DOI:
10.1039/C0NR00440E
(Paper)
Nanoscale, 2010,
2, 2751-2757
Morphology, crystal structure and adsorption performance of hydrothermally synthesized titania and titanate nanostructures
Received
25th June 2010
, Accepted 4th August 2010
First published on
11th October 2010
Abstract
Titania (TiO2) and sodium titanate nanostructures with controllable phases and morphologies were synthesized by a hydrothermal method with titanium disulfide (TiS2) as the starting material. Sodium titanate nanobelts could be synthesized under a relatively low alkaline concentration (1 mol L−1 NaOH) and short duration (6 h). At 3 mol L−1 HCl, rutile TiO2 nanorods were synthesized. Anatase TiO2 nanoparticles were obtained at pH values ranging between 2 and 13. FTIR analysis confirmed the phase change as the pH of the reacting medium increased from highly acidic to highly alkaline. The adsorption of Methylene Blue (MB) on the as-synthesized sodium titanate nanobelts fitted well with the Langmuir monolayer model, with an adsorption capacity as high as 312.5 mg g−1. The kinetics of MB adsorption was found to be a pseudo-second-order kinetic model. In brief, this study demonstrates a simple method to control the phase and morphology of titanium-based oxides. Excellent performance has been shown in the MB adsorption test by the sodium titanate nanostructures.
Introduction
Titania (TiO2) is a wide band-gap semiconductor possessing specific chemical properties and stability. It exists in three distinct phases: anatase, rutile and brookite under atmospheric pressure. Both anatase and brookite are metastable while rutile is a thermodynamically stable phase. TiO2 is widely employed in various applications, especially in photocatalysis1,2 and photovoltaics.3 TiO2 can be synthesized via various methods, such as sol–gel4 and hydrothermal.5 In comparison with other techniques, hydrothermal synthesis is an inexpensive and environment friendly method5 with the ability to control the chemical composition and morphology of the synthesized products.6
The synthesis conditions affecting the phase composition and morphology of hydrothermally synthesized TiO2 are the time and temperature of synthesis, and the pH of the reacting solution. It has been reported that a gradual phase change from anatase to rutile was observed as acidity increased.7,8 Moreover, TiO2 can also be prepared from TiS2 using the hydrothermal method9 under acid conditions or by heat treatment.10 However, to the best of our knowledge, there is no report on subjecting TiS2 under alkaline conditions to synthesizing TiO2 and titanate compounds. There is also no systematic investigation of how a wide range of solution acidity would affect the morphology and crystal structure of the final titanium oxide(s) using TiS2 as the starting material.
In recent years, titanate nanostructures derived via alkaline conditions have attracted much attention11–14 due to their potential in photocatalysis,15,16 ion-exchange,17–19 and adsorption related applications.20,21 Since Kasuga et al. devised a method to synthesize titanate compounds by a hydrothermal method,22 there has been growing interest in producing TiO2-derived titanate structures, including nanowires16,23,24 and nanotubes.12,25,26 However, these titanate products were usually synthesized with very high alkaline concentrations (5–20 mol L−1 NaOH), and the reaction time typically ranged from 12 to 48 h.12,19 In this study, various titania and titanate nanostructures with controllable phases and morphologies were synthesized from TiS2. For the first time, we report that sodium titanate nanostructures could be synthesized at 1 mol L−1 NaOH concentration in a relatively short duration of 6 h. Excellent adsorption performance was demonstrated by the as-synthesized titanate nanostructures.
In a typical procedure, TiS2 (Aldrich, 99.8%) was added to deionised water, and the pH of the reacting solution was subsequently adjusted by adding either HCl or NaOH of different concentrations. The acidity/basicity of the reacting solution ranged from 3 mol L−1 HCl to 10 mol L−1 NaOH. The above colloid was then transferred into a 20 mL-Teflon-lined stainless steel autoclave until 60% of its volume was filled. The autoclave was maintained at 150 °C for 6 h and then air-cooled to room temperature. The samples were collected via centrifugation, gently washed with deionised water and ethanol, and dried in an oven at 80 °C. For comparison purposes, commercial anatase powder (Aldrich, 98%) was also used as starting material and subjected to the same experimental conditions.
Characterization
The X-ray diffraction (XRD) patterns were obtained on a Shimadzu X-ray diffractometer using Cu Kα radiation (λ = 1.54178 Å) at ambient temperature (25 °C). The accelerating voltage and applied current were 40 kV and 40 mA respectively. The morphologies of the samples were examined by field-emission scanning electron microscopy (FESEM, JEOL JSM-6340F) and transmission electron microscopy (TEM, JEOL JEM-2010). The chemical structure of the as-synthesized products was examined using Fourier-transform infrared spectroscopy (FTIR, Perkin Elmer FTIR System Spectrum GX) in the range 400–4000 cm−1.
Dye adsorption test
Methylene Blue (MB) was used to determine the sample's adsorption kinetics and capacities. The adsorption tests were carried out in the dark to prevent the degradation of MB under light. The equilibrium adsorption isotherm was determined with MB solution (50 mL) with varying concentrations from 20 to 80 mg L−1. At each run, the adsorbent (10 mg) was added to the solutions so that the adsorbent loading was 200 mg L−1. After 4320 min, the equilibrium concentration was measured. To understand the adsorption mechanism, the equilibrium adsorption isotherm data were curve-fitted into the well-known Freundlich and Langmuir models:| | 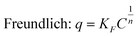 | (1) |
| | 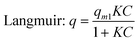 | (2) |
where q (mg g−1) is the amount of adsorbed MB, C (mg L−1) is the concentration of MB at equilibrium, qm1 (mg g−1) is the maximum adsorption capacity, KF and n are Freudlich constants, and K is the Langmuir constant. If the adsorption isotherm exhibits Langmuir behaviour, it indicates monolayer adsorption. On the other hand, a good fit into the Freundlich model indicates a heterogeneous surface binding.
For the kinetics experiment, the synthesized powder (20 mg) was added to MB solution (100 mL) with an initial concentration of 100 mg L−1. The absorbent loading was therefore 200 mg L−1. Stirring was applied throughout the duration of the test (1680 min) at a speed of 300 rpm. To investigate the rate at which MB was adsorbed on the sodium titanate nanostructures, two kinetic models were employed to fit the experimental data: the pseudo-first-order kinetic model and the pseudo-second-order kinetic model. The pseudo-first-order kinetic model can be expressed in its integral form as:
| | | ln(Qe1 − Qt) = lnQe1 − k1t | (3) |
where
Qe1 and
Qt are the amount of
dye adsorbed (mg g
−1) on the adsorbent at equilibrium and at time
t, respectively,
k1 is the rate constant for the pseudo-first-order kinetic model (min
−1), and
t the contact time (minutes). The values of ln(
Qe1 −
Qt) were calculated from kinetic data.
The pseudo-second-order kinetic model can be expressed in its integral form as:
| | 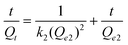 | (4) |
where
k2 (g mg
−1min
−1) is the rate constant for the pseudo-second-order kinetic model, and
Qe2 the equilibrium amount of
dye adsorbed (mg g
−1).
All of the adsorption experiments were carried out at ambient temperature (25 °C) and at a standardized pH value range of 9.5–10. The initial pH value was close to this range, but slightly differed due to the difference in the MB concentration. Therefore minor adjustment was carried out to unify the starting pH of different solutions. The amount of MB adsorbed by the adsorbent, q, was calculated from the difference between the initial and the final MB concentration as follows:
| | 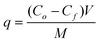 | (5) |
where
q is the amount of MB adsorbed (mg g
−1),
Co and
CF are the initial and final concentrations of MB remaining in the solution (mg L
−1),
V is the volume of the MB solution (L), and
M is the mass of the adsorbent added (g). The
experimental data reported was obtained by averaging the data from five parallel experiments conducted simultaneously under the same experimental conditions. The quantitative determination of the MB concentration in solution at different time intervals was performed by measuring the intensity change of a specific absorption peak of the sample solution under a Perkin-Elmer
UV-Vis-NIR Lambda 900
spectrophotometer.
Results and discussion
Phase composition
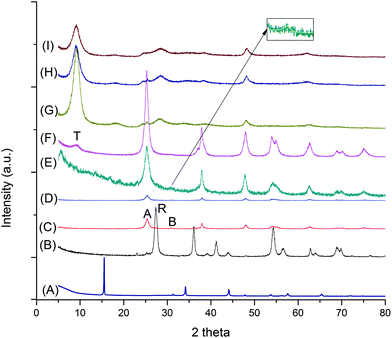
Fig. 1 presents the XRD patterns of the as-synthesized TiO2 nanostructures using TiS2 as the starting material (curve (A), Fig. 1) hydrothermally treated for 6 h under different acidity/basicity. When 3 mol L−1 HCl was used as the reacting solution, rutile was the main phase. In the pH range of 2 to 11, the anatase phase was present, though for the product synthesized at pH 11, a trace amount of brookite phase was also detected (curve (E), Fig. 1). At pH = 13, anatase still remained as the predominant phase, with a small amount of sodium titanate, as seen from the weak peak at θ ≈ 9°. When 1 mol L−1 or higher concentration NaOH was used as the reacting solution, only the sodium titanate phase was present. In comparison, when commercial anatase TiO2 (A-TiO2) powder was used as starting material and synthesized under different acidity/basicity in the pH range of 2 to 13, there was no chemical reaction at all (the XRD data not shown). Only a trace amount of rutile was formed in the samples subjected to 3 mol L−1 HCl and in pH 14 medium (Fig. 2). When 5 mol L−1 NaOH was used as the reacting solution, trace amounts of rutile and titanate products were synthesized (Fig. 2). All strong peaks corresponded to the unreacted starting material A-TiO2. It was only when A-TiO2 was subjected to 10 mol L−1 NaOH reacting solution that it was completely transformed to sodium titanate (Fig. 2). This observation agrees with what has been reported by other researchers,19,21,23–25 where high alkaline concentration (≥ 5 mol L−1 NaOH) is needed to transform the starting material TiO2 to titanate.
 |
| | Fig. 1 XRD patterns of starting material TiS2 (curve (A)) and TiS2-derived products under different acidity/basicity. Curve (B): 3 mol L−1 HCl, (C): pH = 2, (D): pH = 4, (E): pH = 11, (F): pH = 13, (G): pH = 14, (H): 5 mol L−1 NaOH and (I): 10 mol L−1 NaOH. Label A stands for anatase, B for brookite, R for rutile and T for titanate. The inset shows a magnified image of the characteristic brookite peak at θ ≈ 30.7°. | |
 |
| | Fig. 2 XRD patterns of anatase TiO2-derived products under different acidity/basicity. Curve (A): 3 mol L−1 HCl, (B): pH = 14, (C): 5 mol L−1 NaOH, (D): 10 mol L−1 NaOH. Label A stands for anatase, R for rutile and T for titanate. | |
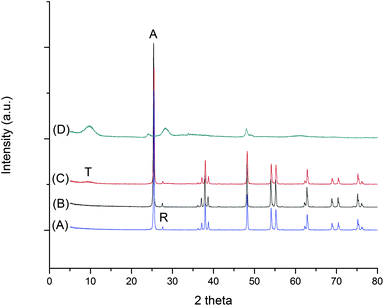
The formation mechanism of different phases from TiS2 under different acidity/basicity is proposed in Fig. 3. When TiS2 is added into aqueous solution, hydrolysis reaction occurs which leads to the formation of Ti(OH)x4−x complex in the solution:
| TiS2 + 4H+ → Ti4+ + 2H2S ↑ (acidic condition) |
| TiS2 + 4NaOH → Ti4+ + 4OH− + 2Na2S (alkaline condition) |
| Ti4+ + x(OH)− → Ti(OH)x4−x (both conditions) |
 |
| | Fig. 3 A schematic of the proposed formation mechanism of titania and titanate samples synthesized under various acidity/basicity. | |
Under highly acidic condition in aqueous medium (e.g. 3 mol L−1 HCl), the number of OH− coordinated to Ti4+ centres is limited, leading to the formation of a more positively charged Ti(OH)x4−x. A more positively charged Ti(OH)x4−x encourages corner-sharing of the TiO6 octahedra, favoring the formation of a rutile phase.27 Similarly, when the solution pH ranges from 2 to 13, there is an increase in the number of OH− coordinated to Ti4+ centres, and hence the complex Ti(OH)x4−x would be less positively charged, which in turn encourages edge-sharing of TiO6 octahedra. Such an arrangement favors the formation of an anatase phase.27 For titanate formation, the dissolution/crystallization process of the precursor takes place.28 Therefore, the bond-breaking of Ti–S (or Ti–O, for TiO2) is the limiting step for TiS2, as the formation of Ti(OH)x4− is required in order for titanate layers to form by olation of Ti(OH)x4−. It has been reported that the bond energy for Ti–S (418 ± 3 kJ mol−1) is lower than Ti–O (672 ± 9 kJ mol−1),29 which may facilitate the formation of titanate in a lower concentration of NaOH (1 mol L−1) as compared to that using TiO2 as precursor.
Fig. 4 presents the FTIR spectra of the as-synthesized TiO2 and titanate products formed under different HCl and NaOH concentrations. Hydroxyl groups could be observed for the sodium titanate products due to the existence of a bending vibration of O–H–O at a wavenumber (ν) of 1630 cm−1 and a strong stretching vibration of O–H group at ν = 3400 cm−1. The relatively high intensity of the peaks associated with the hydroxyl groups suggested that a large amount of hydroxyl groups were located on the surface and in the layers of the sodium titanate products.30 It could also be inferred from Fig. 4 that the amount of hydroxyl groups present decreased as the pH of the synthesis condition decreased, as the intensity of the spectra at ν = 1630 cm−1 and ν = 3400 cm−1 was reduced from the one synthesized at a higher pH to the one at a lower pH. Two sharp peaks at ν = 500 cm−1 and ν = 900 cm−1 observed for sodium titanate products (curves (C)–(E)) were attributed to the stretching of Ti–O found in titanate products.30,31 As expected, the spectra of the rutile (curve (A)) and anatase (curve (B)) products did not contain the peaks at ν = 500 cm−1 and ν = 900 cm−1.
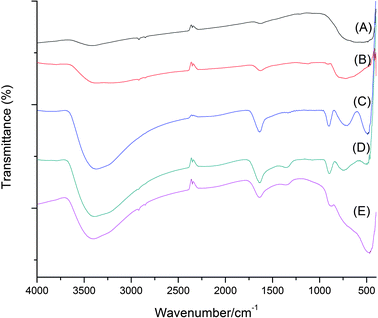 |
| | Fig. 4 FTIR spectra of the as-synthesized powder. Curve (A) is for rutile synthesized in 3 mol L−1 HCl, (B) for anatase synthesized in pH 11, and (C), (D), (E) are for sodium titanate synthesized at pH 14, 5 mol L−1 NaOH and 10 mol L−1 NaOH, respectively. | |
Morphology
The morphologies of the synthesized titania and sodium titanate are shown in Fig. 5, 6 and 7. It is clearly shown that the acidity/basicity of the reacting medium affects the morphology of the as-synthesized products. From Fig. 5a and 6a, rutile TiO2 synthesized using 3 mol L−1 HCl possessed nanorod morphology, with an average length of 100 nm and an average diameter of 10 nm. Anatase TiO2 synthesized in the pH range of 2 to 13 were agglomerated nanoparticles (Fig. 5b and 6c). The morphologies of titanate products synthesized from 1 mol L−1, 5 mol L−1 and 10 mol L−1 NaOH solutions consisted of nanobelt or nanosheet type morphology (Fig. 5c and 7a). The average width of the nanobelts was 7 nm. However, the morphology of sodium titanate synthesized from 10 mol L−1 NaOH with A-TiO2 as the starting material (Fig. 5d and 7c) resembled a nanotubular-like structure, which is in good agreement with many previous reports.18,25,32 The mechanism for the nanotube formation has been explained as the folding of nanosheet under asymmetric surface tension.33,34 However, in the case of TiS2 as the starting material, the same condition (10 mol L−1 NaOH) produced a nanobelt (or nanosheet) structure instead. An apparent explanation could be that the presence of sulfur ions may have modified the surface tension, such that there is insufficient drive for the wrapping action.
 |
| | Fig. 5 FESEM images of hydrothermally synthesized TiO2 and titanates in (a) 3 mol L−1 HCl, (b) pH = 13, (c) pH = 14 using TiS2 as precursor, and (d) titanate nanotubes in 10 mol L−1 NaOH using A-TiO2 as precursor. | |
 |
| | Fig. 6 TEM images of hydrothermally synthesized TiO2 in (a) 3 mol L−1 HCl and (c) pH 11, (b) high-magnification image of (a), and (d) high-magnification image of (c). | |
 |
| | Fig. 7 TEM images of hydrothermally synthesized titanates in (a) 10 mol L−1 NaOH using TiS2 as precursor, and (c) 10 mol L−1 NaOH using A-TiO2 as precursor, (b) high-magnification image of (a), and (d) high-magnification image of (c). | |
Adsorption kinetics
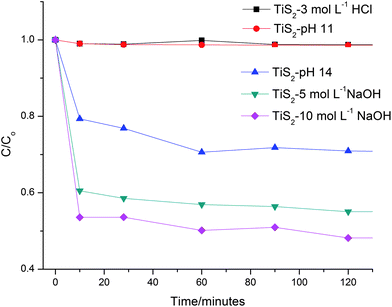
To investigate the rate of adsorption of MB by the various titania and titanate nanostructures, adsorption kinetics tests were carried out. Fig. 8 illustrates the relative MB concentration change with time in the first 120 min when the initial concentration was 100 mg L−1. It is clear that the rates of adsorption of the as-synthesized rutile and anatase samples (TiS2-3 mol L−1 HCl and TiS2-pH 11 respectively) were negligible, whereas substantial amounts of MB were adsorbed by the as-synthesized sodium titanate nanobelts (TiS2-pH 14, TiS2-5 mol L−1 NaOH and TiS2-10 mol L−1 NaOH). After 1680 min, the percentages of MB adsorbed by TiS2-pH 14, TiS2-5 mol L−1 NaOH and TiS2-10 mol L−1 NaOH titanate nanobelts were approximately 37, 45 and 53%, respectively.
Based on the fitting of kinetic data to the pseudo-first-order kinetic model, the R2 values (Table 1) for the adsorption of MB on all the as-synthesized titanate nanostructures were extremely low, ranging from 0.36 to 0.59. This indicates that the adsorption kinetics does not follow the pseudo-first-order kinetic model. On the other hand, when the MB adsorption kinetics was modelled to the pseudo-second-order kinetic model, the R2 values were close to unity for all as-synthesized titanate nanostructures (Table 1). This indicates that the kinetics of adsorption of MB on the as-synthesized titanate nanostructures could be described more accurately by the pseudo-second-order kinetic model. Similar kinetics model has also been observed in the adsorption of MB onto rice husks35 and date pits.36
Table 1 Parameters of pseudo first- and second-order adsorption kinetics for MB adsorption onto various titanate nanostructures
| Adsorbent |
Experimental Qe1a |
Pseudo-first-order kinetics |
Pseudo-second-order kinetics |
|
k
1
|
Q
e1
/cal |
R
2
|
k
2
|
Q
e2
/cal |
R
2
|
|
Qe1 (mg g−1).
k1 (min−1).
Qe1, cal (mg g−1).
k2 (g mg−1min−1).
Qe2, cal (mg g−1).
|
| TiS2-pH 14 |
253.12 |
0.0032 |
169.12 |
0.59 |
4.52 × 10−4 |
166.67 |
0.99 |
| TiS2-5 mol L−1 NaOH |
277.90 |
0.0042 |
107.33 |
0.36 |
1.73 × 10−3 |
227.27 |
0.99 |
| TiS2-10 mol L−1 NaOH |
296.40 |
0.0056 |
97.67 |
0.42 |
1.16 × 10−3 |
263.16 |
0.99 |
Adsorption isotherms
To investigate the MB adsorption capacities of the as-synthesized titanate nanostructures, adsorption isotherm tests were carried out. Fig. 9 presents the adsorption isotherms of MB with various titania and titanate compounds. It is clear that there was negligible adsorption of MB on TiS2-3 mol L−1 HCl. The results of fitting the isotherm curves to Freudlich and Langmuir models were summarized in Table 2. Regression coefficients (R2) for different titanate compounds were all close to unity for the Langmuir model, indicating that the Langmuir model describes well the MB adsorption behaviour. On the other hand, the R2 values for all the titanate compounds were less than 0.9 when the data were fitted with the Freudlich model. This illustrates that the adsorption of MB by the as-synthesized titanate compounds is mainly governed by monolayer adsorption, as described by the Langmuir model. From Table 2, among the titanate compounds tested in the adsorption of MB, TiS2-10 mol L−1 NaOH had the highest qm (297.24 mg g−1) and qm1 (312.50 mg g−1). On the contrary, qm and qm1 of the TiO2-10 mol L−1 NaOH was significantly smaller. It could be inferred that the number of available sites for adsorption on the titanate nanobelts was larger than the number of available sites for adsorption on the titanate nanotubes. The adsorption mechanism may be due to an electrostatic force of attraction between the adsorbent and MB.37–39 It was stated that the dominating surface charge for titanate in alkaline pH was negative. As the isotherm adsorption experiments were carried out at pH 9.5–10, the dominant surface charge for the titanate nanostructures was negative. This phenomenon was also observed in our previous works.20,40 MB, a cationic dye, can be attracted by titanate nanostructures via an electrostatic force of attraction. Compared with existing data in the literature, the sodium titanate nanostructures synthesized in this work exhibited much larger adsorption capacities (Table 3).
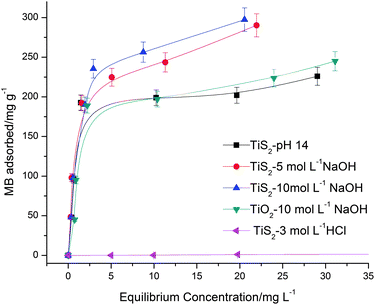 |
| | Fig. 9 Adsorption isotherms of various titania and titanate nanostructures in MB. The y-axis displays the amount of MB adsorbed by the nanostructures, The x-axis displays the equilibrium concentration of MB. | |
| Adsorbent |
Experimental qma |
Langmuir |
Freundlich |
|
K
|
q
m1
|
R
2
|
K
F
|
n
|
R
2
|
|
qm (mg g−1).
K (L mg−1).
q
m1 (mg g−1).
K
F (mg1−n L−n g−1).
|
| TiS2-pH 14 |
254.76 |
0.88 |
227.27 |
0.99 |
98.90 |
3.60 |
0.69 |
| TiS2-5 mol L−1 NaOH |
290.19 |
0.72 |
303.03 |
0.99 |
113.04 |
2.83 |
0.83 |
| TiS2-10 mol L−1 NaOH |
297.24 |
0.70 |
312.50 |
0.99 |
111.09 |
2.45 |
0.84 |
| TiO2-10 mol L−1 NaOH |
244.95 |
0.45 |
256.41 |
0.99 |
84.56 |
2.95 |
0.72 |
Table 3 Comparison of the maximum monolayer adsorption of MB on various adsorbents
Conclusion
Phase- and morphology-controllable titania and titanates nanostructures were hydrothermally synthesized using TiS2 as precursor. While sodium titanate could be synthesized from TiO2 at concentrations of 5–20 mol L−1 NaOH for a relatively long duration, by using TiS2 as starting material, sodium titanate could be synthesized when the concentration of NaOH was 1 mol L−1 with a shorter processing time. This is due to the relatively weaker Ti–S bond strength as compared to Ti–O, which translates to a less drastic condition required to break up the Ti–S bond to enable the formation of titanate layers by olation of Ti(OH)x4−x. It was also observed that the as-synthesized products exhibited different morphologies when different acidity/basicity of the reacting medium was used. The equilibrium isotherm test data fitted well with the Langmuir model, which indicated that the MB was adsorbed to the titanate surface by monolayer adsorption. The adsorption capacities of the as-synthesized sodium titanate nanostructures were also much better than the adsorption capacity of sodium titanate nanotubes synthesized from TiO2. The pseudo-second-order kinetic model was found to describe well the adsorption kinetics. The strong adsorption demonstrated by the synthesized titanate nanostructures deserves further attention as they can be potentially applied in the removal of cationic dyes in water.
Acknowledgements
Financial support from the National Research Foundation of Singapore Government (grant No. MEWR 651/06/160) and Nanyang Technological University (URECA scheme) is gratefully acknowledged.
References
- A. Fujishima, X. Zhang and D. A. Tryk, Surf. Sci. Rep., 2008, 63, 515–582 CrossRef CAS.
- A. G. Agrios and P. Pichat, J. Appl. Electrochem., 2005, 35, 655–663 CrossRef.
- B. O'Regan and M. Gratzel, Nature, 1991, 353, 737 CrossRef CAS.
- M. Kanna and S. Wongnawa, Mater. Chem. Phys., 2008, 110, 166–175 CrossRef CAS.
- K. Byrappa and T. Adschiri, Prog. Cryst. Growth Charact. Mater., 2007, 53, 117–166 CrossRef CAS.
- J. Jitputti, S. Pavasupree, Y. Suzuki and S. Yoshikawa, J. Solid State Chem., 2007, 180, 1743–1749 CrossRef CAS.
- H. Luo, Y. Takata, Y. Lee, J. Zhao, K. Domen and Y. Yan, Chem. Mater., 2004, 16, 846–849 CrossRef CAS.
- J.-K. Oh, J.-K. Lee, S. J. Kim and K.-W. Park, J. Ind. Eng. Chem. (Amsterdam, Neth.), 2009, 15, 270–274 Search PubMed.
- W. Ho, J. C. Yu and S. Lee, J. Solid State Chem., 2006, 179, 1171–1176 CrossRef CAS.
- T. Umebayashi, T. Yamaki, H. Itoh and K. Asai, Appl. Phys. Lett., 2002, 81, 454–456 CrossRef CAS.
- Q. Chen, W. Zhuo, G. Du and L.-M. Peng, Adv. Mater., 2002, 14, 1208–1211 CrossRef CAS.
- D. V. Bavykin, V. N. Parmon, A. A. Lapkin and F. C. Walsh, J. Mater. Chem., 2004, 14, 3370–3377 RSC.
- R. Ma, Y. Bando and T. Sasaki, Chem. Phys. Lett., 2003, 380, 577–582 CrossRef CAS.
- T. W. Kim, S. G. Hur, S. J. Hwang and J. H. Choy, Chem. Commun., 2006, 220–222 RSC.
- T. W. Kim, S. G. Hur, S. J. Hwang, H. Park, W. Choi and J. H. Choy, Adv. Funct. Mater., 2007, 17, 307–314 CrossRef CAS.
- F. Amano, T. Yasumoto, T. Shibayama, S. Uchida and B. Ohtani, Appl. Catal., B, 2009, 89, 583–589 CrossRef CAS.
- C.-K. Lee, C.-C. Wang, L.-C. Juang, M.-D. Lyu, S.-H. Hung and S.-S. Liu, Colloids Surf., A, 2008, 317, 164–173 CrossRef CAS.
- X. Sun and Y. Li, Chem.–Eur. J., 2003, 9, 2229–2238 CrossRef CAS.
- R. Ma, T. Sasaki and Y. Bando, Chem. Commun., 2005, 948–950 RSC.
- Y. X. Tang, Y. K. Lai, D. G. Gong, K.-H. Goh, T.-T. Lim, Z. L. Dong and Z. Chen, Chem.–Eur. J., 2010, 16, 7704–7708 CrossRef CAS.
- H. Y. Niu, J. M. Wang, Y. L. Shi, Y. Q. Cai and F. S. Wei, Microporous Mesoporous Mater., 2009, 122, 28–35 CrossRef CAS.
- T. Kasuga, M. Hiramatsu, A. Hoson, T. Sekino and K. Niihara, Langmuir, 1998, 14, 3160–3163 CrossRef CAS.
- J. Jitputti, Y. Suzuki and S. Yoshikawa, Catal. Commun., 2008, 9, 1265–1271 CrossRef CAS.
- Z.-Y. Yuan and B.-L. Su, Colloids Surf., A, 2004, 241, 173–183 CrossRef CAS.
- T. Kasuga, Thin Solid Films, 2006, 496, 141–145 CrossRef CAS.
- Q. Chen, G. H. Du, S. Zhang and L.-M. Peng, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 587–593 CrossRef CAS.
- A. Testino, I. R. Bellobono, B. Buscaglia, C. Canevali, M. D'Arienzo, S. Polizzi, R. Scotti and F. Morazzoni, J. Am. Chem. Soc., 2007, 129, 3564–3575 CrossRef.
- D. V. Bavykin, J. M. Friedrich and F. C. Walsh, Adv. Mater., 2006, 18, 2807–2824 CrossRef CAS.
-
Y.-R. Luo, in Comprehensive handbook of chemical bond energies, CRC press, 2007, ch. 14, p. 669 Search PubMed.
- M. Qamar, C. R. Yoon, H. J. Oh, N. H. Lee, K. Park, D. H. Kim, K. S. Lee, W. J. Lee and S. J. Kim, Catal. Today, 2008, 131, 3–14 CrossRef CAS.
- N. Sukpirom and M. M. Lerner, Chem. Mater., 2001, 13, 2179–2185 CAS.
- T. Kasuga, M. Hiramatsu, A. Hoson, T. Sekino and K. Niihara, Adv. Mater., 1999, 11, 1307–1311 CrossRef CAS.
- A. Kukovecz, M. Hodos, Z. Konya and I. Kiricsi, Chem. Phys. Lett., 2005, 411, 445–449 CrossRef CAS.
- W. Chen, X. Sun and D. Weng, Mater. Lett., 2006, 60, 3477–3480 CrossRef CAS.
- V. Vadivelan and K. V. Kumar, J. Colloid Interface Sci., 2005, 286, 90–100 CrossRef CAS.
- F. Banat, S. Al-Asheh and L. Al-Makhadmeh, Process Biochem. (Amsterdam, Neth.), 2003, 39, 193–202 Search PubMed.
- M. L. Fetterolf, H. V. Patel and J. M. Jennings, J. Chem. Eng. Data, 2003, 48, 831–835 CrossRef CAS.
- M. Xiao, L. Wang, Y. Wu, X. Huang and Z. Dang, J. Solid State Electrochem., 2008, 12, 1159–1166 CrossRef CAS.
- D. V. Bavykin, K. E. Redmond, B. P. Nias, A. N. Kulak and F. C. Walsh, Aust. J. Chem., 2010, 63, 270–275 CrossRef CAS.
- Y. Lai, Y. Chen, Y. Tang, D. Gong, Z. Chen and C. Lin, Electrochem. Commun., 2009, 11, 2268–2271 CrossRef CAS.
- L. Xiong, Y. Yang, J. Mai, W. Sun, C. Zhang, D. Wei, Q. Chen and J. Ni, Chem. Eng. J. (Amsterdam, Neth.), 2010, 156, 313–320 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2010 |
Click here to see how this site uses Cookies. View our privacy policy here.