Single-walled carbon nanohorns and their applications
Shuyun
Zhu
ab and
Guobao
Xu
*a
aState Key Laboratory of Electroanalytical Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun, Jilin 130022, China. E-mail: guobaoxu@ciac.jl.cn; Fax: +86-431-85262747; Tel: + 86-431-85262747
bGraduate University of the Chinese Academy of Sciences, Beijing, 100864, China
First published on 18th October 2010
Abstract
Single-walled carbon nanohorns (SWCNHs) are horn-shaped single-walled tubules with a conical tip. They are generally synthesized by laser ablation of pure graphite without using metal catalyst with high production rate and high yield, and typically form radial aggregates. SWCNHs are essentially metal-free and very pure, which avoids cumbersome purification and makes them user-friendly and environmentally benign. Currently, SWCNHs have been widely studied for various applications, such as gas storage, adsorption, catalyst support, drug delivery system, magnetic resonance analysis, electrochemistry, biosensing application, photovoltaics and photoelectrochemical cells, photodynamic therapy, fuel cells, and so on. This review outlines the research progress on SWCNHs, including their properties, functionalization, applications, and outlook.
 Shuyun Zhu | Shuyun Zhu received her BSc from Qufu Normal University in 2003 and her MSc from University of Science and Technology of China in 2006. From 2006 to 2009, she was a research associate at Prof. Xu's group at Changchun Institute of Applied Chemistry. She is presently studying for a PhD under the guidance of Professor Guobao Xu at Changchun Institute of Applied Chemistry in the Chinese Academy of Sciences. Her current research interests include single-walled carbon nanohorns, biosensors, and solid-phase microextraction. |
 Guobao Xu | Guobao Xu received his BSc, MSc, and PhD from Jilin University, Xiamen University, and Changchun Institute of Applied Chemistry, respectively. After postdoctoral research at the University of Hong Kong, the Hong Kong Polytechnic University, and NTT Microsystem Integration Laboratories, he joined Changchun Institute of Applied Chemistry as a Professor. His current research interests include electrochemiluminescence, single-walled carbon nanohorns, aptasensors, nanomaterials, and capillary electrophoresis. |
1. Introduction
Single-walled carbon nanohorns (SWCNHs)1 are a new material that are similar to single-walled carbon nanotubes (SWCNTs).2 Their physico-chemical characteristics and some applications have been reviewed.3–5 In this feature article, we review the chemistry of SWCNHs in a wide range with possible future applications.Three different types of single-walled carbon nanohorns (SWCNHs), ‘dahlia-like’, ‘bud-like’, and ‘seed-like’ have been observed. In the first type, the SWCNHs protrude from the aggregate surface (Fig. 1), while in the second and third types, the SWCNHs appear to develop inside the particle itself. SWCNHs have horn-shaped tips, the diameter of 2–5 nm, tubule length of 40–50 nm and cone angle of approximately 20°. About 2000 of SWCNHs assemble to form a spherical aggregate (diameters: 80–100 nm). SWCNHs are composed of single-graphene which are similar to SWCNTs. As-grown SWCNHs are closed tubes and their specific surface area increases by opening the holes. Through the holes, various molecules can enter inside SWCNHs.
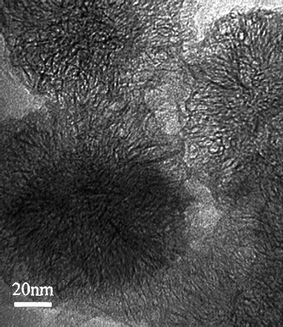
The differences between SWCNH and SWCNT, besides their shape and size, are that there is no use of metal catalysts in their preparation and their high production rate with a high yield of SWCNHs. SWCNHs are prepared through CO2 laser ablation of a graphite target at room temperature under Ar atmosphere at a high rate with a high yield of about 75%,1 and a high purity of about 95%.6 In contrast, the metal catalysts are usually introduced during the production of SWCNTs and carbon nanocages (CNCs).2,7 Afterwards, strong acids are usually used to remove these metal particles, but these strong acids may also damage the graphitic structure and also cause significant loss of carbon materials. Therefore, pure samples of SWCNHs are available more easily than pure samples of SWCNTs and CNCs, which favors the study of their properties and applications.
It has been shown that the nature and pressure of the buffer gas used in the above method plays a key role in the purity and morphology of the product. For example, 95% of the SWCNH aggregates take ‘dahlia-like’ morphology when Ar was used while ‘bud-like’ SWCNH aggregates were formed with a yield of 70 or 80% when He or N2 were used.8 It was also found that CO2 laser ablation of a graphite rod in an Ne atmosphere produced “Ne-dahlia” SWCNH with an aggregate diameter of 50 nm.9 In this approach, a practical production capacity of 1 kg day−1 was achieved, which was about 100 times greater than that attained before by changing the production equipment and using laser ablation.6 In addition, some other methods have also been employed including cavity arc jet methods under open-air conditions,10 a welding arc torch in open air,11 simple pulsed arc discharge,12 and arc in liquid method,13,14 leading to various morphologies and purities.
2. Properties
The Raman spectrum of pristine SWCNHs1 exhibits different characteristics from graphite, glassy carbon, nano-soot, and diamond-like amorphous carbon.16 Two bands with almost equal scattering strengths were detected at 1593 and 1341 cm−1. The unique conical structure of SWCNHs has a great influence on the electronic properties of this material. Theoretical calculations were used to determine the stability, optimum geometry, and electronic properties of SWCNHs. Simulated scanning tunneling microscopy images of the various structures at different bias voltages reflect a net electron transfer towards the pentagon vertex sites.17 The electronic structure of SWCNHs within continuum field-theory gauge model was also studied.18 It was found that only for five pentagons at the tip does a normalized electron state appear at the Fermi level for an unbounded hyperboloid. Pristine SWCNHs showed semiconducting properties, strongly influenced by adsorption of gases, such as oxygen and carbon dioxide19 and the electrical conductivity increased with elevating temperature.20 In addition, the magnetic properties of SWCNHs were investigated using electron spin resonance (ESR) and static magnetic susceptibility measurements, indicating that the individual SWCNH had at least one unpaired electron spin, and a small amount of diamagnetic susceptibility.21 High-resolution 13C nuclear magnetic resonance (NMR) spectroscopy was also used for the investigation of the structure, electronic, and magnetic properties of SWCNHs.22 The results showed that SWCNHs consisted of two components: surface nanohorns (124 ppm, rapid spin–lattice relaxation behavior) and graphite like cone structures (116 ppm, much slower relaxation). Thin films of SWCNHs showed good field emission characteristics such as a low turn-on field and good long-term stability.23 Thermal stability measurements of SWCNHs indicated that the horn-shaped tips became less sharp and the tube walls became corrugated when the heat treatment was 1200–1400 °C.24 In another study, the thermal stability of SWCNHs in O2 indicated SWCNHs had a higher oxidation resistance in the temperature region of 300–550 °C.25X-Ray diffraction (XRD) was used for the study of the structural features of pristine SWCNHs. It indicated the van der Waals distance of the aggregated SWCNHs was ∼0.4 nm, being greater than the interlayer spacing of graphite (0.335 nm).26 Thus SWCNH aggregates should have both microporosity and mesoporosity originating from the above specific structure. In comparison with CNCs in which pore sizes were distributed around several tens of nm,27 the pore sizes of SWCNHs were much smaller. The porosity of SWCNHs has been investigated by a series of reports. SWCNHs have two groups of pores: the inter-nanohorn pores and intra-nanohorn pores.28 SWCNHs are closed in their as-grown state with a surface area of about 300 m2 g−1, but windows can be opened by treating them in an oxygen atmosphere, making the internal pores accessible.29 It was also found the number and size of the window increased on increasing the heating temperature. Moreover, the number and size of nanowindows on the walls of SWCNHs can be controlled by oxidation in oxygen at different temperatures.30 Later, it was found that oxidation with a slow temperature-increase of 1 °C min−1 or in a low oxygen concentration (of 21% in air) could result in hole opening, avoiding c-dust.31 Furthermore, the oxidation treatment of SWCNHs increased the quantity of oxygen-containing functional (OCF) group32 while heat treatment of the SWCNHs under a H2 gas flow resulted in opened SWCNHs with reduced oxygen functionalities (hSWCNHs).33 Acids have also been used for pore opening. Dahlia-like SWCNHs were treated in H2SO4 or an H2O2/H2SO4 mixture,34 or HNO3,35,36 followed by heat treatment, resulting in enhanced microporosity. This was due to an increase of the internal pore volume as well as the interstitial pore volume. The former was generated through oxidative hole opening while the latter was attributed to the intercalation of acid molecules into the interstitial space of the nanohorns.
As-grown SWCNHs are closed, but holes can be created in their walls easily by using the methods mentioned above including heat treatment in oxidative gases and acid treatment. The size and number of holes can be controlled by adjusting the hole-opening conditions. Recently, there are papers reporting that the holes introduced in the rolled graphene structures can be thermally closed. Constant-temperature tight-binding molecular dynamics (TBMD) simulations showed that the holes in the sidewalls were difficult to close by thermal annealing, whereas those at the tips were easy to close.37 It was also found that the holes at the tips of opened SWCNHs exhibited a closed–open–closed evolution when the initial hole size was approximately 0.7–0.9 nm, while the size of the holes were smaller than about 0.7–0.9 nm, they closed quickly and remained closed.38
The opening of bud-like SWCNHs by thermal treatment was also studied. First, bud-like SWCNHs were oxidized in oxygen to produce nano-ordered windows in the walls.39 The oxidation did not change the size and shape of the bud-like SWCNH bundles. So, this was an effective way to produce bud-like SWCNHs with a large micropore volume. Pore structure changes of bud-like SWCNHs produced by laser ablation of carbon in a He atmosphere after oxidation with O2 and CO2 at different temperatures were studied.40 The results showed that O2 opened holes significantly more than CO2. About 77% of the nanohorns were open through oxidization at 693 K. The highest percentage of pore opening with CO2 was 45%. In another study the opening of bud-like SWCNHs was carried out by heat treatment in CO2, indicating that the opening could be controlled by varying the treatment conditions.41 The oxidation of bud-like SWCNHs with CO2 at 1273 K opened a sufficient number of nanohorns, whereas few nanohorns opened at lower temperature. The average size of the generated nanopores increased with temperature and time of treatment. In addition, heat treatment in CO2 decreased the oxygen content in the structure of the generated opened bud-like SWCNHs.
3. Functionalization
Different functionalization of SWCNHs have been described including covalent attachment of organic fragments and non-covalent interactions based on π–π stacking interactions between SWCNHs and aromatic organic units and/or synergistic interactions.3.1. Covalent functionalization
The covalent functionalization of SWCNHs was carried out through 1,3-dipolar cycloaddition of azomethine yields (Fig. 2).42,43 Based on this method, a large number of pyrrolidine units fused to C–C bonds at the side-walls of SWCNHs were introduced.44 The pyrene concentration in the functionalized SWCNHs was measured and found that there was one pyrene moiety for approximately every 45 carbon atoms in SWCNHs. In one study, the 1,3-dipolar cycloaddition reaction of azomethine ylides onto SWCNHs was investigated by theoretical calculations.45 These calculations showed that greater binding energy and reactivity of SWCNHs was associated with carbon atoms located near the conical tip of SWCNHs. A second approach for the covalent functionalization of SWCNHs based on direct nucleophilic addition or via amidation of carboxyl groups with a monoprotected diamine derivative was developed.46 The polymer-SWCNH conjugates were also synthesized with the “grafting to” approach.47,48 The solubility of the conjugates depended on the polymer's solubility. The functionalization of SWCNHs was also achieved via the opening of their conical and highly strained end including the attachment of various organic amines, alcohols, and thiols,49 functionalization with porphyrin (H2P)50,51 and functionalization with pyrene chromophores.52 Aryl diazonium functionalization of SWCNHs was achieved using substituted anilines that loaded on the SWCNH side-walls with desirable functional groups.53 The functionalized SWCNHs were soluble in organic solvents as well as water. The SWCNHs were also covalently functionalized with a positively charged imidazolium based moiety bearing bromide as counter-anion.54 Moreover, the bromide anion of the hybrid material can be exchanged by [PF6−], [ClO4−] and [BF4−] anions, resulting in switching of the solubility of hybrid material from aqueous to organic solvents. In another study, a solvent free microwave-assisted functionalization of SWCNHs was reported following the Bingel reaction conditions.55 The functionalized SWCNHs bore malonate units along their skeleton. Recently, a fast and facile protocol for the covalent functionalization of SWCNHs was also described.56 The approach was realized by using in situ bulk free radical polymerization of methacrylic acid and the functionalized SWCNHs were water soluble.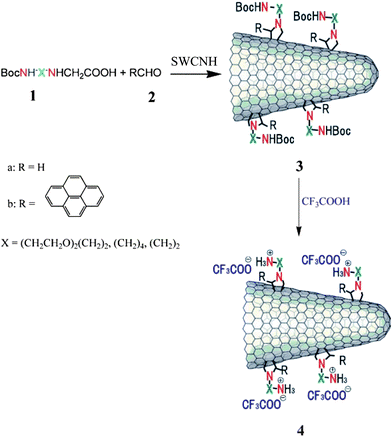 | ||
| Fig. 2 1, 3-dipolar cycloaddition of azomethine ylides to SWCNHs (from ref. 42). | ||
3.2. Noncovalent functionalization
Supramolecular approaches utilizing noncovalent π–π stacking interactions between the sidewalls of SWCNHs with aromatic organic materials and/or synergistic electrostatic interactions have been developed in order to avoid the perturbation of the continuous π-electronic network of SWCNHs. Thus, a pattered assembly of a SWCNH monolayer on oxide surface was formed by using a bifunctional molecule bearing a pyrene moiety (π–π stacking interactions with SWCNHs) on one end and silane (covalent bonding with hydroxyl groups of an oxide surface) on the other.57 The same methodology was employed for the synthesis of a SWCNH/nanoparticle binary nanomaterial using a pyrene-succinimidyl ester bifunctional molecule.58 In another work, tetracationic water-soluble porphyrin (H2P4+) was immobilized onto the skeleton of SWCNHs by π–π stacking interactions without disrupting their π-electronic network.59 Similarly, an anionic porphyrin (H2P2−) was integrated with SWCNHs, mediated by positively pyrene (pyr+) units (Fig. 3).60 In addition, association of negatively charged tetrathiafulvalene carboxylate (TTF−) units with positively charged pyrene (pyr+) noncovalently associated on the surface of SWCNHs to form the nanosized SWCNH-pyr+-TTF− architecture (Fig. 4).61 Theoretical calculations including density functional theory (DFT) and time dependent density functional theory (TDDFT) calculations were also carried out on SWCNH nanohybrids with these photo- or electro-active substituents, H2P, pyrene, and TTF.62 It was found that the relevant excitations in the individual substituents retained their characteristics in the nanohybrid systems. Recently, a water soluble nanohybrid was synthesized through the noncovalent polymer functionalization of SWCNHs using a block polyelectrolyte.63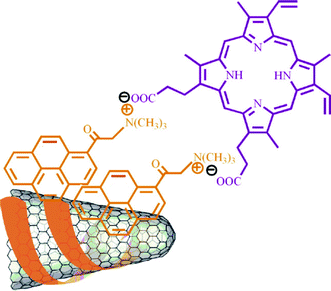 | ||
| Fig. 3 SWCNHs-pyr+- H2P2− nanoensemble (from ref. 60). | ||
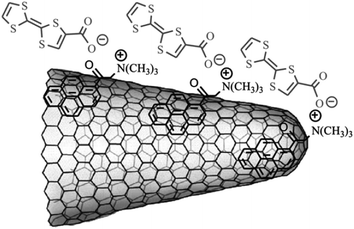 | ||
| Fig. 4 Partial structure of SWCNH-pyr+-TTF−-nanoarchitecture (from ref. 61). | ||
4. Application
4.1. Gas storage media
SWCNHs with high purity and specific morphology are novel candidates for gas storage, because the gas molecules can be stored in both the cylindrical inner nanospace and interstitial channels. In particular, compressed SWCNHs exhibited high methane storage capacity, reaching 160 cm3 cm−3 of nanocarbon at 3.5 MPa and 303 K, exceeding the US Department of Energy (DOE) target of 150 cm3 cm−3 of adsorbent that has been set for practical use of storage media.64 At low pressures, the compressed SWCNHs exhibited an uptake of methane that was comparable with the SWCNT square array. While at pressures > 4 MPa, the uptake of methane at SWCNH approached that of a SWCNT triangular array. Later, it was found that the addition of lanthanide nitrate in the compressed SWCNHs improved the methane capacity through charge transfer effects.65 In another report, the amount of methane adsorbed inside SWCNHs was shown to be exceptionally large, and corresponded to the large methane adsorption capacity of compressed pellets.66The physical adsorption of hydrogen on the SWCNHs was also investigated by the gravimetric experiments. The results indicated that there were two physical adsorption sites of interstitial and internal spaces on SWCNH assemblies (Fig. 5) and lowering the temperature increased the adsorbed hydrogen density.67 Moreover, hydrogen adsorption isotherms in SWCNHs measured at 20 K indicated that the average density of confined hydrogen inside SWCNHs was higher than that of liquid hydrogen in the bulk and nearly approached the density of solid hydrogen at the triple point.68 In another study, the adsorption of both hydrogen and deuterium had been measured at 77 K and compared with the simulations for these hydrogen isotopes’ adsorption on the SWCNH model.69 According to the simulations, the hydrogen isotopes are preferentially adsorbed in the cone part of SWCNH with a strong potential field, and quantum effects caused the density of adsorbed H2 inside the SWCNH to be 8–26% smaller than that of D2. The difference between the amounts of H2 and D2 adsorbed became larger as the pressure decreases.
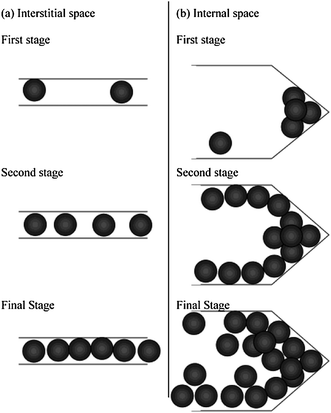 | ||
| Fig. 5 Hydrogen adsorption processes in (a) interstitial and (b) internal spaces of SWCNH assembly (from ref. 67). | ||
4.2. Adsorption
Noble gases, like xenon, were adsorbed onto SWCNHs. The adsorption isotherms of xenon indicated that the ability to adsorb such molecules of SWCNHs was stronger than that of planar graphite.70 The experimental results indicated that the interstitial channels between SWCNHs allowed the Xe gas atom to penetrate while the interstitial channels in SWCNT bundles were too small to allow for Xe penetration.71 Both the specific surface area and the highest-binding-energy values determined for SWCNHs indicated that this novel material had very highly desirable characteristics as an adsorbent.Benzene was also adsorbed onto both SWCNHs and oxidized SWCNHs (oxSWCNHs).72 The SWCNHs adsorbed benzene only on the outside walls, while the oxSWCNHs adsorbed benzene also on the inside walls since the holes through the walls, enabling benzene to reach the inside tubules. In another study, adsorption of benzene and xylene appeared to occur in three sites of SWCNHs as thermogravimetric analysis (TGA) had shown.73 The energetically deepest sites were inside the walls at the tips and convex parts whose large curvatures led to greater contact areas with adsorbed molecules. The second deepest sites were on the other parts of the wall surfaces away from the tips and convex parts, and the energetically shallowest sites were in the central region of the hollow spaces inside the SWCNHs. Liquid ethanol can also be adsorbed onto SWCNHs effectively.25 The amount of liquid ethanol adsorbed by SWCNHs was about 3.5 times larger than that of a super high surface area carbon.
Water adsorption onto SWCNHs has also been investigated. The experimental evidence indicated the formation of clusters in the internal and interstitial nanospaces of SWCNHs, in contrast to the monolayer formation observed for nonpolar molecules.31 In another study adsorption on open SWCNH treated by oxidation at 823 K included adsorption in interstitial nanospaces and intratube nanospaces.74 The amount of adsorption in the interstitial nanospaces was 20% of that in the intratube nanospaces.
Adsorption of tetracyano-p-quinodimethane (TCNQ) onto SWCNHs was compared with that of hSWCNHs.75 The results indicated that the hSWCNH surface had more adsorption sites for TCNQ than that of SWCNH, which was most likely because the hSWCNHs had holes and defects generated by the high-temperature treatments in the oxygen, though some holes were closed and defects were partially annealed by the 1200 °C heat treatment in hydrogen gases (Fig. 6).
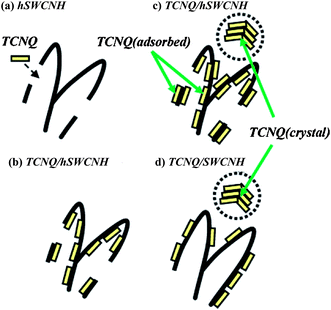 | ||
| Fig. 6 Schematic illustration of adsorption process of TCNQ onto hSWCNHs and SWCNHs. TCNQ molecules (yellow platelets) first enter inside hSWCNHs through holes (a) and then adsorb inside hSWCNHs (b), followed by adsorption onto the outside surface of hSWCNHs and TCNQ (crystal) formation away from hSWCNHs (c). TCNQ (adsorbed) and TCNQ (crystal) also formed for SWCNHs (d) (from ref. 75). | ||
Moreover, the adsorption properties of an aqueous electrolyte (RbBr2) indicated that the hydration number of rubidium ions and the Rb–H2O distance were smaller when adsorbed in SWCNHs than in bulk solution.74,76
4. 3. Catalyst support
Nanoparticle–carbon nanotube (CNT) hybrid materials have been used as catalysts.77 The application requires uniform nanoparticles attached to the nanotube surface. However, many techniques for deposition of metal on CNTs have led to the formation of nanowires78 or coatings77 due to the weak interactions between the graphene walls of CNTs and most metals. SWCNHs loaded on a catalyst support are another hot topic. The unique structure of SWCNHs would improve the durability of catalysts.79 Deposition of Pd small nanoclusters on oxSWCNHs has been reported. The synthesis involved reduction of Pd ions in the presence of a polymer and then adsorption of the resulted metal clusters into both the internal and interstitial pores of oxSWCNHs.80 Pd nanoparticles were deposited on oxSWCNHs from an aqueous solution of PdCl2 by in situ reduction of [PdCl4]2− with ethanol in the presence of poly(vinylpyrrolidone) (PVP) as a stabilizing polymer. The applied strategy provided small Pd clusters with a diameter of ∼2.3 nm, robustly attached to the nanotubular carbon. Later, catalytic activities of palladium nanoparticles-tailored SWCNHs (Pd-SWCNHs) or oxSWCNHs (Pd-oxSWCNHs) for H2–O2 gas phase reactions and liquid phase reactions such as Miyasura–Suzuki coupling were reported.81 The catalytic activities were examined in terms of turnover number (TON), which is the total mole amount of products per that of catalytic elements and showed an essential catalytic activity of catalysts. The turnover frequency (TOF) was obtained by normalizing TON with time and indicated an essential catalytic reaction rate for catalytic elements. The TON and TOF of Pd-oxSWCNHs reached almost 68![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 and 34000 mol mol−1 h−1, respectively. To the best of our knowledge, this was the highest TOF obtained in a Pd-catalyzed coupling process with heterogeneous catalysts so far. The nanoporosities and catalytic activities of Pd-SWCNHs and Pd-oxSWCNHs were further studied in another report.82 For a water formation reaction at 273 K, the catalytic activity of Pd-SWCNHs was higher than that of Pd-oxSWCNHs, while for a water formation at 298 K, the catalytic activity of Pd-oxSWCNHs became higher than that of Pd-SWCNHs.
000 and 34000 mol mol−1 h−1, respectively. To the best of our knowledge, this was the highest TOF obtained in a Pd-catalyzed coupling process with heterogeneous catalysts so far. The nanoporosities and catalytic activities of Pd-SWCNHs and Pd-oxSWCNHs were further studied in another report.82 For a water formation reaction at 273 K, the catalytic activity of Pd-SWCNHs was higher than that of Pd-oxSWCNHs, while for a water formation at 298 K, the catalytic activity of Pd-oxSWCNHs became higher than that of Pd-SWCNHs.
SWCNHs were also employed as a platinum (Pt) catalyst support in electrodes of polymer electrolyte fuel cells (PEFCs). The Pt catalyst was supported on the SWCNHs by using a colloidal method.83 The size of Pt particles was about 2 nm and less than half of that supported on the conventional carbon black. The current density for the SWCNH cell was larger than that for the carbon black cell, which demonstrated that the suitability of the SWCNHs as an electrode material for PEFCs. Later, one-step synthesis of the Pt-loaded SWCNHs by ‘arc in liquid nitrogen’ method was demonstrated using Pt-contained graphite anode.84 The size distribution of Pt particles could be controlled by adjusting the concentration of Pt in the graphite anode. It was also verified that the as-grown Pt-loaded products produced by this method could be useful for the power generation by PEFCs. In another report, Pt nanoparticles which could act as a catalyst in a fuel cell were incorporated into the nanospaces of SWCNHs.85 The obtained Pt particle sizes were about 1.5–2.5 nm in the interior of the nanohorn. They were almost spherical, and were located near the defects. The Pt particles prepared by this method had an upper limit to their sizes and quantities. Therefore, a simple method for preparing size-controlled fine particles was presented. The papers mentioned above all need a longer time for the preparation of Pt nanoparticles. The deposition of Pt nanoparticles onto oxSWCNHs was achieved within several minutes by microwave irradiation of an aqueous mixture of ethylene glycol, Pt salt, and pristine SWCNHs.86 Recently, high-power direct methanol fuel cells (DMFCs) were made by using SWCNHs to support the Pt catalyst.87 The mean diameter of the Pt particles supported on the treated-SWCNHs (oxidized with H2O2) was 2.9 nm, which was roughly 2/3 that of the particles on untreated-SWCNHs, for 60 wt% Pt content (Fig. 7). The highest power density of 76 mW cm−2 at 0.4 V was obtained for a passive-type DMFC operated at 40 °C by these improvements. Moreover, production of hydrogen using methane and water at low temperature, mediated by Eu–Pt supported on SWCNHs had also been reported.88 In addition, palladium and platinum nanoparticles were decorated onto the skeleton of SWCNHs by the self-regulated reduction of palladium acetate and chloroplatinic acid, respectively.89 X-Ray spectroscopy indicated that the size of metal nanoparticles was in the range of 2–5 nm. The hybrid materials had catalytic activity toward the formation of carbon–carbon bonds via the Heck, Suzuki and Stille reactions.
 | ||
| Fig. 7 TEM images of the supported catalyst: (a) 20 wt % Pt on as-SWCNHs, (b) 20 wt % Pt on treated-SWCNHs, (c) 60 wt % Pt on as-SWCNHs, and (d) 60 wt % Pt on treated-SWCNHs (from ref. 87). | ||
4.4. Use as carriers in drug delivery system
The extensive surface areas and multitudes of horn interstices of SWCNHs enable large amounts of guest molecules to be adsorbed onto them. The adsorption of selected chemical compounds represents promising carriers in drug delivery systems (DDSs). Compared with CNCs, SWCNHs can only adsorb some smaller molecules because of their smaller pore size. However, SWCNHs can avoid the cytotoxicity resulting from metal catalysts used during the production of CNCs. In addition, the SWCNTs assemble to form bundles with micrometre-order length while thousands of SWCNHs assemble to form a spherical aggregate with diameters of ∼ 100 nm. Materials with diameter of ∼100 nm have enhanced permeability and retention under the passive tumor-targeting conditions and tend to accumulate at tumor tissues. The antiinflammatory glucocorticoid dexamethasone (DEX) has been adsorbed onto as-grown SWCNHs, oxSWCNHs and hSWCNHs.90 Adsorption analyses indicated that the latter two had higher affinity for DEX than the former. A large amount of DEX was adsorbed onto oxSWCNHs, which was approximately 6 times larger than that obtained for as-grown SWCNHs. DEX-oxSWCNHs released DEX slowly both in buffer solution and cell cultural medium. The well known anticancer agent cisplatin (CDDP) can also be incorporated into and released from oxSWCNHs (Fig. 8).91 It was found that release rate of CDDP from oxSWCNHs in PBS and a culture medium was slower, and the released CDDP was effective in terminating the growth of human lung-cancer cells. This suggested that oxSWCNHs were potentially useful as a drug carrier. The quantity of CDDP released from CDDP@oxSWCNHs was also compared with that from CDDP@hSWCNHs.92 The quantity of CDDP released from CDDP@oxSWCNHs in PBS was only 15%, while that from CDDP@hSWCNHs was 70%. The slow release rate was attributed to the transformation of –COOH and –OH at the hole edges of oxSWCNHs into –COONa and –ONa groups which hindered CDDP passing through the holes from inside hSWCNHs. Furthermore, the hole-opening condition of SWCNHs were optimized in terms of the least CDDP quantity deposited outside of oxSWCNHs.93 The optimum target temperature was 500 °C with an increase rate of 1 °C min−1. The obtained CDDP@oxSWCNHs released about 80% of CDDP, slowly over 50 h, which is favorable for CDDP carriers in vivo. Recently oxSWCNHs themselves were reported to have an in vivo antineoplastic effect and thus CDDP@oxSWCNHs had a higher antitumor efficiency than the intact CDDP both in vitro and in vivo.94 In another work, various potent erythropoietin (EPO) carriers were tested, revealing high adsorption on SWCNTs, while that of SWCNHs was low.95 This showed that not only the adsorption space, which was thought to be of importance to act as an effective drug delivery tool, but also the structure of the adsorbent was of importance.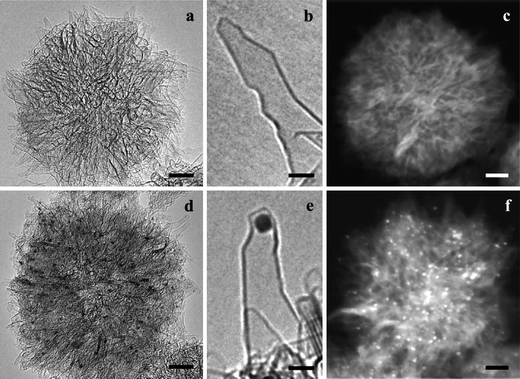 | ||
| Fig. 8 (a, b) HRTEM images of oxSWCNHs (scale bars of 10 and 2 nm, respectively). (c) Z-Contrast image of oxSWCNHs aggregate (10 nm). (d, e) HRTEM images of CDDP@oxSWCNHs (10 and 2 nm) in which black spots are cisplatin clusters. (f) Z-Contrast image of CDDP@oxSWCNHs in which bright spots are cisplatin clusters (10 nm) (from ref. 91). | ||
To avoid potential health hazards caused by occupational exposure to SWCNHs in DDSs, the toxicity of SWCNHs should be investigated from various aspects. The toxicities of as-grown SWCNHs were investigated both in vivo and in vitro. The test results showed that as-grown SWCNHs had low toxicities.96 The pulmonary toxicity of SWCNHs was also investigated.97 The results suggested that SWCNHs were a relatively innocuous nanomaterial when delivered to mice in vivo using aspiration as a delivery mechanism. Moreover, water soluble SWCNHs prepared by amination was taken up by mammalian cells and did not show significant cytotoxicity.98
4.5. Application in magnetic resonance analysis
Further research on SWCNH applications in the living body and the assessment of the associated toxicological hazards will require non-anatomical in vivo observations of the motion and accumulation of SWCNHs. Magnetic resonance imaging (MRI) could be used for these observations if an MRI contrast agent was attached to SWCNHs. The potential of Gd2O3-deposited oxSWCNHs as a positive MRI contrast agent was demonstrated.99 Ultrafine Gd2O3 nanoparticles on oxSWCNHs worked as a positive MR contrast agent. A sharp, bright image was observed even at a low concentration of Gd2O3-oxSWCNHs. In addition to the Gd3+ complexes, superparamagnetic magnetite (i.e. magnetite Fe3O4) nanoparticles with diameters less than about 25 nm are another effective contrast agent for MRI. Superparamagnetic magnetite must be strongly attached to CNTs to keep it from breaking off when the CNTs are moving through the living body. Many chemical modifications of the edges and outside walls of CNTs and the physical adsorption of various materials have been investigated.100–102 But no method has been found for the strong attachment of magnetite nanoparticles onto CNTs. Fe(OAC)2 was first deposited on oxSWCNHs and then heated to 400 °C. This yielded superparamagnetic magnetite (Fe3O4, 6 nm in diameter) strongly attached to oxSWCNHs (MAGoxSWCNHs), making possible MRI of MAGoxSWCNHs in living mice.103 After these low toxicity MAGoxSWCNHs were injected intravenously, MRI showed MAGoxSWCNHs accumulation on spleen and kidneys, a fact verified by histopathological examinations.4.6. Electrochemical applications of SWCNHs
SWCNHs were used as electrode materials for the electrochemical applications. In contrast to CNTs, SWCNHs are metal-free. This can avoid controversial results due to the impurities in CNTs. First, SWCNH paste electrode was used for amperometric determination of concentrated hydrogen peroxide (H2O2), and was compared with other carbon electrodes.104 The calibration plots were linear from 0.5 to 100 mM at an activated SWCNH paste electrode and edge plane graphite (EPG) electrode. In another work, a new approach based on the direct growth of SWCNHs on carbon microfiber substrates had been developed for the straightforward fabrication of free-standing (binderless) electrodes for electrochemical applications.105 Preliminary studies demonstrated their possible application in lithium-ion batteries. Furthermore, by pulsed laser deposition (PLD)-decorating the SWCNHs-coated electrodes with Pt nanoparticles, they were also shown to act as highly effective electrodes for either O2 reduction or methanol oxidation, two electrochemical reactions that are central to fuel cell technology. Recently, SWCNHs were used as solid-phase extraction adsorbent.106 They can extract 4-nitrophenol rapidly and efficiently. The solid-phase extraction of 4-nitrophenol was monitored by an electrochemical method. The combination of solid-phase extraction to SWCNH-modified glassy carbon electrode (GCE), medium exchange, and linear sweep voltammograms allowed fast, sensitive, and selective determination of 4-nitrophenol. Compared with bare GCE and MWCNT-modified GCE, the sensitivity for the determination of 4-nitrophenol at SWCNH-modified GCE increased by 51.6 times and 5.55 times, respectively. Also, a potential application of SWCNHs to electrodes for supercapacitors has been recently reported.107 The SWCNHs can be considered as a promising candidate for electrode material of supercapacitors, owing to their porous structure and novel physical properties. Recently, SWCNHs were applied for the modification of GCE. This modified electrode exhibited high electro-catalytic activities towards the oxidation of uric acid (UA), dopamine (DA), and ascorbic acid (AA) by significantly decreasing their oxidation overpotentials and enhancing the peak currents (Fig. 9).108 Linear sweep voltammetry (LSV) was used for the simultaneous determination of UA, DA, and AA in their ternary mixture. The proposed method improved sensitivity for the determination of UA by more than one order of magnitude. The present method was applied to the determination of UA, DA, and AA in urine samples by using standard adding method and the results were satisfactory. A sandwich nanohybrid of SWCNHs-TiO2-porphrin was prepared via the dentate binding of the carboxylate groups onto TiO2 nanoparticles as bridges for highly sensitive amperometric biosensing of chloramphenicol (CAP).109 The SWCNHs-TiO2-porphrin modified electrode showed excellent electrocatalytic activity toward reduction of CAP due to the synergic effect among SWCNHs, TiO2 and porphrin. The designed biosensor exhibited good analytical performance in the detection of CAP such as rapid response, wide linear range, low detection limit, good fabrication reproducibility, and acceptable accuracy.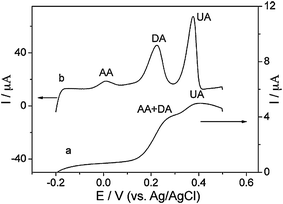 | ||
| Fig. 9 LSVs at the GCE (a) and SWCNH-modified GCE (b) in 0.1 M pH 7.0 PBS containing 100 μM UA, DA, and AA. Scan rate: 100 mV s−1 (from ref. 108). | ||
4.7. Biosensing application of SWCNHs
CNTs have been successfully used as “molecular wires” to realize direct electron transfer between redox enzymes and electrode surfaces, which can fabricate new kinds of mediator-free biosensors.110–113 However, most CNT synthesis methods inevitably introduce some impurities. Tedious purification is necessary, and metal catalyst residues still existed in CNTs after purification. The impurities resulted in conflicting reports.114–116 SWCNHs are essentially metal-free and can be used directly for biosensing studies. SWCNHs were first utilized to fabricate a glucose biosensor.117 The biosensor was constructed by encapsulating glucose oxidase in the Nafion-SWCNHs composite. The glucose biosensor based on the Nafion-SWCNHs nanocomposite possessed high sensitivity, low detection limit, and good selectivity. The high sensitivity of the biosensor implied that SWCNHs can provide superior conductivity for electron transfer, and the Nafion-SWCNHs nanocomposite film provided a biocompatible microenvironment to maintain enzymatic activity. Later SWCNHs were used as a novel and biocompatible matrix for fabricating H2O2 biosensors. The direct immobilization of acid-stable and thermostable soybean peroxidase (SBP) on a SWCNH modified electrode surface can realize the direct electrochemistry of the enzyme. Based on direct electrochemistry of SBP, a mediator-free H2O2 biosensor was constructed.15 Heme protein myoglobin (Mb) was adsorbed onto the surface of noncovalently functionalized SWCNHs using poly(styrene sulfonate) (PSS) to fabricate the electrochemical sensor. The proposed biosensor exhibited good electrocatalysis to the reduction of H2O2 (Fig. 10). The noncovalent functionalization of SWCNHs will facilitate applications of SWCNHs in fields of biosensor and electrochemistry.118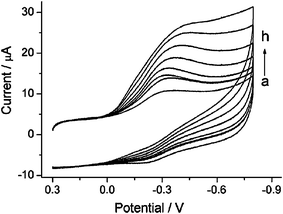 | ||
| Fig. 10 Cyclic voltammograms of the Mb/PSS-SWCNH/GCE in the presence of 0 μM (a), 3 μM (b), 5 μM (c), 10 μM (d), 100 μM (e), 150μM (f), 300 μM (g), and 350 μM (h) H2O2 in 0.1 M (pH 5.5) acetate buffer solution. Scan rate: 0.2 V s−1 (from ref. 118). | ||
4.8. Applications of SWCNHs for photovoltaics and photoelectrochemical cells
Carbon-based nanostructured materials, such as fullerenes and CNTs, constitute a unique platform for energy conversion systems, for example, to photovoltaics and optoelectronics. In comparison with the metal/or amorphous carbon contaminated SWCNTs, the metal-free SWCNHs with high purity can be utilized toward new photoactive hybrid materials. For example, copper(II)-2,2′:6′,2′′-terpyridine (CuIItpy) was coordinated with the –COOH at conical terminals of SWCNHs, resulting a novel SWCNHs-COO–CuIItpy metallo-nanocomplex (Fig. 11).119 Steady-state and time-resolved fluorescence emission measurements demonstrated that electron transfer occurred from the singlet excited state of CuIItpy to SWCNHs. Photoexcitation of CuIItpy resulted in the one-electron reduction of nanohorns with a simultaneous one-electron oxidation of the CuIItpy unit (SWCNHs·−–COO–(CuIItpy)·+) as revealed by transient absorption measurements. The formation of the photoinduced charge-separated states within the SWCNHs·−–COO–(CuIItpy)·+ was formed with the addition of an electron mediator, such as hexyl-viologen dication (HV2+) and an electron–hole shifter in polar solvents. Recently, SWCNHs containing amino (SWCNH-NH2) were covalently and non-covalently functionalized with tetrabenzoic acid porphyrin (H2P(CO2H)4) to form dispersible nanohybrids in aqueous solutions.120 The H2P(CO2H)4 fluorescence intensity quenching by SWCNH-NH2 suggested charge separation via the excited singlet state of the H2P moieties, generating radical cations localized in the H2P moieties and electrons trapped in SWCNHs. With the addition of methylviologen dication (MV2+) and a hole-shifting reagent, electron pooling was observed by the light-illumination of the H2P(CO2H)4 moieties in SWCNH-NH2 nanohybrids, indicating that the electron generated by the charge-separation migrates to MV2+ in aqueous solution.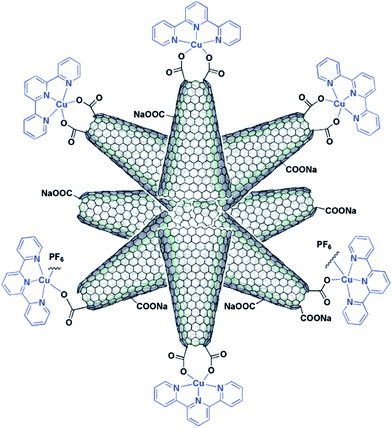 | ||
| Fig. 11 Illustrative structure of the SWCNHs-COO- CuIItpy metallo-nanocomplex (from ref. 119). | ||
4.9. Application of modified SWCNHs to the photodynamic therapy
Carbonaceous tubules absorb light in the near-infrared region, and can cause cell death by a localized photothermal or photohyperthermia (PHT) effect. As a result, SWCNHs can also be applied to photodynamic therapy (PDT). A double PDT and PHT cancer phototherapy system using oxSWCNHs loaded on zinc phthalocyanine (ZnPc) and protein bovine serum albumin (BSA) was fabricated (Fig. 12).121 In this system, ZnPc was the PDT agent, oxSWCNHs was the PHT agent, and BSA enhanced biocompatibility. The phototherapy effect was confirmed both in vitro and in vivo. When ZnPc-oxSWCNHs-BSA was injected into tumors that were subcutaneously transplanted into mice, subsequent laser irradiation caused the tumors to disappear. In contrast, the tumors still grew when only ZnPc was injected. Thus, SWCNHs may be a valuable new tool for use in cancer therapy. Later, the conditions for electron- and energy-transfer mechanisms useful for the photosynthetic model and photodynamic therapy were investigated through the photophysical studies of ZnPc-oxSWCNHs-BSA nanoensembles.122 Whole photochemical processes are illustrated in Scheme 1.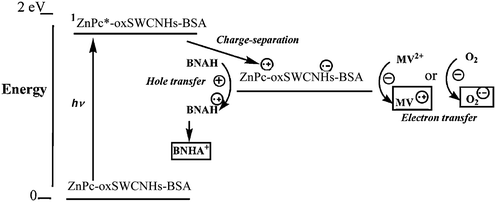 | ||
| Scheme 1 Photoinduced processes for ZnPc-oxSWCNHs-BSA in the presence of MV2+ and 1-benzyl-1,4-dihydro-nicotinamide (BANH) in aqueous solution (from ref. 122). | ||
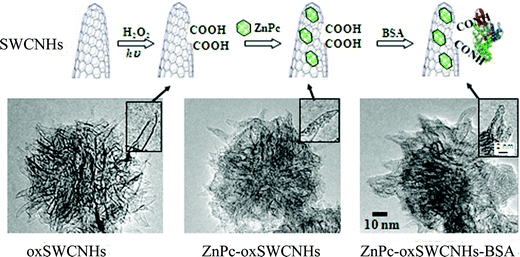 | ||
| Fig. 12 Preparation of ZnPc-oxSWCNHs-BSA. (A) Diagram showing steps in the production of ZnPc-oxSWCNHs-BSA. The SWCNHs were visualized by TEM at each stage of production. (B) oxSWCNHs. (C) ZnPc-oxSWCNHs. (D) ZnPc-oxSWCNHs-BSA. Insets: Magnified images (from ref. 121). | ||
4.10. Other applications
Dielectrophoresis (DEP) has been applied towards the realization of SWCNHs as gas sensor devices.123 It was found that the conductance of the DEP-fabricated SWCNH sensor increased or decreased upon exposure to ppm-levels of NO2 or NH3, respectively. The intrinsic NO2 sensitivity of the SWCNHs was lower than that of SWCNTs but comparable with the intrinsic sensitivity of MWCNTs. In another report, SWCNHs produced by a gas-injected arc-in-water method was also fabricated as a gas sensor.124 Its sensing properties were examined for the detection of NH3 and O3 at room temperature. It was confirmed that the electrical resistance of the SWCNH film increased with adsorption of NH3, whereas the adsorption of O3 induced a decrease of the resistance. The gas sensor using SWCNHs showed higher sensitivity than the sensor using SWCNTs of the same electric resistance at the present condition with relatively a large inter-electrode gap. In addition, a sensor based on SWCNH film was used to detect ozone water.125 The magnetic properties of SWCNH doped MgB2 had been studied towards new superconductive materials.126 The SWCNH is also expected to display good tribological properties due to its spherical shape with a sub-micro diameter. So, the tribological properties of SWCNH-doped grease127 and polyimide composites128 were investigated.5. Conclusions and outlook
Due to their unique structures and properties, SWCNHs have been successfully used for various applications. Despite the continuous efforts and researches on SWCNHs, the field is still in its infancy. Great opportunities for further applications are foreseen. The size of the holes on the oxSWCNHs can be controlled by adjusting the temperature. The opened holes can be closed controllably. So, the oxSWCNHs with different holes can be used for selective adsorption or storage. The next step will be to develop an alternative storage process, “taking in” and “taking out” the captured substances. The experimental results indicated that oxSWCNHs can be useful as a drug carrier. The low toxicities and in vivo anticancer effect of oxSWCNHs can be potentially used for clinical applications, particularly by combining with molecular recognition techniques. Due to their high surface area and unique structure, SWCNHs were successfully used as support for synthesis of size-controlled fine Pt particles with good stability. The technique can be applied to a variety of other metals such as Au and Pd. The metal nanoparticles–SWCNH hybrid may find applications in catalysis, surface enhanced Raman spectroscopy, electrochemistry, and so on. Up to now, few papers have been reported about the electrochemical, electrochemiluminescent, and biosensing applications of SWCNHs. SWCNHs are theoretically excellent electrode materials due to their edge plane defect sites and high purity. Therefore, new SWCNH-based electrochemical biosensors should be fabricated in the near future for the determination of proteins, DNA or small molecules. Moreover, SWCNHs have good adsorbent ability due to their high surface area and unique structures. So, SWCNHs can be used in various extraction applications such as solid-phase microextraction (SPME) which can be combined with the liquid chromatogram (LC), gas chromatogram (GC), microchip capillary electrophoresis, and micro total analysis systems. SWCNHs present significant opportunities to basic science and nanotechnology, and also pose significant challenges for future work in these fields. New and innovative SWCNH-based hybrid materials shall be prepared and studied for their further applications. The versatility and commercialization of SWCNHs will make this an even more active research area.Acknowledgements
The authors are very grateful to Professor S. Iijima (Solution Oriented Research for Science and Technology in Japan Science and Technology Agency) for the generous offer of SWCNHs. This work is kindly supported by the National Natural Science Foundation of China (No. 20875086 & 20505016), the Ministry of Science and technology of the People's Republic of China (No.2006BAE03B08), and the Department of Sciences & Technology of Jilin Province (20070108 and 20082104).References
- S. Iijima, M. Yudasaka, R. Yamada, S. Bandow, K. Suenaga, F. Kokai and K. Takahashi, Chem. Phys. Lett., 1999, 309, 165–170 CrossRef CAS.
- S. Iijima and T. Ichihashi, Nature, 1993, 363, 603–605 CrossRef CAS.
- M. Yudasaka, S. Iijima and V. H. Crespi, Top. Appl. Phys., 2008, 111, 605–629 CAS.
- M. Yudasaka, S. Iijima, Carbon Nanohorns in Carbon Nanotubes and Related Structures, ed. D. M. Guldi and N. Martin, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2010, Ch. 13, pp. 385–404 Search PubMed.
- G. Pagona, G. Mountrichas, G. Rotas, N. Karousis, S. Pispas and N. Tagmatarchis, Int. J. Nanotechnol., 2009, 6, 176–195 Search PubMed.
- T. Azami, D. Kasuya, R. Yuge, M. Yudasaka, S. Iijima, T. Yoshitake and Y. Kubo, J. Phys. Chem. C, 2008, 112, 1330–1334 CrossRef CAS.
- Y. W. Ma, Z. Hu, K. F. Huo, Y. N. Lu, Y. M. Hu, Y. Liu, J. H. Hu and Y. Chen, Carbon, 2005, 43, 1667–1672 CrossRef CAS.
- D. Kasuya, M. Yudasaka, K. Takahashi, F. Kokai and S. Iijima, J. Phys. Chem. B, 2002, 106, 4947–4751 CrossRef CAS.
- T. Azami, D. Kasuya, T. Yoshitake, Y. Kubo, M. Yudasaka, T. Ichihashi and S. Iijima, Carbon, 2007, 45, 1364–1369 CrossRef.
- M. Ikeda, H. Takikawa, T. Tahara, Y. Fujimura, M. Kato, K. Tanaka, S. Itoh and T. Sakakibara, Jpn. J. Appl. Phys., Part 2, 41(7B), L852–L854 Search PubMed.
- H. Takikawa, M. Ikeda, K. Hirahara, Y. Hibi, Y. Tao, P. A. Ruiz Jr., T. Sakakibara, S. Itoch and S. Iijima, Phys. B, 2002, 323, 277–279 CrossRef CAS.
- T. Yamaguchi, S. Bandow and S. Iijima, Chem. Phys. Lett., 2004, 389, 181–185 CrossRef CAS.
- N. Sano, J. Nakano and T. Kanki, Carbon, 2004, 42, 686–688 CrossRef CAS.
- N. Sano, J. Phys. D: Appl. Phys., 2004, 37, L17–L20 CrossRef CAS.
- L. H. Shi, X. Q. Liu, W. X. Niu, H. J. Li, S. Han, J. A. Chen and G. B. Xu, Biosens. Bioelectron., 2009, 24, 1159–1163 CrossRef CAS.
- S. Bandow, A. M. Rao, G. U. Sumanasekera, P. C. Eklund, F. Kokai, K. Takahashi, M. Yudasaka and S. Iijima, Appl. Phys. A: Mater. Sci. Process., 2000, 71, 561–564 CrossRef CAS.
- S. Berber, Y. K. Kown and D. Tománek, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 62, R2291–R2294 CrossRef CAS.
- D. V. Kolesnikov and V. A. Osipov, JETP Lett., 2004, 79, 532–536 CrossRef CAS.
- K. Urita, S. Seki, S. Utsumi, D. Noguchi, H. Kanoh, H. Tanaka, Y. Hattori, Y. Ochiai, N. Aoki, M. Yudasaka, S. Iijima and K. Kaneko, Nano Lett., 2006, 6, 1325–1328 CrossRef CAS.
- A. Zahab, L. Spina, P. Poncharal and C. Marliere, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 62, 10000–10003 CrossRef CAS.
- S. Bandow, F. Kokai, K. Takahashi, M. Yudasaka and S. Iijima, Appl. Phys. A: Mater. Sci. Process., 2001, 73, 281–285 CrossRef CAS.
- H. Imai, P. K. Babu, E. Oldfield, A. Wieckowski, D. Kasuya, T. Azami, Y. Shimakawa, M. Yudasaka, Y. Kubo and S. Iijima, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 125405 CrossRef.
- J. M. Bonard, R. Gaal, S. Garaj, L. Thien-Nga, L. Forro, K. Takahashi, F. Kokai, M. Yudasaka and S. Iijima, J. Appl. Phys., 2002, 91, 10107–10109 CrossRef CAS.
- M. Yudasaka, T. Ichihashi, D. Kasuya, H. Kataura and S. Iijima, Carbon, 2003, 41, 1273–1280 CrossRef CAS.
- J. A. Nisha, M. Yudasaka, S. Bandow, F. Kokai, K. Takahashi and S. Iijima, Chem. Phys. Lett., 2000, 328, 381–386 CrossRef.
- S. Bandow, F. Kokai, K. Takahashi, M. Yudasaka, L. C. Qin and S. Iijima, Chem. Phys. Lett., 2000, 321, 514–519 CrossRef CAS.
- J. N. Wang, L. Zhang, J. J. Niu, F. Yu, Z. M. Sheng, Y. Z. Zhao, H. Chang and C. Pak, Chem. Mater., 2007, 19, 453–459 CrossRef CAS.
- K. Murata, K. Kaneko, F. Kokai, K. Takahashi, M. Yudasaka and S. Iijima, Chem. Phys. Lett., 2000, 331, 14–20 CrossRef CAS.
- K. Murata, K. Kaneko, W. A. Steele, F. Kokai, K. Takahashi, D. Kasuya, K. Hibarahara, M. Yudasaka and S. Iijima, J. Phys. Chem. B, 2001, 105, 10210–10216 CrossRef CAS.
- K. Murata, K. Hirahara, M. Yudasaka, S. Iijima, D. Kasuya and K. Kaneko, J. Phys. Chem. B, 2002, 106, 12668–12669 CrossRef CAS.
- J. Fan, M. Yudasaka, J. Miyawaki, K. Ajima, K. Murata and S. Iijima, J. Phys. Chem. B, 2006, 110, 1587–1591 CrossRef CAS.
- E. Bekyarova, Y. Hanzawa, K. Kaneko, J. Silvestre-Albero, A. Sepulveda-Escribano, F. Rodriguez-Reinoso, D. Kasuya, M. Yudasaka and S. Iijima, Chem. Phys. Lett., 2002, 366, 463–468 CrossRef CAS.
- J. Miyawaki, M. Yudasaka and S. Iijima, J. Phys. Chem. B, 2004, 108, 10732–10735 CrossRef CAS.
- C. M. Yang, D. Kasuya, M. Yudasaka, S. Iijima and K. Kaneko, J. Phys. Chem. B, 2004, 108, 17775–17782 CrossRef CAS.
- C. M. Yang, H. Noguchi, K. Murata, M. Yudasaka, A. Hashimoto, S. Iijima and K. K. aneko, Adv. Mater., 2005, 17, 866–870 CrossRef CAS.
- R. Yuge, T. Ichihashi, J. Miyawaki, T. Yoshitake, S. Iijima and M. Yudasaka, J. Phys. Chem. C, 2009, 113, 2741–2744 CrossRef CAS.
- J. Miyawaki, R. Yuge, T. Kawai, M. Yudasaka and S. Iijima, J. Phys. Chem. C, 2007, 111, 1553–1555 CrossRef CAS.
- J. Fan, R. Yuge, J. Miyawaki, T. Kawai, S. Iijima and M. Yudasaka, J. Phys. Chem. C, 2008, 112, 8600–8603 CrossRef CAS.
- E. Bekyarova, K. Kaneko, D. Kasuya, K. Murata, M. Yudasaka and S. Iijima, Langmuir, 2002, 18, 4138–4141 CrossRef CAS.
- E. Bekyarova, K. Kaneko, D. Kasuya, K. Takahashi, F. Kokai, M.Yudasaka and S. Iijima, Phys. B, 2002, 323, 143–145 CrossRef CAS.
- E. Bekyarova, K. Kaneko, M. Yudasaka, D. Kasuya, S. Iijima, A. Huidobro and F. Rodriguez-Reinoso, J. Phys. Chem. B, 2003, 107, 4479–4484 CrossRef CAS.
- N. Tagmatarchis, A. Maigne, M. Yudasaka and S. Iijima, Small, 2006, 2, 490–494 CrossRef CAS.
- C. Cioffi, S. Campidelli, F. G. Brunetti, M. Meneghetti and M. Prato, Chem. Commun., 2006, 2129–2131 RSC.
- G. Pagona, G. Rotas, I. D. Petsalakis, G. Theodorakopoulos, J. Fan, A. Maigne, M. Yudasaka, S. Iijima and N. Tagmatarchis, J. Nanosci. Nanotechnol., 2007, 7, 3468–3472 CrossRef CAS.
- I. D. Petsalakis, G. Pagona, G. Theodorakopoulos, N. Tagmatarchis, M. Yudasaka and S. Iijima, Chem. Phys. Lett., 2006, 429, 194–198 CrossRef CAS.
- C. Cioffi, S. Campidelli, C. Sooambar, M. Marcaccio, G. Marcolongo, M. Meneghetti, D. Paolucci, F. Paolucci, C. Ehli, G. M. A. Rahman, V. Sgobba, D. Guldi and M. Prato, J. Am. Chem. Soc., 2007, 129, 3938–3945 CrossRef.
- G. Mountrichas, S. Pispas and N. Tagmatarchis, Chem.–Eur. J., 2007, 13, 7595–7599 CrossRef CAS.
- G. Mountrichas, N. Tagmatarchis and S. Pispas, J. Nanosci. Nanotechnol., 2009, 9, 3775–3779 CrossRef CAS.
- G. Pagona, N. Tagmatarchis, J. Fan, M. Yudasaka and S. Iijima, Chem. Mater., 2006, 18, 3918–3920 CrossRef CAS.
- G. Pagona, A. S. D. Sandanayaka, Y. Araki, J. Fan, N. Tagmatarchis, G. Charalambidis, A. G. Coutsolelos, B. Boitrel, M. Yudasaka, S. Iijima and O. Ito, Adv. Funct. Mater., 2007, 17, 1705–1711 CrossRef CAS.
- G. Pagona, A. S. D. Sandanayaka, T. Hasobe, G. Charalambidis, A. G. Coutsolelos, M. Yudasaka, S. Iijima and N. Tagmatarchis, J. Phys. Chem. C, 2008, 112, 15735–15741 CrossRef CAS.
- A. S. D. Sandanayaka, G. Pagona, J. Fan, N. Tagmatarchis, M. Yudasaka, S. Iijima, Y. Araki and O. Ito, J. Mater. Chem., 2007, 17, 2540–2546 RSC.
- G. Pagona, N. Karousis and N. Tagmatarchis, Carbon, 2008, 46, 604–610 CrossRef CAS.
- N. Karousis, T. Ichihashi, S. Chen, H. Shinobara, M. Yudasaka, S. Iijima and N. Tagmatarchis, J. Mater. Chem., 2010, 20, 2959–2964 RSC.
- S. P. Economopoulos, G. Pagona, M. Yudasaka, S. Iijima and N. Tagmatarchis, J. Mater. Chem., 2009, 19, 7326–7331 RSC.
- G. Mountrichas, S. Pispas, T. Ichihasi, M. Yudasaka, S. Iijima and N. Tagmatarchis, Chem.–Eur. J., 2010, 16, 5927–5933 CAS.
- J. Zhu, M. Yudasaka, M. F. Zhang, D. Kasuya and S. Iijima, Nano Lett., 2003, 3, 1239–1243 CrossRef CAS.
- J. Zhu, D. Kase, K. Shiba, D. Kasuya, M. Yudasaka and S. Iijima, Nano Lett., 2003, 3, 1033–1036 CrossRef CAS.
- G. Pagona, A. S. D. Sandanayaka, Y. Araki, J. Fan, N. Tagmatarchis, M. Yudasaka, S. Iijima and O. Ito, J. Phys. Chem. B, 2006, 110, 20729–20732 CrossRef CAS.
- G. Pagona, J. Fan, A. Maigne, M. Yudasaka, S. Iijima and N. Tagmatarchis, Diamond Relat. Mater., 2007, 16, 1150–1153 CrossRef CAS.
- G. Pagona, A. S. D. Sandanayaka, A. Maigne, J. Fan, G. C. Papavassiliou, I. D. Petsalakis, B. R. Steele, M. Yudasaka, S. Iijima, N. Tagmatarchis and O. Ito, Chem.–Eur. J., 2007, 13, 7600–7607 CrossRef CAS.
- I. D. Petsalakis, G. Pagona, N. Tagmatarchis and G. Theodorakopoulos, Chem. Phys. Lett., 2007, 448, 115–120 CrossRef CAS.
- G. Mountrichas, T. Ichihashi, S. Pispas, M. Yudasaka, S. Iijima and N. Tagmatarchis, J. Phys. Chem. C, 2009, 113, 5444–5449 CrossRef CAS.
- E. Bekyarova, K. Murata, M. Yudasaka, D. Kasuya, S. Iijima, H. Tanaka, H. Kahoh and K. Kaneko, J. Phys. Chem. B, 2003, 107, 4681–4684 CrossRef CAS.
- K. Murata, A. Hashimoto, M. Yudasaka, D. Kasuya, K. Kaneko and S. Iijima, Adv. Mater., 2004, 16, 1520–1522 CrossRef CAS.
- K. Murata, J. Miyawaki, M. Yudasaka, S. Iijima and K. Kaneko, Carbon, 2005, 43, 2826–2830 CrossRef CAS.
- K. Murata, K. Kaneko, H. Kanoh, D. Kasuya, K. Takahashi, F. Kokai, M. Yudasaka and S. Iijima, J. Phys. Chem. B, 2002, 106, 11132–11138 CrossRef CAS.
- H. Tanaka, H. Kanoh, M. El-Merraoui, W. A. Steele, M. Yudasaka, S. Iijima and K. Kaneko, J. Phys. Chem. B, 2004, 108, 17457–17465 CrossRef CAS.
- H. Tanaka, H. Kanoh, M. Yudasaka, S. Iijima and K. Kaneko, J. Am. Chem. Soc., 2005, 127, 7511–7516 CrossRef CAS.
- A. J. Zambano, S. Talapatra, K. Lafdi, M. T. Aziz, W. McMilin, G. Shaughnessy, A. D. Migone, M. Yudasaka, S. Iijima, F. Kokai and K. Takahashi, Nanotechnology, 2002, 13, 201–204 CrossRef CAS.
- S. Talapatra, A. Z. Zambano, S. E. Weber and A. D. Migone, Phys. Rev. Lett., 2000, 85, 138–141 CrossRef CAS.
- J. Fan, M. Yudasaka, Y. Kasuya, D. Kasuya and S. Iijima, Chem. Phys. Lett., 2004, 397, 5–10 CrossRef CAS.
- M. Yudasaka, J. Fan, J. Miyawaki and S. Iijima, J. Phys. Chem. B, 2005, 109, 8909–8913 CrossRef CAS.
- S. Utsumi, H. Kanoh, M. Yudasaka and S. Iijima, Adsorption, 2005, 11, 21–28 CrossRef.
- R. Yuge, M. Yudasaka, A. Maigne, M. Tomonari, J. Miyawaki, Y. Kubo, H. Imai, T. Ichihashi and S. Iijima, J. Phys. Chem. C, 2008, 112, 5416–5422 CrossRef CAS.
- T. Ohkubo, Y. Hattori, T. Konishi, H. Sakai, M. Abe, D. Kasuya, M. Yudasaka, S. Iijima, T. Fujikawa and K. Kaneko, Phys. Scr., 2005, 685–687 CrossRef CAS.
- Y. Zhang, N. W. Franlin, R. J. Chen and H. Dai, Chem. Phys. Lett., 2000, 331, 35–41 CrossRef CAS.
- S. Fullam, D. Cottell, H. Rensmo and D. Fitzmaurice, Adv. Mater., 2000, 12, 1430–1432 CrossRef CAS.
- Y. Y. Shao, G. P. Yin and Y. Z. Gao, J. Power Sources, 2007, 171, 558–566 CrossRef CAS.
- E. Bekyarova, A. Hashimoto, M. Yudasaka, Y. Hattori, K. Murata, H. Kanoh, D. Kasuya, S. Iijima and K. Kaneko, J. Phys. Chem. B, 2005, 109, 3711–3714 CrossRef CAS.
- T. Itoh, H. Danjo, W. Sasaki, K. Urita, E. Bekyarova, M. Arai, T. Imamoto, M. Yudasaka, S. Ijima, H. Kanoh and K. Kaneko, Carbon, 2008, 46, 172–175 CrossRef CAS.
- T. Itoh, K. Urita, E. Bekyarova, M. Arai, M. Yudasaka, S. Ijima, T. Ohba, K. Kaneko and H. Kanoh, J. Colloid Interface Sci., 2008, 322, 209–214 CrossRef CAS.
- T. Yoshitake, Y. Shimakawa, S. Kuroshima, H. Kimura, T. Ichihashi, Y. Kubo, D. Kasuya, K. Takahashi, F. Kokai, M. Yudasaka and S. Iijima, Phys. B, 2002, 323, 124–126 CrossRef CAS.
- N. Sano and S. Ukita, Mater. Chem. Phys., 2006, 99, 447–450 CrossRef CAS.
- R. Yuge, T. Ichihashi, Y. Shimakawa, Y. Kubo, M. Yudasaka and S. Ijima, Adv. Mater., 2004, 16, 1420–1423 CrossRef.
- S. Yoshida and M. Sano, Chem. Phys. Lett., 2006, 433, 97–100 CrossRef CAS.
- M. Kosaka, S. Kuroshima, K. Kobayashi, S. Sekino, T. Ichihashi, S. Nakamura, T. Yoshitake and Y. Kubo, J. Phys. Chem. C, 2009, 113, 8660–8667 CrossRef CAS.
- K. Murata, M. Yudasaka and S. Iijima, Carbon, 2006, 44, 818–820 CrossRef CAS.
- N. Karousis, T. Ichihashi, M. Yudasaka, S. Iijima and N. Tagmatarchis, J. Nanosci. Nanotechnol., 2009, 9, 6047–6054 CrossRef CAS.
- T. Murakami, K. Ajima, J. Miyawaki, M. Yudasaka, S. Iijima and K. Shiba, Mol. Pharm., 2004, 1, 399–405 CrossRef CAS.
- K. Ajima, M. Yudasaka, T. Murakami, A. Maigne, K. Shiba and S. Iijima, Mol. Pharm., 2005, 2, 475–480 CrossRef CAS.
- K. Ajima, M. Yudasaka, A. Maigne, J. Miyawaki and S. Iijima, J. Phys. Chem. B, 2006, 110, 5773–5778 CrossRef CAS.
- K. Ajima, A. Maigne, M. Yudasaka and S. Iijima, J. Phys. Chem. B, 2006, 110, 19097–19099 CrossRef CAS.
- K. Ajima, T. Murakami, Y. Mizaoguchi, K. Tsuchida, T. Ichihashi, S. Iijima and M. Yudasaka, ACS Nano, 2008, 2, 2057–2064 CrossRef CAS.
- N. Venkatesan, J. Yoshimitsu, Y. Ito, N. Shibata and K. Takada, Biomaterials, 2005, 26, 7154–7163 CrossRef CAS.
- J. Miyawaki, M. Yudasaka, T. Azami, Y. Kubo and S. Iijima, ACS Nano, 2008, 2, 213–226 CrossRef CAS.
- R. M. Lynch, B. H. Voy, D. F. Glass, S. M. Mahurin, B. Zhao, H. Hu, A. M. Saxton, R. L. Donnell and M. D. Cheng, Nanotoxicology, 2007, 1, 157–166 CrossRef CAS.
- H. Isobe, T. Tanaka, R. Maeda, E. Noiri, N. Solin, M. Yudasaka, S. Iijima and E. Nakamura, Angew. Chem., Int. Ed., 2006, 45, 6676–6680 CrossRef CAS.
- J. Miyawaki, M. Yudasaka, H. Imai, H. Yorimitsu, K. Isobe, E. Nakamura and S. Iijima, J. Phys. Chem. B, 2006, 110, 5179–5181 CrossRef CAS.
- J. Chen, M. A. Hamon, H. Hu, Y. Chen, A. M. Rao, P. C. Eklund and R. C. Haddon, Science, 1998, 282, 95–98 CrossRef CAS.
- B. W. Smith, M. Monthioux and D. E. Luzzi, Nature, 1998, 396, 323–326 CrossRef CAS.
- R. J. Chen, Y. Zhang, D. Wang and H. Dai, J. Am. Chem. Soc., 2001, 123, 3838–3839 CrossRef CAS.
- J. Miyawaki, M. Yudasaka, H. Imai, H. Yorimitsu, H. Isobe, E. Nakamura and S. Iijima, Adv. Mater., 2006, 18, 1010–1014 CrossRef CAS.
- S. Y. Zhu, L. S. Fan, X. Q. Liu, L. H. Shi, H. J. Li, S. Han and G. B. Xu, Electrochem. Commun., 2008, 10, 695–698 CrossRef CAS.
- B. Aissa, Z. Hamoudi, H. Takahashi, K. Tohji, M. Mohamedi and M. A. E. Khakani, Electrochem. Commun., 2009, 11, 862–866 CrossRef CAS.
- S. Y. Zhu, W. X. Niu, H. J. Li, S. Han and G. B. Xu, Talanta, 2009, 79, 1441–1445 CrossRef CAS.
- C. M. Yang, Y. J. Kim, M. Endo, H. Kanoh, M. Yudasaka, S. Iijima and K. Kaneko, J. Am. Chem. Soc., 2007, 129, 20–21 CrossRef CAS.
- S. Y. Zhu, H. J. Li, W. X. Niu and G. B. Xu, Biosens. Bioelectron., 2009, 25, 940–943 CrossRef CAS.
- W. W. Tu, J. P. Lei, L. Ding and H. X. Ju, Chem. Commun., 2009, 4227–4229 RSC.
- J. X. Wang, M. X. Li, Z. J. Shi, N. Q. Li and Z. N. Gu, Anal. Chem., 2002, 74, 1993–1997 CrossRef CAS.
- J. Wang, Electroanalysis, 2005, 17, 7–14 CrossRef CAS.
- J. J. Gooding, R. Wibowo, J. Q. Liu, W. Yang, D. Losic, S. Orbon, F. J. Mearns, J. G. Shapter and D. B. Hibbert, J. Am. Chem. Soc., 2003, 125, 9006–9007 CrossRef CAS.
- F. Patolsky, Y. Weizmann and I. Willner, Angew. Chem., Int. Ed., 2004, 43, 2113–2117 CrossRef CAS.
- C. E. Banks, A. Crossley, C. Salter, S. J. Wilkins and R. G. Compton, Angew. Chem., Int. Ed., 2006, 45, 2533–2537 CrossRef CAS.
- B. Sljukic, C. E. Banks and R. G. Compton, Nano Lett., 2006, 6, 1556–1558 CrossRef CAS.
- C. P. Jones, K. Jurkschat, A. Crossley, R. G. Compton, B. L. Riehl and C. E. Banks, Langmuir, 2007, 23, 9501–9504 CrossRef CAS.
- X. Q. Liu, L. H. Shi, W. X. Niu, H. J. Li and G. B. Xu, Biosens. Bioelectron., 2008, 23, 1887–1890 CrossRef CAS.
- X. Q. Liu, H. J. Li, F. A. Wang, S. Y. Zhu, Y. L. Wang and G. B. Xu, Biosens. Bioelectron., 2010, 25, 2194–2199 CrossRef CAS.
- G. Rotas, A. S. D. Sandanayaka, N. Tagmatarchis, T. Ichihashi, M. Yudasaka, S. Iijima and O. Ito, J. Am. Chem. Soc., 2008, 130, 4725–4731 CrossRef CAS.
- A. S. D. Sandanayaka, O. Ito, T. Tanaka, H. Isobe, E. Nakamura, M.Yudasaka and S. Iijima, New J. Chem., 2009, 33, 2261–2266 RSC.
- M. F. Zhang, T. Murakami, K. Ajima, K. Tsuchida, A. S. D. Sandanayaka, O. Ito, S. Iijima and M. Yudasaka, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 14773–14778 CrossRef CAS.
- A. S. D. Sandanayaka, O. Ito, M. F. Zhang, K. Ajima, S. Iijima, M. Yudasaka, T. Murakami and K. Tsuchida, Adv. Mater., 2009, 21, 4366–4371 CrossRef CAS.
- J. Suehiro, N. Sano, G. B. Zhou, H. Imakiire, K. Imasaka and M. Hara, J. Electrost., 2006, 64, 408–415 CrossRef CAS.
- N. Sano, M. Kinugasa, F. Otsuki and J. Suehiro, Adv. Powder Technol., 2007, 18, 455–466 Search PubMed.
- N. Sano and F. Ohtsuki, J. Electrost., 2007, 65, 263–268 CrossRef CAS.
- E. Ban, R. Sakaquchi, Y. Matsuoka, T. Goto, K. Watanabe and G. Nishijima, Phys. C, 2005, 426–431, 1249–1253 CrossRef CAS.
- K. Kobayashi, S. Hironaka, A. Tanaka, K. Umeda, S. Iijima, M. Yudasaka, D. Kasuya and M. Suzukoi, J. Jpn. Pet. Inst., 2005, 48, 121–126 Search PubMed.
- A. Tanaka, K. Umeda, M. Yudasaka, M. Suzukia, T. Ohana, M. Yumura and S. Iijima, Tribol. Lett., 2005, 19, 135–142 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |