosphoramidites of the four deoxynucleosides (4a–t), both coating strands were readily assembled via automated DNA synthesis on a conventional synthesizer. ‘Filler’ molecule 5, used for blocking any remaining gold surface, was prepared on support 6, accessible in three steps from hexaethylene glycol, succinic anhydride, and commercial controlled pore glass. Gold nanoparticles of 15 nm (7) and 60 nm diameter (8) coated with a combination of DNA strands (1 or 2) and 5 proved stable at an NaCl concentration of 0.3 M.
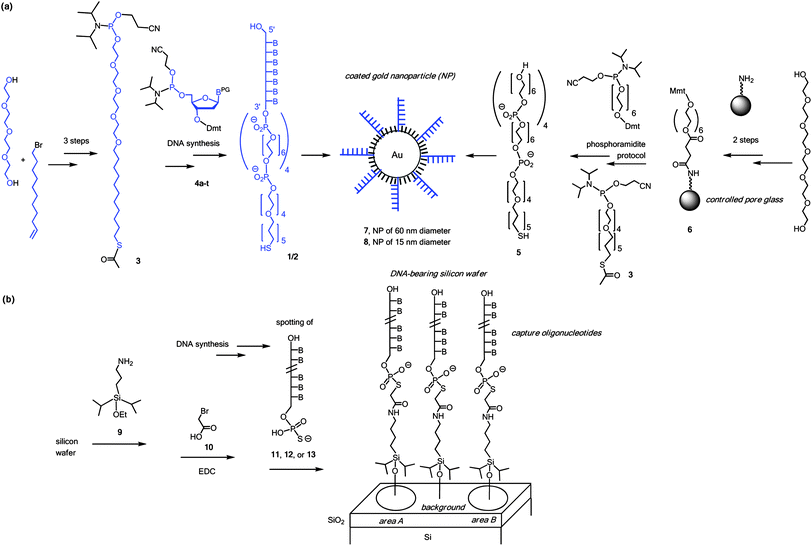 | ||
| Fig. 1 Surface chemistries employed. (a) Synthesis of components, and generation of coated nanoparticles; (b) generation of silicon wafers with DNA. Components beyond the molecular scale are shown in cartoon format and are not drawn to scale. B = nucleobase, Dmt = dimethoxytrityl, Mmt = monomethoxytrityl, PG = protecting group for exocyclic amine of nucleobase. | ||
Different strategies for covalently linking the capture strands to the wafer were tested. Tris(alkoxy)silanes, such as APTES, gave rough surfaces, probably due to three-dimensional growth, as observed in AFM images of the surface (not shown). Among the monoalkoxydialkyl silanes, 3-aminopropyldiisopropylethoxysilane (9) gave more stable functionalized surfaces than its dimethylsilane counterpart, while both gave smooth surface topologies. We used α-bromoacetic acid (10) as bifunctional linker reagent in an adaptation of a methodology developed for diagnostic microarrays.13 Capture strands terminating in a 5′-phosphorothioate group (11–13) were prepared by standard DNA synthesis, employing 3′-phosphoramidites, followed by a chemical phosphorylation step and sulfurization with Beaucage's reagent.14 Capture strands terminating in a thiol gave less satisfactory results, most probably due to their propensity to form disulfides. Detailed protocols are given in the ESI†.
Fig. 2 shows the sequences of coating strands 1 and 2, capture strands 11–13, and splint strands 14 and 15. The latter were employed (rather than a two-strand system) to gain flexibility in tuning duplex stabilities and because three-strand systems have proven reliable in nanoparticle assembly studies.11 Areas A and B were generated by spotting 500 nL of solutions of 11 or 13, whereas the control regions on the same wafer were treated with unrelated sequence 12. After washing with copious amounts of ethanol, splint strands 14 and 15 were added, followed by washing with ammonium acetate buffer. Then, the mixture of DNA-coated nanoparticles 7 and 8 in phosphate buffer was added to the surface by carefully dropping the wafer onto the suspension.
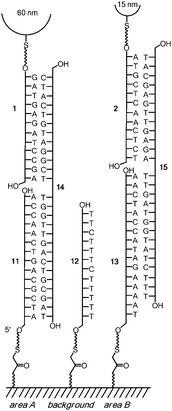 | ||
| Fig. 2 Sequences of oligonucleotides used for nanoparticle sorting. Only one strand per surface area is shown for simplicity. | ||
As noted previously,12 this geometry is critical for avoiding the settling of any residual nanoparticle aggregates onto the surface. Hybridization was allowed to proceed for 10 min at 40 °C, followed by 10 min at 25 °C. A large excess of buffer was added, followed by washing, drying and imaging by AFM.
Fig. 3 shows representative AFM images of all three areas (A, B, and background, the latter coated with an unrelated DNA sequence). Table 1 lists densities of either particle population on each of the areas. The standard deviations were calculated from counts on four non-adjacent areas of 6.25 µm2 each.
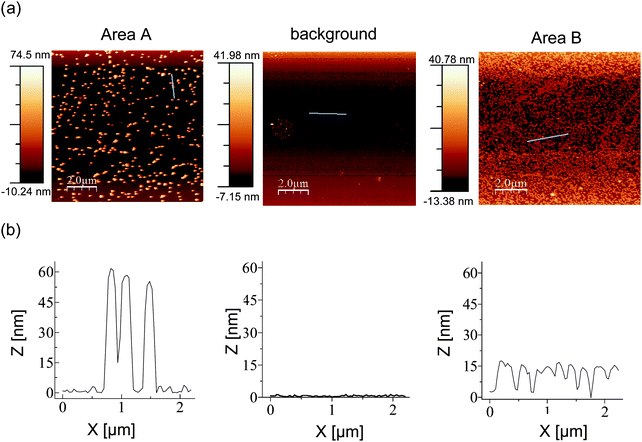 | ||
| Fig. 3 AFM images of representative areas of the surface of a silicon wafer after sorting. (a) AFM images with lines selected for height profiles shown in blue, (b) height profiles. | ||
| Area | 15 nm | 60 nm |
|---|---|---|
| A | 8 ± 2 | 63 ± 2 |
| Background | 10 ± 2 | 2 ± 1 |
| B | 350 ± 20 | 3 ± 1 |
As expected,12 the layer of the 15 nm particles is of greater density than that formed by the much larger, 60 nm nanoparticles. Area A attracted an occasional 15 nm particle, leading to a selectivity of approx. 8:1, whereas very few large particles were detected in area B, where the small gold colloids were programmed to bind. For area B, an excellent selectivity ratio of >100:1 resulted. These findings were reproduced in two independent assays.
The density of 15 nm nanoparticles achieved on area B is 56 ± 3 particles µm−2, which is less than that observed in an earlier study employing a single diameter of colloids (15 nm) and a dendrimer-coated silicon substrate.15 This is probably due to the presence of filler compound 5 on our nanoparticles. However, the density found for either of our two particle populations (15 nm and 60 nm diameter) on their respective areas with complementary capture DNA is greater than that reported for the immobilization of DNA-coated gold colloids of 40 nm diameter on a gold surface.16
We emphasize that selectivities on the level shown here require careful control over the surface chemistries. Nanoparticle preparations were validated via hybridization. Validation experiments involved two nanoparticle populations, one prepared from 3′-thiol-DNA, one from 5′-thiol-DNA, and a splint strand complementary to both. Only samples that passed the hybridization test were employed on surfaces. Further, the surface modification on the wafer was checked by using fluorophore-labelled capture strands, as well as qualitative tests, such as contact angle measurements, and microscopic inspection. Visual inspection was also employed for nanoparticle suspensions and surfaces, particularly checking for the absence of the pink hue typical of aggregated nanoparticles.
In conclusion, we report the streamlined synthesis of DNA-coated gold nanoparticles, surface passified by the use of an optimized filler molecule. Using capture DNA immobilized via a linker prepared from monoethoxysilane, α-bromoacetic acid and phosphorothioates, we were able to demonstrate DNA-based sorting of small and large nanoparticles to the surface areas bearing complementary strands. We expect that dip-pen nanolithography,17,18 when combined with the current surface chemistries and possibly other positioning approaches,19 will produce patterned nanoparticle-bearing surfaces suitable for new applications. Additional functionality may be added by using nanoparticles with tailor-made surface modifications.20
Acknowledgements
The authors thank Dr Tetiana Kurkina and Dr Kannan Balasubramanian (Max-Planck-Institute for Solid State Research, Stuttgart) for access to an AFM and help with the acquisition of images, Drs K. Müller and M. Meng for help with exploratory AFM work, and T. Lommel and A. Singh for helpful discussions. This work was supported by The Center of Functional Nanostructures (CFN) at the University of Karlsruhe/K.I.T., subproject C4.3.Notes and references
- (a) N. C. Seeman, Nature, 2003, 421, 427 CrossRef; (b) U. Feldkamp and C. M. Niemeyer, Angew. Chem., Int. Ed., 2006, 45, 1856 CrossRef CAS; (c) K. V. Gothelf, Org. Biomol. Chem., 2005, 3, 4023 RSC; (d) F. A. Aldaye, A. L. Palmer and H. F. Sleiman, Science, 2008, 321, 1795 CrossRef CAS.
- J. Wengel, Org. Biomol. Chem., 2004, 2, 277 RSC.
- G. Gotesman and R. Naaman, Langmuir, 2008, 24, 5981 CrossRef CAS.
- (a) C. A. Mirkin, R. L. Letsinger, R. C. Mucic and J. J. Storhoff, Nature, 1996, 382, 607 CrossRef CAS; (b) A. P. Alivisatos, K. P. Johnsson, X. Peng, T. E. Wilson, C. J. Loweth, M. P. J. Bruchez and P. G. Schultz, Nature, 1996, 382, 609 CrossRef CAS.
- M. Fischler and U. Simon, J. Mater. Chem., 2009, 19, 1518 RSC.
- L. M. Dillenback, G. P. Goodrich and C. D. Keating, Nano Lett., 2006, 6, 16–23 CrossRef CAS.
- C. M. Niemeyer, B. Ceyhan and P. Hazarika, Angew. Chem., Int. Ed., 2003, 42, 5766 CrossRef CAS.
- F. Seela, A. M. Jawelekar, L. Chi and D. Zhong, Chem. Biodiversity, 2005, 2, 84 CrossRef CAS.
- N. L. Rosi and C. A. Mirkin, Chem. Rev., 2005, 105, 1547 CrossRef CAS.
- (a) J.-M. Nam, C. S. Thaxton and C. A. Mirkin, Science, 2003, 301, 1884 CrossRef CAS; (b) J.-M. Nam, S. I. Stoeva and C. A. Mirkin, J. Am. Chem. Soc., 2004, 126, 5932 CrossRef CAS.
- (a) L. M. Demers, S.-J. Park, T. A. Taton, Z. Li and C. A. Mirkin, Angew. Chem., Int. Ed., 2001, 40, 3071 CrossRef CAS; (b) S.-W. Chung, D. S. Ginger, M. W. Morales, Z. Zhang, V. Chandrasekhar, M. A. Ratner and C. A. Mirkin, Small, 2005, 1, 64 CrossRef CAS.
- U. R. Plutowski, S. S. Jester, S. Lenhert, M. M. Kappes and C. Richert, Adv. Mater., 2007, 19, 1951 CrossRef CAS.
- (a) M. C. Pirrung, J. D. Davis and A. L. Odenbaugh, Langmuir, 2000, 16, 2185 CrossRef CAS; (b) M. C. Pirrung, Angew. Chem., 2002, 114, 1326 CrossRef.
- R. P. Iyer, W. Egan, J. B. Regan and S. L. Beaucage, J. Am. Chem. Soc., 1990, 112, 1253 CrossRef CAS.
- E. Koplin, C. M. Niemeyer and U. Simon, J. Mater. Chem., 2006, 16, 1338 RSC.
- C. M. Niemeyer, B. Ceyhan, S. Gao, L. Chi, S. Peschel and U. Simon, Colloid Polym. Sci., 2001, 279, 68 CrossRef CAS.
- L. M. Demers, S.-J. Park, A. Taton, Z. Li and C. A. Mirkin, Angew. Chem., Int. Ed., 2001, 40, 3071 CrossRef CAS.
- B. Basnar and I. Willner, Small, 2009, 5, 28 CrossRef CAS.
- G. Braun, M. Diechrierow, S. Wilkinson, F. Schmidt, M. Huben, E. Weinhold and N. O. Reich, Bioconjugate Chem., 2008, 19, 476 CrossRef CAS.
- U. H. F. Bunz and V. M. Rotello, Angew. Chem., Int. Ed., 2010, 49, 3268 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Protocols for the synthesis of all components, preparation of surfaces and nanoparticles, as well as sorting assay. See DOI: 10.1039/c0nr00612b |
| This journal is © The Royal Society of Chemistry 2010 |