Functionalization of manganite nanoparticles and their interaction with biologically relevant small ligands: Picosecond time-resolved FRET studies†
Anupam
Giri
,
Abhinandan
Makhal
,
Barnali
Ghosh
*,
A. K.
Raychaudhuri
* and
Samir Kumar
Pal
*
Unit for Nano Science & Technology, S. N. Bose National Centre for Basic Sciences, Block JD, Sector III, Salt Lake, Kolkata, 700 098, India. E-mail: arup@bose.res.in; barnali@bose.res.in; skpal@bose.res.in
First published on 11th October 2010
Abstract
We report molecular functionalization of the promising manganite nanoparticles La0.67Sr0.33MnO3 (LSMO) for their solubilization in aqueous environments. The functionalization of individual NPs with the biocompatible citrate ligand, as confirmed by Fourier transform infrared (FTIR) spectroscopy, reveals that citrates are covalently attached to the surface of the NPs. UV-VIS spectroscopic studies on the citrate functionalized NPs reveals an optical band in the visible region. Uniform size selectivity (2.6 nm) of the functionalization process is confirmed from high resolution transmission electron microscope (HRTEM). In the present study we have used the optical band of the functionalized NPs to monitor their interaction with other biologically important ligands. Förster resonance energy transfer (FRET) of a covalently attached probe 4-nitrophenylanthranilate (NPA) with the capped NPs confirm the attachment of the NPA ligands to the surface functional group (–OH) of the citrate ligand. The FRET of a DNA base mimic, 2-aminopurine (2AP), with the NPs confirms the surface adsorption of 2AP. Our study may find relevance in the study of the interaction of individual manganite NPs with drug/ligand molecules.
Introduction
Functionalization of magnetic nanoparticles is crucial for their practical use in nanobiotechnology.1–3 In particular, the use of properly functionalized magnetic nanoparticles in clinical medicine has intensified.4,5 With proper surface coating, these magnetic nanoparticles can be dispersed into water, forming monodisperse water-based suspensions.6–8 Formation of nanoparticles involving biological macromolecules is a very effective route towards biocompatible magnetic materials.9–12 Mixed-valent manganites have been recognized to be very useful magnetic materials for their potential in colossal magnetoresistance13,14 and spintronics.15 In recent times significant efforts have been made to use manganite nanoparticles for prospective applications in cancer therapy involving the hyperthermal effect.16 In this regard several attempts have been made to solubilize the manganite nanoparticles in aqueous solution by biocompatible macromolecules including dextran, bovine serum albumin and fatty amines, resulting a suspension of the manganite nanoparticles in solution.17,18 However, to date no attempt has been made to functionalize individual manganite nanoparticles with small biocompatible ligands. The individually functionalized nanoparticles are important in order to study their interaction with other biologically relevant ligand/drug molecules.19,20Herein, we report the functionalization of one of the most promising manganite nanoparticles (NPs), La0.67Sr0.33MnO3 (called LSMO hereafter) with a biocompatible citrate ligand. The size selective solubilization of the functionalized NPs in aqueous solution has also been demonstrated from HRTEM. UV-VIS spectroscopic study of the functionalized NPs shows an intense peak at the visible region as a consequence of electronic interaction of the citrate ligand with the surface atoms of the NPs. In our studies we have exploited the visible band of the functionalized NPs in order to study the interaction of the NPs with small biologically relevant ligands namely 2-aminopurine (2AP) and 4-nitrophenylanthranilate (NPA). Picosecond time-resolved Förster resonance energy transfer (FRET) studies on the ligands with the functionalized NPs, confirm the dipolar interactions of the ligands with the NPs and reveal the ligand binding sites of the NPs in aqueous solution. The FRET distances from the attached ligand to the host LSMO NPs also confirm the existence of the isolated functionalized NPs in the aqueous solution.
Materials and methods
Trisodium citrate, 4-nitrophenylanthranilate (NPA), 2-aminopurine (2AP), potassium bromide (KBr) and phosphate buffer, were obtained from Sigma-Aldrich (USA) and were used as received without further purification. All the aqueous solutions were prepared in 10 mM phosphate buffer of pH 7 using water from the Millipore system. The details of the preparation of LSMO nanoparticles by using modified sol–gel technique are described in the ESI.† The structural and magnetic characterization of the as-prepared nanoparticles are also described in the ESI. The as-prepared LSMO nanoparticles were rendered water-soluble using the reactivity of carboxylate group of citrate with the Mn centre in LSMO, by two hours of extensive mixing using the cyclo-mixer. The un-reacted NPs were filtered out.Optical absorption of the resulting greenish-yellow solution was measured with Shimadzu Model UV-2450 spectrophotometer. Steady-state emissions were measured with a Jobin Yvon Model Fluoromax-3 fluorimeter. Picosecond-resolved fluorescence transients were measured by using a commercially available spectrophotometer (Life Spec-ps) from Edinburgh Instruments, UK for 375 nm excitation (80 ps instrument response function, IRF). For 300 nm excitation we used the third harmonic laser beam of 900 nm (0.5 nJ per pulse) using a mode locked Ti-sapphire laser with an 80 MHz repetition rate (Tsunami, Spectra Physics), pumped by a 10 W Millennia (Spectra Physics) followed by a pulse-picker (rate 8 MHz) and a third harmonic generator (Spectra Physics, model 3980). The third harmonic beam is used for excitation of the sample inside the Time-Correlated-Single-Photon-Counting (TCSPC) instrument (IRF = 50 ps) and the second harmonic beam is collected for the start pulse. A JEOL JEM-2100 high-resolution transmission electron microscopy (HRTEM) equipped with an energy dispersive X-ray (EDAX) spectrometer was used to characterize the particle size and individual structure of the water solubilized NPs and to analyse their elemental composition. TEM samples were prepared by dropping sample stock solutions onto a 300-mesh carbon coated copper grid and dried overnight in air. A JASCO FT/IR-6300 spectrometer was used for the Fourier transform infrared spectroscopy (FTIR) to confirm the covalent attachment of the citrate molecules with the nanoparticle. For FTIR measurements, powdered citrate–LSMO samples were mixed with KBr powder and pelletized. The background correction was made by using a reference blank of KBr pellet.
By ligation with 4-nitrophenylanthranilate (NPA) chromophore, we establish that there is a free citrate –OH group available for further functionalization, after capping the LSMO NPs. Because NPA has an active p-nitrophenyl ester group that can react with nucleophiles, it is expected to react with the free –OH group of the citrate capped LSMO. In a 2 mL 0.50 M citrate–LSMO solution, 250 μL of 1 mM NPA (in acetonitrile) was added at room temperature with stirring, in five aliquots with an interval of 30 min. The yellow solution was filtered to remove the side product p-nitrophenol and excess NPA. The filtered solution was washed extensively by chloroform to remove excess soluble NPA and the side product. A DNA base (adenine) mimic, 2-aminopurine (2AP) has been used to investigate non-covalent surface adsorption of a ligand on the functionalized NPs. 2AP, an analogue of guanine and adenine, is a fluorescent molecular marker used in nucleic acid research.21 It pairs most commonly with thymine as an adenine-analogue, but can also pair with cytosine as a guanine-analogue,22 for this reason it is often used in the laboratory for mutagenesis. Adsorption of 2AP to the surface of citrate–LSMO was done by mixing 2AP ([2AP] = 20 μM) and citrate–LSMO, with a concentration ratio [2AP]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[citrate–LSMO] = 1:1000, for at least 2 h, and then using this for FRET studies. Details of the picosecond time-resolved FRET studies and the parameters involved in the equations are described in the ESI.† Once the value of the Förster distance (R0) is known, the donor–acceptor distance (r) can be easily calculated using the formula,
:[citrate–LSMO] = 1:1000, for at least 2 h, and then using this for FRET studies. Details of the picosecond time-resolved FRET studies and the parameters involved in the equations are described in the ESI.† Once the value of the Förster distance (R0) is known, the donor–acceptor distance (r) can be easily calculated using the formula,
| r6 = [R60(1 − E)]/E | (1) |
| E = 1 − (FDA/FD) | (2a) |
| E = 1 − (τDA/τD) | (2b) |
The distance measured using eqn (2a) and (2b) are expressed as rSS (steady state measurement) and rTR (time-resolved measurement), respectively. The potential danger of using eqn (2a) for the estimation of donor–acceptor distance has been discussed in our earlier literature.23 We have used amplitude weighted 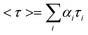 lifetimes where αi is the relative amplitude contribution to the lifetime τi.
lifetimes where αi is the relative amplitude contribution to the lifetime τi.
Results and discussions
To obtain direct evidence for the functionalization of LSMO NPs, FTIR measurements were performed on both the as-prepared and functionalized samples. The FTIR spectra of LSMO NPs, citrate–LSMO conjugates and trisodium citrate are shown in Fig. 1a. For trisodium citrate the characteristic bands (centre of gravity at 1417 cm−1) correspond to the symmetric stretching of –COO−, at 1592 cm−1 for the antisymmetric stretching of –COO−,24–27 and at 3455 cm−1 due to stretching vibration of –OH28 are clearly shown in Fig. 1a(I). When the citrate ligand bound to NPs surface (Fig. 1a(II)) the antisymmetric stretching of COO− at 1595 cm−1 almost remain same but the symmetric COO− stretching mode of citrate becomes red-shifted and appears sharply at 1398 cm−1. It has been shown that Mn remains in the form of Mn(III) at the surface of LSMO NPs.29 The red-shifted symmetric COO− stretching mode clearly confirms the bi-dentate binding of two carboxylate oxygens with the surface Mn3+ ions of LSMO NPs. In order to confirm that Mn is the main interacting species, we have studied the interactions of the citrate ligands with La2O3 and LaMnO3, which reveals a visible band only for LaMnO3. Our observation is consistent with the fact that Mn is the main interacting species which can interact with the ligand. It is also evident that the position of –OH stretching vibration of citrate remains same but become more broaden after interacting with the NPs surface. So, from FTIR study it is evident that the COO− functional group/groups present in citrate are covalently bonded to the NPs surface and the remaining polar functional groups make the NPs water soluble. Since LSMO is a stable oxide30 in water and due to the presence of citrate as capping agent, there will be no change to the surface property of LSMO upon solubilization into water.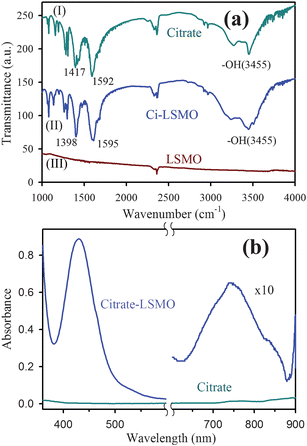 | ||
| Fig. 1 (a) shows FTIR spectra of (I) pure tri-sodium citrate crystals, (II) functionalized citrate–LSMO and (III) as-prepared bulk LSMO, recorded with a KBr pellet. (b) shows UV-VIS spectra of citrate functionalized LSMO NPs in solution. | ||
Fig. 1b illustrates the UV-VIS spectrum of the citrate capped LSMO. A high intensity broad band centred at 430 nm is clearly shown, along with a smaller band centred at 742 nm. The near visible absorption of La0.9Sr0.1MnO3 single crystal has been reported in the literature,31 to be at 1.75 eV (710 nm). The near visible band with a peak at 1.75 eV is attributed to an e1g (Mn3+)–e2g(Mn3+) transition between states split by Jahn–Teller interaction. The smaller band at 742 nm for citrate–LSMO arises because of the above (d–d) transition and is shifted towards lower energy (compared to La0.9Sr0.1MnO3 single crystal at 710 nm) due to the presence of COO− (of citrate), which is a low field ligand compared to32 O2− (of LSMO). The origin of the high intensity band at 430 nm is attributed to ligand (citrate) to metal (Mn3+) charge transfer (LMCT), and the transition occurs from filled ligand pπ orbitals to empty metal (Mn3+) 3d orbitals.32,33
The magnetic properties of citrate–LSMO were studied by using standard field dependent magnetization measurements. The starting as-prepared NP is a room temperature ferromagnetic material.31 The details of the magnetic measurements of the as-prepared bulk NPs are described in the ESI.† The M(H) curve of the functionalized NPs in water at room temperature and at 80 K shows a linear behaviour with low magnetization values, indicative of paramagnetic behaviour. So, the ferromagnetic NPs (as-prepared) become paramagnetic upon size selective (av. 2.6 nm) functionalization (see below) and consequent solubilization by citrate ligand.30,34
Fig. 2a represents the TEM image of the solubilized citrate–LSMO NPs. The image reveals that the solubilized NPs are almost spherical in shape and follow a uniform size distribution. The average sizes of the solubilized NPs, as estimated from the TEM image, has been found to be 2.6 nm. The corresponding HRTEM image (upper inset of Fig. 2a) confirms the crystallinity of the NPs in their structure. The interplanar distance of the fringes is measured to be about 0.29 nm, corresponding to the distance between (104) planes of the LSMO crystal lattice. The selective area electron diffraction (SAED) pattern simultaneously obtained from the TEM measurements (lower inset of Fig. 2a) suggests the single crystalline structure of citrate–LSMO. The elemental composition of the citrate–LSMO conjugates was examined using an EDAX spectrometer attached to TEM operated at 200 kV. EDAX spectrum (Fig. 2b) of the solubilized NPs confirms the presence of La, Sr and Mn as elemental composition. The image in the inset of Fig. 2b represents the corresponding fast Fourier transform (FFT) pattern.
 | ||
| Fig. 2 (a) TEM image of citrate–LSMO NPs, upper inset shows a HRTEM image of the crystalline structure of citrate–LSMO NPs, lower inset shows the selective area electron diffraction (SAED) pattern of the citrate–LSMO NPs. (b) shows EDAX spectrum of the citrate–LSMO NPs, an FFT image of the funtionalized NPs is shown in the inset. | ||
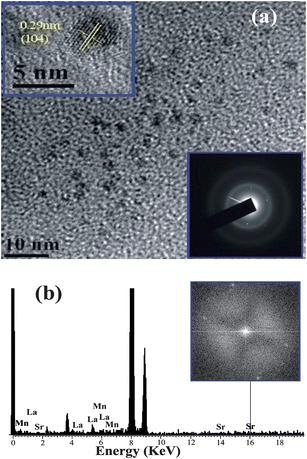
The direct bonding of citrate ligands to the LSMO NPs surfaces ensured that the overall size of the NPs remain small, with a thin solubilizing shell. The –OH functional groups of citrate–LSMO were labelled covalently with 4-nitrophenylanthranilate (NPA) chromophore35 and an efficient FRET occurs. The chromophore, NPA, has an active p-nitrophenyl ester group that can react with nucleophiles.36 NPA was reacted with the hydroxyl groups of the citrate ligands anchored on the LSMO NPs surface, where the hydroxyl groups have been used as nucleophiles. Fig. 3a reveals a huge spectral overlap between the emission spectrum of citrate–NPA (donor) and the absorption spectrum of citrate–LSMO (acceptor) suggesting an efficient Förster resonance energy transfer (FRET) between the donor and acceptor. Inset of Fig. 3b, represents the steady state photo-luminescence (PL) spectra of both citrate–LSMO–NPA, and citrate–NPA and it is found that the NPA emission undergoes drastic quenching in presence of the LSMO NPs, that clearly indicates the energy transfer occurs between the chromophore (NPA) and the nanoparticles. We further confirm the FRET, measuring the excited state lifetime of NPA from picosecond time-resolved study. Fig. 3b shows the picosecond time-resolved PL transients of citrate–NPA and citrate–LSMO–NPA at 450 nm. The picosecond time-resolved fluorescence decay of citrate–NPA (donor) revealed multi exponential time constants of 0.99 ns (21%), 7.16 ns (32%), 0.07 ns (46%) giving an average time constant (<τ>) of 2.53 ns as shown in Table 1. For the donor–acceptor system (citrate–LSMO–NPA) the time constants obtained as 0.94 ns (20%), 5.14 ns (10%) and 0.08 ns (69%) giving an average time constant (<τ>) of 0.75 ns (Table 1). The substantial shortening in the NPA fluorescence lifetime upon conjugation with NPs indicates conclusively that efficient FRET occurs from the NPA donor to the citrate–LSMO acceptor. We have estimated the extinction coefficient (3827.58 mol−1 cm−1) of the acceptor Ci-LSMO using the UV-VIS absorption (at 430 nm) of the functionalized NPs in water. Taking quantum yield of NPA37 in absence of acceptor as 0.51 and based on the spectral overlap, we have estimated a FRET efficiency of 70% in our FRET system using eqn (2b). The calculated Förster distance R0, for the NPA–(citrate–LSMO) complex is 2.90 nm. The donor–acceptor distance (r) calculated using eqn (1) is 2.51 nm (Table 1). The distance is consistent with the covalent attachment of NPA ligand with –OH group of the citrate at the surface of LSMO NPs.
 | ||
| Fig. 3 (a) shows the spectral overlap between donor (citrate–NPA) emission and acceptor (citrate–LSMO) absorption, (b) shows quenching of the donors excitation lifetime in the presence of the acceptor, inset shows steady-state quenching of the donor emission. An excitation wavelength of 320 nm and 375 nm is used for steady state and time resolved experiments, respectively. | ||
| Systems | Lifetimes/ns | E TR (%) | r TR/nm | E SS (%) | r SS/nm |
|---|---|---|---|---|---|
| Citrate–NPA | τ 1 = 0.99 (21%) | — | — | — | — |
| τ 2 = 7.16 (32%) | |||||
| τ 3 = 0.07 (46%) | |||||
| τ av = 2.53 | |||||
| Citrate–LSMO–NPA | τ 1 = 0.94 (20%) | 70.00 | 2.51 | 80.00 | 2.31 |
| τ 2 = 5.14 (10%) | |||||
| τ 3 = 0.08 (69%) | |||||
| τ av = 0.75 | |||||
| Citrate–2AP | τ 1 = 8.03 (100%) | — | — | — | — |
| Citrate–LSMO–2AP | τ 1 = 10.50 (14%) | 80.60 | 2.24 | 96.00 | 1.66 |
| τ 2 = 0.11 (86%) | |||||
| τ av = 1.55 |
Fig. 4a shows the spectral overlap between the emission spectrum of citrate–2AP (donor) and the absorption spectrum of citrate–LSMO (acceptor) suggesting the possibility of efficient Förster resonance energy transfer (FRET) between the donor and the acceptor, when 2AP becomes adsorbed at the surface of the citrate–LSMO NPs. Inset of Fig. 4b represents the steady state photo luminescence (PL) quenching of the donor (citrate–2AP) in presence of LSMO NPs. Picosecond time- resolved PL transients (Fig. 4b) of both donor and donor–acceptor systems monitored at 400 nm, shows significant shortening in the 2AP fluoresence lifetime upon adsorption at the NPs surface. The picosecond time-resolved fluorescence decay of citrate–2AP (donor) revealed single exponential time constant of 8.03 ns. For the donor–acceptor system (citrate–LSMO–2AP) the time constants are obtained as 10.5 ns (14%), 0.11 ns (86%) giving an average time constant (<τ>) of 1.55 ns (Table 1). The extensive quenching in the 2AP fluorescence lifetime upon surface adsorption conclusively indicates that efficient FRET occurs from the 2AP donor to the citrate–LSMO acceptor.
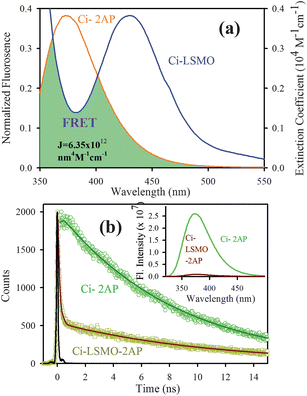 | ||
| Fig. 4 (a) shows the spectral overlap between donor (citrate–2AP) emission and acceptor (citrate–LSMO) absorption, (b) shows quenching of the donors excitation lifetime in the presence of the acceptor, inset shows steady-state quenching of the donor emission. An excitation wavelength of 300 nm is used for both experiments. | ||
Taking quantum yield of 2AP38 in absence of acceptor as 0.66 and based on the spectral overlap, here also we have estimated a FRET efficiency of 80.60% using eqn (2b). The measured Förster distance, R0, for the (citrate–LSMO–2AP) complex is 2.84 nm. The donor–acceptor distance (r) calculated using eqn (1) is 2.24 nm (Table 1), which indicates a very high efficiency of energy transfer and clearly supports the idea that 2AP was in close proximity to the solubilized LSMO NP surface. Despite the fact that the area integral of the donor–acceptor overlap (J value) is much larger for NPA, 2AP shows more efficient energy transfer. As the rate of energy transfer strongly depends upon the donor–acceptor distance (kFRET ∞ 1/r6), 2AP (r = 2.24 nm) shows more efficient energy transfer with the functionalized NPs than NPA (r = 2.51 nm). It has to be noted that the fluorescence quenching mechanism of 2AP and NPA could be due to electron transfer in the system, which normally demands surface-to-surface contact of the donor and acceptor.39–41 However in our system the donors are separated (∼1 nm) from the surface of the acceptor NPs. Moreover, there are several reports regarding the fluorescence quenching of 2AP caused by charge transfer with other DNA bases, however the associated fluorescence life times are much shorter (21–35 ps)38,42,43 compared to an observed value of 110 ps (Table 1). Again NPA is a well known resonance energy transferring probe9,37,44 and there is no report of electron transfer or charge transfer involving NPA. The overall picture that is revealed from our studies is schematically shown in Scheme 1.
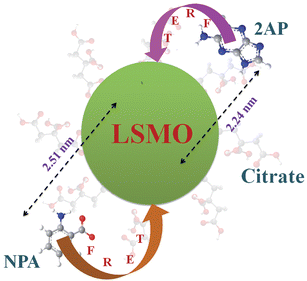 | ||
| Scheme 1 Functionalization of the manganite nanoparticles (NPs) with citrate ligands is shown. Covalent attachment of the fluorescent probe NPA and non-covalent adduction of one of DNA base mimics 2AP are also shown. The efficient energy transfer (FRET) from the fluorescent ligands to the NPs and corresponding donor–acceptor distances is also indicated. | ||
It has to be noted that a number of applications of the FRET model in the dye–nanoparticle system have been evidenced in recent literature.45–47 Rationalization of the nonradiative energy transfer from fluorescent ligands to the nanoparticles is also tricky, as the measured distances are often comparable to the diameter of the nanoparticles, and a more appropriate model is nano surface energy transfer (NSET48). However, the physical parameters [Fermi frequency (ωf) and Fermi wavevector (kf) of acceptor metal NPs] to incorporate the NSET model in the present system are not available in the published literature.
Conclusions
In summary, we have functionalized individual LSMO nanoparticles (NPs) by using citrate ligands as capping agents. While FTIR spectroscopy confirms the covalent attachment of the citrate ligand to the surface of the NPs, UV-VIS spectroscopy reveals strong electronic interaction of the ligands with the electronic states of the NPs, resulting in a strong absorption band in the visible region. The uniform size selection (av. 2.6 nm) upon functionalization of the LSMO NPs in the aqueous solutions has been confirmed from HRTEM. The magnetic property of the NPs in aqueous solutions shows significant change compared to that of the as-prepared material. The FRET from a covalently attached ligand NPA to the core of the NPs reveals a distance of 2.51 nm, indicating the covalent attachment of the fluorescent ligand with the –OH functional group of the capped citrate–LSMO. A fluorescent analogue of adenine, 2AP also shows drastic quenching of its excited state lifetime in presence of the solubilized NPs and the calculated FRET distance (2.24 nm) indicates its surface adsorption at the NPs. The functionalized manganite NPs may find applications in the field of nano-biotechnology, as the solubilizing layer (citrate) provides multiple functional groups (hydroxyl and carboxylic acids) for covalent conjugation with other biological macromolecules such as small peptides, DNA, RNA and biocompatible polymers etc.Acknowledgements
A. G. and A. M. thank UGC (India) and CSIR (India) respectively for their fellowships. B. G. wants to thank DST (India) for funding under WOS-A scheme (SR/WOS-A/PS-15/2008). We thank DST (India) for the funding (SR/SO/BB-15/2007).Notes and references
- G. M. Whitesides, The ‘right’ size in nanobiotechnology, Nat. Biotechnol., 2003, 21, 1161–1165 CrossRef CAS.
- C. M. Niemeyer, Nanoparticles, proteins, and nucleic acids: Biotechnology meets materials science, Angew. Chem., Int. Ed., 2001, 40, 4128–4158 CrossRef CAS.
- J. C. Love, L. A. Estroff, J. K. Kriebel, R. G. Nuzzo and G. M. Whitesides, Self-assembled monolayers of thiolates on metals as a form of nanotechnology, Chem. Rev., 2005, 105, 1103–1170 CrossRef CAS.
- C. Xu, K. Xu, H. Gu, R. Zheng, H. Liu, X. Zhang, Z. Guo and B. Xu, Dopamine as a robust anchor to immobilize functional molecules on the iron oxide shell of magnetic nanoparticles, J. Am. Chem. Soc., 2004, 126, 9938–9939 CrossRef CAS.
- C. Xu, K. Xu, H. Gu, X. Zhong, Z. Guo, R. Zheng, X. Zhang and B. Xu, Nitrilotriacetic acid-modified magnetic nanoparticles as a general agent to bind histidine-tagged proteins, J. Am. Chem. Soc., 2004, 126, 3392–3393 CrossRef CAS.
- S. H. Sun, Recent advances in chemical synthesis, self-assembly, and applications of FePt nanoparticles, Adv. Mater., 2006, 18, 393–403 CrossRef CAS.
- L. Fu, V. P. Dravid and D. L. Johnson, Self-assembled (SA) bilayer molecular coating on magnetic nanoparticles, Appl. Surf. Sci., 2001, 181, 173 CrossRef CAS.
- J. Park, J. Joo, S. G. Kwon, Y. Jang and T. Hyeon, Synthesis of monodisperse spherical nanocrystals, Angew. Chem., Int. Ed., 2007, 46, 4630–4660 CrossRef CAS.
- P. K. Verma, A. Giri, N. T. Thanh, L. D. Tung, O. Mondal, M. Pal and S. K. Pal, Superparamagnetic fluorescent nickel-enzyme nanobioconjugates: Synthesis and characterization of a novel multifunctional biological probe, J. Mater. Chem., 2010, 20, 3722–3728 RSC.
- R. K. Mitra, P. K. Verma, D. Wulferding, D. Menzel, T. Mitra, A. M. Todea, P. Lemmens, A. Mueller and S. K. Pal, A molecular magnet confined in the nanocage of a globular protein, Chem. Phys. Chem, 2010, 11, 389 CrossRef CAS.
- C. M. Niemeyer, Functional hybrid devices of proteins and inorganic nanoparticles, Angew. Chem., Int. Ed., 2003, 42, 5796–5800 CrossRef CAS.
- C. M. Niemeyer, Nanoparticles, proteins, and nucleic acids: Biotechnology meets materials science, Angew. Chem., Int. Ed., 2001, 40, 4128–4158 CrossRef CAS.
- C. N. R. Rao & B. RaveauColossal Magnetoresistance, Charge Odering and Related Properties of Manganese Oxides, 1998 Search PubMed.
- Y. Tokura Colossal Magnetoresistive Oxide, Gordon & Breach, London, 2000 Search PubMed.
- I. Bergenti, V. Dediu, M. Cavallini, E. Arisi, A. A.Riminucci and C. Taliani, Properties of thin manganite films grown on semiconducting substrates for spintronics applications, Curr. Appl. Phys., 2007, 7, 47–50 CrossRef.
- D. K. Kim, M. S. Amin, S. Elborai, S. H. Lee, Y. Koseoglu, M. Zahn and M. Muhammed, Energy absorption of superparamagnetic iron oxide nanoparticles by microwave irradiation, J.Appl.Phys., 2005, 97, 105101.
- K. R. Bhayani, S. N. Kale, S. Arora, R. Rajagopal, H. Mamgain, R. Kaul-Ghanekar, D. C. Kundaliya, S. D. Kulkarni, R. Pasricha, S. D. Dhole, S. B. Ogale and K. M. Paknikar, Protein and polymer immobilized La0.7Sr0.3MnO3 nanoparticles for possible biomedical applications, Nanotechnology, 2007, 18, 345101 CrossRef.
- R. Rajagopal, J. Mona, S. N. Kale, T. Bala, R. Pasricha, P. Poddar, M. Sastry, B. L. V. Prasad, D. C. Kundaliya and S. B. Ogale, La0.7Sr0.3MnO3 nanoparticles coated with fatty amine, Appl. Phys. Lett., 2006, 89, 023107 CrossRef.
- A. K. Gupta and M. Gupta, Synthesis and surface engineering of iron oxide nanoparticles for biomedical applications, Biomaterials, 2005, 26, 3995–4021 CrossRef CAS.
- S. J. Rosenthal, I. Tomlinson, E. M. Adkins, S. Schroeter, S. Adams, L. Swafford, J. McBride, Y. Wang, L. J. DeFelice and R. D. Blakely, Targeting cell surface receptors with ligand-conjugated nanocrystals, J. Am. Chem. Soc., 2002, 124, 4586–4594 CrossRef CAS.
- M. Jean John and K. B. Hall, 2-Aminopurine fluorescence quenching and life times: Role of base stacking, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 37–41 CrossRef CAS.
- L. C. Sowers, G. V. Fazakerley, R. Eritza, B. E. Kaplan and M. F. Goodman, Base pairing and mutagenesis: Observation of a protonated base pair between 2-aminopurine and cytosine in an oligonucleotide by proton NMR, Proc. Natl. Acad. Sci. U. S. A., 1986, 83, 5434–5438 CAS.
- P. Majumder, R. Sarkar, A. K. Shaw, A. Chakraborty and S. K. Pal, Ultrafast dynamics in a nano-cage of enzymes: Solvation and fluorescence resonance energy transfer in reverse micelle, J. Colloid Interface Sci., 2005, 290, 462 CrossRef CAS.
- X. Zou, E. Ying and S. Dong, Seed-mediated synthesis of branched gold nanoparticles with the assistance of citrate and the IR surface-enhanced Raman scattering properties, Nanotechnology, 2006, 17, 4758–4764 CrossRef CAS.
- T. Zhu, K. Vasilev, M. Kreiter, S. Mittler and W. Knoll, Surface modification of citrate-reduced colloidal gold nanoparticles with 2-mercaptosuccinic acid, Langmuir, 2003, 19, 9518–9525 CrossRef CAS.
- M. Dakanali, E. T. Kefalas, C. P. Raptopoulou, A. Terzis, T. Mavromoustakos and A. Salifoglou, Synthesis and spectroscopic and structural studies of a new cadmium(II)–citrate aqueous complex. Potential relevance to cadmium(II)–citrate speciation and links to cadmium toxicity, Inorg. Chem., 2003, 42, 2531–2537 CrossRef CAS.
- P. Uznanski and E. Bryszewska, Synthesis of silver nanoparticles from carboxylate precursors under hydrogen pressure, J. Mater. Sci., 2010, 45, 1547–1552 CrossRef CAS.
- R. M. Silverstein & F. X. WebsterSpectroscopic Identification of Organic Compounds, 6th edn, John Wiley & Sons, Inc., 1998 Search PubMed.
- R. Rajagopal, J. Mona, S. N. Kale, T. Bala, R. Pasricha, P. Poddar, M. Sastry, B. L. V. Prasad, D. C. Kundaliya and S. B. Ogale, La0.7Sr0.3MnO3 nanoparticles coated with fatty amine, Appl. Phys. Lett., 2006, 89, 023107 CrossRef.
- Y. Tian, D. Chen and X. Jiao, La1−xSrIMnO3 (x = 0, 0.3, 0.5, 0.7) nanoparticles nearly freestanding in water: Preparation and magnetic properties, Chem. Mater., 2006, 18, 6088–6090 CrossRef CAS.
- N. N. Loshkareva, Y. P. Sukhorukov, E. A. Neifel’d, V. E. Arkhipov, A. V. Korolev, V. S. Gaviko, E. V. Panfilova, V. P. Dyakina, Y. M. MukovskiÏ and D. A. Shulyatev, Centers of charge nonuniformity in absorption spectra of lanthanum manganites, J. Exp. Theor. Phys., 2000, 90, 389–396 CrossRef.
- J. E. Huheey, E. A. Keiter & R. L. KeiterInorganic Chemistry – Principles of Structure and Reactivity, 4th edn, Pearson Education Search PubMed.
- J. B. Vincent, K. Folting, J. C. Huffman and G. Christou, Use of tetra-N-butylammonium permanganate for inorganic syntheses in nonaqueous solvents. preparation and structure of a manganese(III) dimer containing bridging phenoxo oxygen atoms, Inorg. Chem., 1986, 25, 996–999 CrossRef CAS.
- Y. Labaye, O. Crisan, L. Berger, J. M. Greneche and J. M. D. Coey, Surface anisotropy in ferromagnetic nanoparticles, J. Appl. Phys., 2002, 91, 8715 CrossRef CAS.
- J. Broos, A. J. W. G. Visser, J. F. J. Engbersen, W. Verboom, A. v. Hoek and D. N. Reinhoudt, Flexibility of enzymes suspended in organic solvents probed by time-resolved fluorescence anisotropy. evidence that enzyme activity and enantioselectivity are directly related to enzyme flexibility, J. Am. Chem. Soc., 1995, 117, 12657–12663 CrossRef CAS.
- A. Luyai, Y. Lasanajak, D. Smith, R. Cummings and X. Song, Facile Preparation of fluorescent neoglycoproteins using p-nitrophenyl anthranilate as a heterobifunctional linker, Bioconjugate Chem., 2009, 20, 1618–1624 CrossRef CAS.
- S. Shankara Narayanan and S. K. Pal, Structural and functional characterization of luminescent silver–protein nanobioconjugates, J. Phys. Chem. C, 2008, 112, 4874–4879 CrossRef CAS.
- O. J. G. Somsen, v. A. Hoek and v. H. Amerongen, Fluorescence quenching of 2-aminopurine in dinucleotides, Chem. Phys. Lett., 2005, 402, 61–65 CrossRef CAS.
- M. Grätzel, Photoelectrochemical cells, Nature, 2001, 414, 338 CrossRef CAS.
- V. K. Thorsmølle, B. Wengera, J. Teuscher, C. Bauer and J.-E. Moser, Dynamics of photoinduced interfacial electron transfer and charge transport in dye-sensitized mesoscopic semiconductors, Chimia, 2007, 61, 631–634 CrossRef CAS.
- A. Furube, L. Du, K. Hara, R. Katoh and M. Tachiya, Ultrafast plasmon-induced electron transfer from gold nanodots into TiO2 nanoparticles, J. Am. Chem. Soc., 2007, 129, 14852–14853 CrossRef CAS.
- E. L. Rachofsky, R. Osman and J. B. A. Ross, Probing structure and dynamics of DNA with 2-aminopurine: effects of local environment on fluorescence, Biochemistry, 2001, 40, 946–956 CrossRef CAS.
- O. F. A. Larsen, I. H. M. van Stokkum, F. L. de Weerd, M. Vengris, C. T. Aravindakumar, R. v. Grondelle, N. E. Geacintov and v. H. Amerongen, Ultrafast transient-absorption and steady-state fluorescence measurements on 2-aminopurine substituted dinucleotides and 2-aminopurine substituted DNA duplexes, Phys. Chem. Chem. Phys., 2004, 6, 154–160 RSC.
- N. Hagag, E. R. Birnbaum and D. W. Darnall, Resonance energy transfer between cysteine-34, tryptophan-2 14, and tyrosine-41 1 of human serum albumin, Biochemistry, 1983, 22, 2420–2427 CrossRef CAS.
- T. Chuan Lim, J. V. Bailey, Y.-P. Ho and T.-H. Wang, Intercalating dye as an acceptor in quantum-dot-mediated FRET, Nanotechnology, 2008, 19, 075701 CrossRef.
- J. Bujdák, D. Chorvát and N. Iyi, Resonance energy transfer between rhodamine molecules adsorbed on layered silicate particles, J. Phys. Chem. C, 2010, 114, 1246–1252 CrossRef CAS.
- I. L. Medintz, H. T. Uyeda, R. Goldman Ellen and A. H. Mattoussi, Quantum dot bioconjugates for imaging, labelling and sensing, Nat. Mater., 2005, 4, 435 CrossRef CAS.
- C. S. Yun, A. Javier, T. Jennings, M. Fisher, S. Hira, S. Peterson, B. Hopkins, N. O. Reich and G. F. Strouse, Nanometal surface energy transfer in optical rulers, breaking the FRET barrier, J. Am. Chem. Soc., 2005, 127, 3115–3119 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterisation procedure of the NPs; details of time-resolved measurements; DA distance measurements from FRET studies. See DOI: 10.1039/c0nr00490a |
| This journal is © The Royal Society of Chemistry 2010 |