Gram-scale, low-cost, rapid synthesis of highly stable Mg–ACC nanoparticles and their long-term preservation
Jun
Jiang
,
Min-Rui
Gao
,
Yun-Hao
Qiu
and
Shu-Hong
Yu
*
Division of Nanomaterials and Chemistry, Hefei National Laboratory for Physical Sciences at the Microscale, Department of Chemistry, University of Science and Technology of China, Hefei 230026, P. R. China. E-mail: shyu@ustc.edu.cn; Fax: +0086 551 3603040
First published on 23rd September 2010
Abstract
A simple chemistry route is reported for the gram-scale, low-cost, rapid synthesis of highly stable Mg–ACC nanoparticles. The possible structure of Mg–ACC can be defined as Mg0.15Ca0.85CO3·H2O0.85. The molar ratio of Mg2+:Ca2+:CO32− and the concentrations of the reactants (CaCl2, Na2CO3, and MgCl2) play important roles in the Mg:Ca molar ratio of the obtained Mg–ACC nanoparticles. In particular, Mg–ACC can be preserved for over one year without crystallization by either storing its dry powder at −5 °C or storing it in ethanol at 5 °C. The ability to synthesize Mg–ACC nanoparticles on a large scale is useful for biomineralization studies and industrial applications.
Calcium carbonate is one of the most important biological and industrial materials because of its abundance in nature and its wide application in industry. The polymorphs of calcium carbonate include three anhydrous crystalline phases (aragonite, vaterite, and calcite) and three metastable forms: monohydrate, hexahydrate and amorphous calcium carbonate),1 of which amorphous calcium carbonate (ACC) is the least stable polymorph and has been widely found in organisms.2 A variety of calcium carbonate biominerals are now thought to be crystallized via an amorphous precursor pathway.3 In addition, magnesium is expected to influence biogenic CaCO3, by incorporating into the calcite lattice,4,5 inducing the formation of aragonite,6 stabilizing amorphous calcium carbonate and so on.5 Interestingly, we have come to learn that almost all known biogenic ACC contains magnesium,7 which presents in large quantities in seawater8 (about 50–60 mM Mg2+, relative to 12 mM Ca2+). Therefore, Mg–ACC plays a key role in the biomineralization of CaCO3. Due to the high plasticity of Mg–ACC, the large-scale synthesis of Mg–ACC and its long-term preservation are fascinating for biomineralization and industrial applications.
Ajikumar and co-workers reported that Mg–ACC particles can be prepared by the gas diffusion technique.9 The ability of the magnesium ion, which can stabilize ACC, has been demonstrated by this method.10 However, the yield and the stabilization of Mg–ACC are not good using this synthesis method.
In this Communication, we report the gram-scale, low-cost, rapid synthesis of highly stable Mg–ACC nanoparticles by rapid mixing of solutions of CaCl2, Na2CO3, and MgCl2. The possible composition of Mg–ACC has been proposed to be Mg0.15Ca0.85CO3·H2O0.85 (when concentration of the reactants is 0.0454 mol L−1). The molar ratio of Mg2+:Ca2+:CO32− and the concentrations of the precursor solutions (CaCl2, Na2CO3, and MgCl2) play important roles in the Mg:Ca molar ratio of the obtained Mg–ACC. In particular, Mg–ACC can be preserved for over one year by storing its dry powder at −5 °C or storing it in ethanol at 5 °C.
In a typical experimental procedure, calcium chloride (0.1 mol L−1, 25 mL) and magnesium chloride hexahydrate (0.5 mol L−1, 5 mL) were mixed by mechanical stirring. Then, anhydrous sodium carbonate (0.1 mol L−1, 25 mL) was added rapidly to the mixed solution with mechanical stirring at an ambient temperature of 25 °C. The precipitated colloidal phase was filtered immediately and washed with ethanol. The precipitates were dried in a vacuum desiccator for one day. Calcium chloride (Mw = 110.99), anhydrous sodium carbonate (Mw = 105.99) and magnesium chloride hexahydrate (Mw = 203.30) were commercially available and of analytical grade and used without further purification.
X-Ray power diffraction (XRD) were carried out on a Philips X'Pert PRO SUPER X-ray diffractometer equipped with graphite monochromatized Cu Kα radiation. Field-emission scanning electron microscopy (FESEM) was carried out with a field-emission scanning electron microanalyzer (JEOL-6700F). Transmission electron microscopy (TEM) was performed on JEOL-2010 operated at an acceleration voltage of 200 kV. FT-IR spectra were measured on a Bruker Vector-22 FT-IR spectrometer from 4000 to 400 cm−1 at room temperature. The Mg:Ca molar ratio was analyzed by the inductively coupled plasma atomic emission spectroscopy (ICP-AES) technique using an Atomscan Advantage (Thermo Jarrell Ash Corporation, USA) instrument. Thermogravimetric analysis (TGA) was carried out on a DTG-60H thermal analyzer (Shimadzu) with a heating rate of 10 K min−1 from room temperature to 800 °C in an air flow. Differential scanning calorimetry (DSC) was performed on a VP-DSC calorimeter (Microcal) with a heating rate of 10 K min−1 from room temperature to 600 °C in an air flow.
The obtained magnesium amorphous calcium carbonate (Mg–ACC) powder was synthesized by rapid mixing of solutions of CaCl2, Na2CO3, and MgCl2. The XRD pattern shows no diffraction peak for the powder (Fig. 1), which means that the sample is not crystalline.
 | ||
| Fig. 1 XRD pattern of Mg–ACC powder. | ||
FT-IR confirms the amorphous phase further. In the temperature-variable FT-IR spectra (Fig. 2), at room temperature (20 °C), the out-of-plane bending (ν2) shifts to 863 cm−1, the symmetric stretch at 1076 cm−1 broadens and the asymmetric stretch (ν3) splits into two parts at around 1417 and 1483 cm−1 (Fig. 2a), which is consistent with the typical spectrum of ACC.11 With increasing the temperature to 250 °C, the sample is still not crystalline as shown in Fig. 2b. If the measuring temperature increases to 300 °C (Fig. 2c), the sample begins to crystallize, which is confirmed by the presence of a 874 cm−1 (ν2) mode.12 With a further increase of the measuring temperature to 400 °C (Fig. 2d), the result is almost the same as that measured at 300 °C. It is important to point out that the crystallization temperature is between 250 °C and 300 °C, which indicates that the Mg–ACC is comparatively stable.
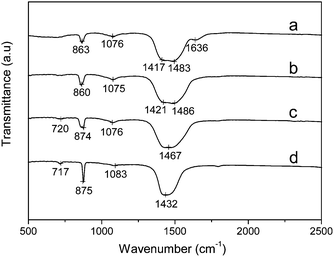 | ||
| Fig. 2 Temperature-dependent FT-IR spectra of Mg–ACC powder: (a) 20 °C; (b) 250 °C; (c) 300 °C; (d) 400 °C. | ||
SEM imaging revealed the occurrence of nanoparticles (Fig. 3b) and a TEM image of the obtained Mg–ACC is shown in Fig. 3c. Again, it confirms that these ACC particles are on the nanoscale and tend to aggregate.
 | ||
| Fig. 3 Mg–ACC obtained by rapid mixing of solutions of CaCl2, Na2CO3, and MgCl2. (a) A photograph of the Mg–ACC powder dispersed in ethanol homogeneously; (b) an SEM image of the obtained Mg–ACC; (c) a TEM image of the obtained Mg–ACC. | ||
The Ca:Mg molar ratio of the Mg–ACC powder is 5.66![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, which was measured by ICP-AES. TG-DTA was used to further analyze the composition of Mg–ACC. The TG curve of the products shows a four-step weight loss (Fig. 4). The weight losses before 102 °C (8 wt%) and between 102 and 395 °C (13 wt%) are attributed to the loss of adsorbed water and crystallized water in the product, respectively.13 MgCO3 decomposed at around 395 °C and CaCO3 decomposed after 550 °C as indicated by the TG-DTA curve (Fig. 4). Correspondingly, two weight losses measured to be 5.8 wt% and 32 wt% in the TG curve, are due to the loss of CO2 gas from the product. The ratio of Mg:Ca:CO32−:H2O was found to be 0.15:0.85:1:0.85. The possible composition of Mg–ACC can be proposed as Mg0.15Ca0.85CO3·H2O0.85 and the result is quite similar to the biogenic ACC investigated by Addadi and coworkers.14
:1, which was measured by ICP-AES. TG-DTA was used to further analyze the composition of Mg–ACC. The TG curve of the products shows a four-step weight loss (Fig. 4). The weight losses before 102 °C (8 wt%) and between 102 and 395 °C (13 wt%) are attributed to the loss of adsorbed water and crystallized water in the product, respectively.13 MgCO3 decomposed at around 395 °C and CaCO3 decomposed after 550 °C as indicated by the TG-DTA curve (Fig. 4). Correspondingly, two weight losses measured to be 5.8 wt% and 32 wt% in the TG curve, are due to the loss of CO2 gas from the product. The ratio of Mg:Ca:CO32−:H2O was found to be 0.15:0.85:1:0.85. The possible composition of Mg–ACC can be proposed as Mg0.15Ca0.85CO3·H2O0.85 and the result is quite similar to the biogenic ACC investigated by Addadi and coworkers.14
 | ||
| Fig. 4 Thermogravimetric and differential thermal curves of Mg–ACC powder. | ||
In a typical experiment, Mg–ACC was synthesized by rapid mixing of the solutions CaCl2: 0.1 mol L−1, 25 mL; Na2CO3: 0.1 mol L−1, 25 mL and MgCl2·6H2O: 0.5 mol L−1, 5 mL, and the molar ratio of the reactants (Ca2+:CO32−:Mg2+) is 1:1:1 (Fig. 5a and Fig. 6a). With the decrease in volume of the MgCl2·6H2O to 3 mL, Mg–ACC could still be obtained (Fig. 5b and Fig. 6b). The Mg:Ca molar ratio of the obtained Mg–ACC is 1:8.64 (by ICP-AES), which is lower than the typical sample (1:5.66). If the volume of the MgCl2·6H2O decreases to 2.5 mL, the product begins to crystallize as shown in Fig. 5c and Fig. 6c. Decreasing the volume of the MgCl2·6H2O to 0 mL results in calcite (Fig. 5d and Fig. 6d). When the molar ratio of the reactants (Ca2+:CO32−:Mg2+) is lower than 5:5:3 in this system, Mg–ACC cannot be obtained. Therefore, the molar ratio of Mg2+:Ca2+:CO32− plays a key role in the synthesis of Mg–ACC.
 | ||
| Fig. 5 XRD patterns of calcium carbonate samples obtained from different volumes of MgCl2·6H2O. (a) 5 mL; (b) 3 mL; (c) 2.5 mL; (d) 0 mL. The sample was prepared by rapid mixing of solutions of the reactants (CaCl2, Na2CO3, and MgCl2). | ||
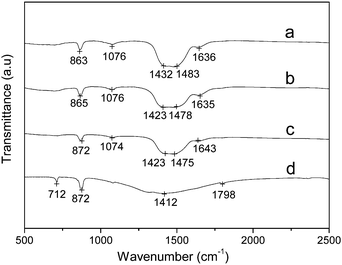 | ||
| Fig. 6 FT-IR spectra of calcium carbonate samples obtained from different volumes of MgCl2·6H2O. (a) 5 mL; (b) 3 mL; (c) 2.5 mL; (d) 0 mL. The sample was prepared by rapid mixing of solutions of the reactants (CaCl2, Na2CO3, and MgCl2). | ||
When the concentration of the reactants is 0.0227 mol L−1, the molar ratio (Ca2+:CO32−:Mg2+) is 1:1:1, and the Ca:Mg molar ratio of the obtained Mg–ACC (Fig. 7a) is 8.56:1 (by ICP-AES). With the increase of the concentration of the reactants to 0.0454 mol L−1, the molar ratio (Ca2+:CO32−:Mg2+) remained constant, but the Ca:Mg molar ratio of the obtained Mg–ACC (Fig. 7b) was 5.66:1 (by ICP-AES). With a further increase of the concentration of the reactants to 0.0909 mol L−1 the molar ratio (Ca2+:CO32−:Mg2+) remained constant, but the Ca:Mg molar ratio of the obtained Mg–ACC (Fig. 7c) was 5.0:1 (by ICP-AES). Based on these experimental results, we have found that when the molar ratio of the reactants remains constant (Ca2+:CO32−:Mg2+ = 1:1:1), the Ca:Mg molar ratio of the obtained Mg–ACC is inversely proportional to the concentration of the reactants as shown in Table 1. The higher the concentration of the reactants the more ions in the mixture, thus a possible explanation is that the reaction rate is directly related to the Ca:Mg molar ratio of the Mg–ACC in this system. The faster the reaction rate, the more magnesium is doped into the obtained Mg–ACC.
 | ||
| Fig. 7 FT-IR spectra of calcium carbonate samples obtained from different concentration of the reactants as the molar ratio (Ca2+:CO32−:Mg2+ = 1:1:1) remains constant: (a) 0.0227 mol L−1; (b) 0.0454 mol L−1; (c) 0.0909 mol L−1. | ||
:1:1) remains constant
| Sample | Molar ratio of the reactants (Ca2+:CO32−:Mg2+) | Concentration of the reactants [Mg2+] = [Ca2+] = [CO32−] | Ca:Mg | FT-IR |
|---|---|---|---|---|
| 1 | 1:1:1 |
0.0227 mol L−1 | 8.56:1 |
Mg–ACC |
| 2 | 1:1:1 |
0.0454 mol L−1 | 5.66:1 |
Mg–ACC |
| 3 | 1:1:1 |
0.0909 mol L−1 | 5.00:1 |
Mg–ACC |
It is well known that Mg–ACC is a metastable phase which can act as a precursor to crystalline CaCO3 phases such as calcite, vaterite or aragonite. However, to date, long-term preservation methods for Mg–ACC have not been realized.
Herein, we introduce two methods for the long-term preservation of Mg–ACC. Mg–ACC was synthesized by rapid mixing of solutions of CaCl2: 0.1 mol L−1, 25 mL; Na2CO3: 0.1 mol L−1, 25 mL; MgCl2 ·6H2O: 0.5 mol L−1, 5 mL. Then, in the first method the obtained Mg–ACC powder was stored at −5 °C. According to Ogino et al.15 this low temperature can retard the transformation of ACC into crystalline CaCO3. When we used this method, the sample did not crystallize even after one year (Fig. 8a). The second method involves preserving the obtained Mg–ACC powder in ethanol at 5 °C. When this second method was used, the sample also did not crystallize after one year (Fig. 8b). The ethanol itself inhibits the dissolution of Mg–ACC since the alcohol greatly reduces the solubility of ionic solids like CaCO3. Thus, the solution-mediated phase transition of Mg–ACC to calcite or aragonite can barely take place.16 Therefore, the ethanol and the low temperature play important roles in the long-term preservation of Mg–ACC. These two valid preservation methods might be used widely in the long-term preservation of Mg–ACC in the future.
 | ||
| Fig. 8 (a) FT-IR spectra of Mg–ACC preserved by two methods over one year: (a) −5 °C; (b) in ethanol at 5 °C. | ||
In summary, we have reported a simple chemistry route for the gram-scale, low-cost, rapid synthesis of highly stable Mg–ACC nanoparticles. The possible composition of Mg–ACC can be defined as Mg0.15Ca0.85CO3·H2O0.85. The molar ratio of Mg2+:Ca2+:CO32− and the concentration of the reactants CaCl2, Na2CO3, and MgCl2 play important roles in the Mg:Ca molar ratio of the obtained Mg–ACC. As-prepared Mg–ACC nanoparticles can be preserved for over one year without crystallization by storing either its dry powder at −5 °C or storing it in ethanol at 5 °C. Due to its high plasticity, Mg–ACC can undergo induced crystallization to produce particles with desirable morphologies and polymorphs. Therefore, the successful synthesis of Mg–ACC nanoparticles on a large scale would be useful for biomineralization studies and industrial applications.
Acknowledgements
This work is supported by the National Basic Research Program of China (2010CB934700), the National Natural Science Foundation of China (NSFC, No. 50732006), and the International Science & Technology Cooperation Program of China (2010DFA41170).References
- (a) Y. Levi-Kalisman, S. Raz, S. Weiner, L. Addadi and I. Sagi, Adv. Funct. Mater., 2002, 12, 43 CrossRef CAS; (b) F. C. Meldrum, Int. Mater. Rev., 2003, 48, 187 Search PubMed.
- (a) M. Faatz, F. Grohn and G. Wegner, Adv. Mater., 2004, 16, 996 CrossRef CAS; (b) S. Raz, P. C. Hamilton, F. H. Wilt, S. Weiner and L. Addadi, Adv. Funct. Mater., 2003, 13, 480 CrossRef CAS; (c) X. R. Xu, J. T. Han, D. H. Kim and K. Cho, J. Phys. Chem. B, 2006, 110, 2764 CrossRef CAS; (d) X. R. Xu, J. T. Han and K. Cho, Chem. Mater., 2004, 16, 1740 CrossRef CAS; (e) J. T. Han, X. R. Xu, D. H. Kim and K. Cho, Adv. Funct. Mater., 2005, 15, 475 CrossRef CAS; (f) J. Aizenberg, D. A. Muller, J. L. Grazul and D. R. Hamann, Science, 2003, 299, 1205 CrossRef CAS; (g) E. Loste and F. Meldrum, Chem. Commun., 2001, 901 RSC.
- (a) M. Li, B. Lebeau and S. Mann, Adv. Mater., 2003, 15, 2032 CrossRef CAS; (b) M. Li and S. Mann, Adv. Funct. Mater., 2002, 12, 773 CrossRef CAS.
- (a) S. Raz, S. Weiner and L. Addadi, Adv. Mater., 2000, 12, 38 CrossRef CAS; (b) F. C. Meldrum and S. T. Hyde, J. Cryst. Growth, 2001, 231, 544 CrossRef CAS; (c) X. Cheng, P. L. Varona, M. J. Olszta and L. B. Gower, J. Cryst. Growth, 2007, 307, 395 CrossRef CAS.
- E. Loste, R. M. Wilson, R. Seshadri and F. C. Meldrum, J. Cryst. Growth, 2003, 254, 206 CrossRef CAS.
- (a) F. Lippman, Fortschr. Miner., 1960, 38, 156 Search PubMed; (b) R. L. Folk, J. Sediment. Petrol., 1974, 44, 40 Search PubMed.
- S. Weiner, Y. Levi-Kalisman, S. Raz and L. Addadi, Connect. Tissue Res., 2003, 44, 214 CAS.
- L. B. Gower, Chem. Rev., 2008, 108, 4551 CrossRef CAS.
- P. K. Ajikumar, L. G. Wong, G. Subramanyam, R. Lakshminarayanan and S. Valiyaveettil, Cryst. Growth Des., 2005, 5(3), 1129 CrossRef CAS.
- (a) R. S. K. Lam, J. M. Charnock, A. Lenniec and F. C. Meldrum, CrystEngComm, 2007, 9, 1226 RSC; (b) Y. Nishino, Y. Oaki and H. Imai, Cryst. Growth Des., 2009, 9, 223 CrossRef CAS.
- (a) L. Addadi, S. Raz and S. Weiner, Adv. Mater., 2003, 15, 959 CrossRef CAS; (b) C. Günther, A. Becker, G. Wolf and M. Epple, Z. Anorg. Allg. Chem., 2005, 631, 2830 CrossRef; (c) F. M. Michel, J. MacDonald, J. Feng, B. L. Phillips, L. Ehm, C. Tarabrella, J. B. Parise and R. J. Reeder, Chem. Mater., 2008, 20, 4720 CrossRef CAS.
- F. A. Andersen and L. Brecevic, Acta Chem. Scand., 1991, 45, 1018 CrossRef CAS.
- Y. Oaki, S. Kajiyama, T. Nishimura, H. Imai and T. Kato, Adv. Mater., 2008, 20, 3633 CrossRef CAS.
- Y. Politi, T. Arad, E. Klein, S. Weiner and L. Addadi, Science, 2004, 306, 1161 CrossRef CAS.
- T. Ogino, T. Suzuki and K. Sawada, Geochim. Cosmochim. Acta, 1987, 51, 2757 CAS.
- H. S. Lee, T. H. Ha and K. Kim, Mater. Chem. Phys., 2005, 93, 376 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |