Luminescent quantum clusters of gold in transferrin family protein, lactoferrin exhibiting FRET†
Paulrajpillai Lourdu
Xavier
a,
Kamalesh
Chaudhari
b,
Pramod Kumar
Verma
c,
Samir Kumar
Pal
c and
Thalappil
Pradeep
*a
aDST Unit on Nanoscience (DST UNS), Department of Chemistry and Sophisticated Analytical Instrument Facility, Indian Institute of Technology Madras, Chennai, 600 036, India. E-mail: pradeep@iitm.ac.in
bDepartment of Biotechnology, Indian Institute of Technology Madras, Chennai, 600 036, India
cUnit for Nanoscience and Technology, Department of Chemical, Biological and Macromolecular Sciences, Satyendra Nath Bose National Centre for Basic Sciences, Block JD, Sector III, Salt Lake, Kolkata, 700 098, India
First published on 30th September 2010
Abstract
We report the synthesis of highly luminescent, water soluble quantum clusters (QCs) of gold, which are stabilized by an iron binding transferrin family protein, lactoferrin (Lf). The synthesized AuQC@Lf clusters were characterized using UV-Visible spectroscopy, X-ray photoelectron spectroscopy (XPS), transmission electron microscopy (TEM), photoluminescence (PL), matrix assisted laser desorption ionization mass spectrometry (MALDI-MS), FTIR spectroscopy and circular dichroism (CD) spectroscopy along with picosecond-resolved lifetime measurements. Detailed investigations with FTIR and CD spectroscopy have revealed changes in the secondary structure of the protein in the cluster. We have also studied Förster resonance energy transfer (FRET) occurring between the protein and the cluster. The ability of the clusters to sense cupric ions selectively at ppm concentrations was tested. The stability of clusters in widely varying pH conditions and their continued luminescence make it feasible for them to be used for intracellular imaging and molecular delivery, particularly in view of Lf protection.
1. Introduction
The ardent quest for materials with novel properties has driven us to synthesize nanomaterials such as quantum dots (QDs), nanorods, etc.1,2 Applications and safety of these materials have become important aspects of science and engineering in the recent past. Luminescent nanomaterials like QDs are potential candidates for biological and optoelectronic applications but their proven cytotoxicity in the unprotected form3–5 urges us to look for novel and biocompatible materials. Noble metal quantum clusters (NMQCs) are one such class of materials which have been of recent interest in our group.6–9 NMQCs exhibit highly polarizable transitions which scale in size with EFermi/N1/3 where EFermi is Fermi energy of the bulk metal and N is the number of atoms. Luminescence in them arises from intraband transitions, and these conduction-electron transitions are the low-number limit of the plasmons—the collective dipole oscillations occurring when a continuous density of states is reached.10 Quantum clusters of gold are distinctly different from bulk and metallic nanoparticles, containing very few atoms. They have a sub-nanometre core size with discrete energy levels and show molecule-like properties in their absorption and photoluminescence features. Such clusters of gold and silver are synthesized by various methods with suitable ligands containing thiol and amine groups11–14 such as short peptides (e.g. glutathione)6,11 and proteins7,15,16 (e.g. bovine serum albumin (BSA)). Applications of proteins and peptides to synthesize inorganic nanomaterials was discussed in detail by Dickerson and co-workers.17 Protein protected luminescent NMQCs are attractive due to simplicity in their preparation and potential applications. In this context, the reducing ability of tyrosine and other aromatic amino acids under basic pH (>11) was exploited followed by stabilization of the core by the cysteine residues of the protein.15Lactoferrin (Lf) is a multifunctional protein of the transferrin family, which are iron-binding glycoproteins. Lf has a molecular weight of ∼83 kDa and it is cationic with an isoelectric point of 8.5–9.0. It is found in biological fluids such as milk, blood, cervical mucus, seminal fluids, saliva, tears, and within the specific granules of polymorphonuclear leukocytes. It can be found in three different forms depending on the percentage of iron content, Native lactoferrin (NLf, Fe3+ < 20%), hololactoferrin (HLf, iron saturated) and apolactoferrin (ALf, iron depleted form). It exhibits antimicrobial, antiviral, immunomodulation, antioxidation anti-inflammation, antistress, and analgesic effects, as well as causes enhancement in lipid metabolism. It is one of the few proteins which can cross the blood–brain barrier. Extensive studies have been done in the recent past on the properties, mammalian receptors and applications of Lf.18–23,30 Lf has receptors on several Gram negative bacteria too.24 The in vivo clinical applications of transferrin family proteins are well known.25 Lactoferrin has been used to functionalize iron oxide nanoparticles.26 Recently it has been used in receptor mediated drug delivery, cancer therapy and gene delivery targeting brain as well as for oral drug delivery.27–30 Multifunctional thin films of lactoferrin have also been produced.31 The EMEA (European Medicines Agency) and FDA (Food and Drug Administration) have granted permission to use it as orphan drug in treating cystic fibrosis.32,33
Owing to the composition of lactoferrin (34 cysteine residues and 22 tyrosine residues),18 we thought this protein could be a candidate to make biocompatible and functionalized quantum clusters of gold and can be used for some interesting applications. To the best of our knowledge this is the first time lactoferrin was used as both reducing and stabilizing agent for the production and stabilization of gold nanoparticles as well as quantum clusters. In the recent past, studies done in our group have shown that quantum clusters reduced and stabilized with BSA are nontoxic and can be used for targeted cancer cell imaging.6,7 Hence there is possibility that these lactoferrin stabilized clusters can also be used for similar applications.
2. Experimental
2.1 Reagents, materials and instruments
Tetrachloroauric acid trihydrate (HAuCl4·3H2O) was purchased from CDH, India. Native bovine lactoferrin, ∼17.5% iron saturated, purity >96% of Taradon Laboratory was a kind gift from the company Tablets India Pvt Ltd, Chennai, India. The purity of the protein was tested by SDS–PAGE and MALDI–TOF MS and no impurities were detected (Fig. S1 of the ESI†). Millipore water was used throughout the experiments. Sodium hydroxide pellets from Qualigens Pvt Ltd and Citric Acid from Merk Pvt Ltd were used for the experiments. Potassium bromide (spectroscopic grade) used for infrared studies, was purchased from Merck. Sinapinic acid was used as the matrix for MALDI-TOF MS. All chemicals were used as such without further purification.2.2 Instrumentation
UV/Vis spectra were measured with a Perkin Elmer Lambda 25 instrument in the range 200–1100 nm. Luminescence measurements were carried out on a Jobin Vyon NanoLog instrument. The band pass for excitation and emission was set as 2 nm. Luminescence transients were measured and fitted by using a commercially available spectrophotometer (LifeSpec-ps) from Edinburgh Instruments, UK (80 ps instrument response function (IRF)). The clusters were excited at 445 nm and the emission decays were collected at 650 nm. For the FRET study, we have used a femtosecond-coupled TCSPC (time-correlated single-photon counting) set-up. The sample was excited at 300 nm using the third harmonic of a mode-locked Ti-sapphire laser (Tsunami, Spectra-Physics) pumped by 10 W Millennia (Spectra Physics). The high repetition 80 MHz of the Ti:sapphire laser was lowered to 8 MHz using a pulse picker (Spectra-Physics, model 3980) to accommodate the timing capability of the electronics system. The fundamental output wavelength of the laser was tuned to 900 nm and frequency doubled to 450 nm. The second harmonic was monitored by a THORLABS DET10A/M Si based photodiode. This photodiode signal was then sent to a HORIBA JOBIN YVON TB-01 triggering electronics to generate the start pulse. Finally, all of the fluorescence transients were measured at magic angle. X-ray photoelectron spectroscopy (XPS) measurements were conducted using an Omicron ESCA Probe spectrometer with polychromatic Mg Kα X-rays (hυ = 1253.6 eV). The samples were spotted as drop-cast films on a sample stub. A constant analyzer energy of 20 eV was used for the measurements. High resolution transmission electron microscopy of clusters was carried out with a JEOL 3010 instrument. The samples were drop casted on carbon-coated copper grids and allowed to dry under ambient conditions. Matrix-assisted laser-desorption ionization mass spectrometry (MALDI-MS) studies were conducted using a Voyager-DE PRO Biospectrometry Workstation from Applied Biosystems. A pulsed nitrogen laser of 337 nm was used for the MALDI-MS studies. Mass spectra were collected in positive-ion mode and were averaged for 100 shots. FTIR spectra were measured with a Perkin Elmer Spectrum One instrument. KBr crystals were used as the matrix for preparing samples. The second derivative of FTIR spectrum was taken using “spectrum one” software provided by Perkin Elmer. Far-UV CD spectrum was obtained using a J-600 Spectropolarimeter (JASCO).2.3 The PL quantum yield measurements AuQC@Lf
The PL quantum yields of AuQC@Lf samples were determined by the integrated luminescence intensity method by comparison of the luminescence emission with the standard reference sample of fluorescein in 0.1M NaOH at 27 °C. The luminescence emission of a sample can be written in a formula with related parameters.| F = KΦCσlI |
where the terms with suffixes r and s mean reference fluorescein and AuQC, respectively. Since the product, Cσ represents the absorption of the sample, the term of Crσr/Csσscan be replaced by Ar/As where A is the absorption coefficient of the sample at the excitation wavelength. When the values of Ar and As are measured, Φs of AuQC can be obtained from,
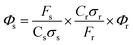
We measured Ar and As at 494 nm in a UV-visible spectrometer and for both, the absorption coefficients were set as 0.1. Then the photoluminescence intensities of these samples were measured.
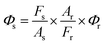
2.4 Energy transfer efficiency measurement
The steady-state transfer efficiency, E is measured using the relative fluorescence intensity of the donor, in the absence (FD) and presence (FDA) of the acceptor as,For donor–acceptor (D–A) systems decaying with multiexponential lifetimes, the time-resolved energy transfer efficiency (E) is calculated from the amplitude weighted lifetimes
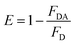
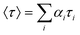 of the donor in absence (τD) and presence (τDA) of the acceptor as,
of the donor in absence (τD) and presence (τDA) of the acceptor as,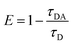
2.5 Synthesis
AuQC@NLf was prepared as follows. It follows the procedure in Scheme 1. Briefly, tetrachloroauric acid trihydrate (HAuCl4·3H2O) and NLf were mixed and stirred using a magnetic stirrer for 2 min. To this, 1 M NaOH was added, its final concentration in the mixture was 5%. This reaction mixture was stirred for 24 h, which yields highly luminescent quantum clusters when the Au![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :NLf ratio was of 17:1, by molarity. ALf and HLf were prepared by the method given by Roland et al.34 ALf was prepared by dialyzing NLf against 0.1 M citric acid at pH 2.3. Extensive dialysis against distilled water was done to remove excess citric acid and Fe3+ ions. HLf was prepared by dialyzing NLf against excess Fe3+ ions in the presence of CO32− ions. Excess Fe3+ ions were removed by dialysis against deionized water. The iron saturation level was monitored by UV-visible spectroscopy. AuQC@ALf and AuQC@HLf were prepared by the same method as mentioned above for AuQC@NLf. QCs were formed only in the concentration range of 2.5 mM to 10 mM of Au3+ for 150 μM of lactoferrin (molar ratio 8:1 to 33:1) and these clusters protected with NLf are referred to as AuQC@NLf. Nanoparticles formed at higher concentration of Au3+ are referred to as AuNP@NLf.
:NLf ratio was of 17:1, by molarity. ALf and HLf were prepared by the method given by Roland et al.34 ALf was prepared by dialyzing NLf against 0.1 M citric acid at pH 2.3. Extensive dialysis against distilled water was done to remove excess citric acid and Fe3+ ions. HLf was prepared by dialyzing NLf against excess Fe3+ ions in the presence of CO32− ions. Excess Fe3+ ions were removed by dialysis against deionized water. The iron saturation level was monitored by UV-visible spectroscopy. AuQC@ALf and AuQC@HLf were prepared by the same method as mentioned above for AuQC@NLf. QCs were formed only in the concentration range of 2.5 mM to 10 mM of Au3+ for 150 μM of lactoferrin (molar ratio 8:1 to 33:1) and these clusters protected with NLf are referred to as AuQC@NLf. Nanoparticles formed at higher concentration of Au3+ are referred to as AuNP@NLf.
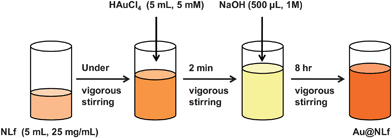 | ||
| Scheme 1 Synthetic procedure to make AuQC@NLf. | ||
3. Results and discussion
3.1 UV-Visible spectroscopic studies
The clusters were formed only in specific window of concentrations. The absorption spectra of clusters formed at various Au:NLf ratios are presented in Fig. 1A. At higher concentrations of HAuCl4:NLf (83:1, 67:1 molar ratios), surface plasmon resonance of non-luminescent gold nanoparticles, absorbtion at 530 nm can be seen. Cluster formation begins at the HAuCl4:NLf ratio of 33:1, hence the product obtained at this concentration was a mixture of luminescent clusters and nanoparticles. As we reduced the concentration of HAuCl4:NLf (17:1, 8:1), the plasmonic peak intensity gradually reduced and disappeared, but the features of aromatic amino acids in protein retained at ∼280 nm. The clusters do not show any distinct absorption features, as in the case of bovine serum albumin (BSA) protected quantum clusters.7,15 At this optimized concentration of HAuCl4:NLf of 17:1, the resulting product was luminescent with red emission. However, further reduction in the concentration HAuCl4:NLf to 3:1 did not yield luminescent clusters. When the experiment was done in the absence of NaOH, only nanoparticles were formed. The clusters were seen after 8 h of incubation itself. However, after 24 h of incubation, no change was seen in the spectrum. Intense luminescence from the clusters was observable to the naked eye. As a result, the emission could be photographed (Fig. 1B). As can be seen, as a function of Au concentration, different shades of red are observed. However, as the concentration is increased beyond the critical value, nanoparticles are seen, which do not exhibit any observable emission. These clusters were luminescent in solid state also (Fig. S2 of the ESI†). At low temperature enhancement in luminescence intensity was observed (Fig. S3 of the ESI†).
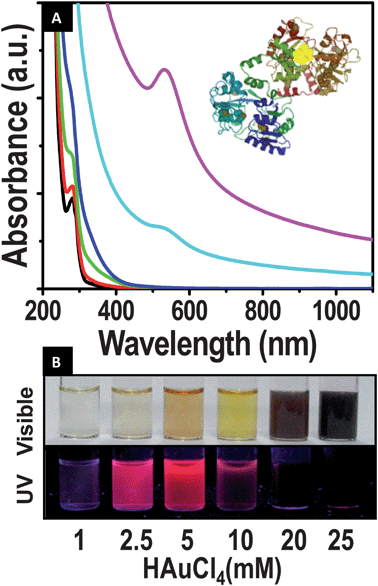 | ||
| Fig. 1 (A) UV-visible spectra of the evolution of clusters. Concentration of NLf was kept constant (150 μM) and the molar ratio of NLf:gold was varied. The spectra were collected after 24 h of the reaction; 1:3 (black), 1:8(red), 1:17 (green), 1:33 (blue), 1:67 (cyan) and 1:83 (magenta). Clusters were formed only at certain gold:Lf ratios. Schematic of the cluster in protein is shown in the inset. (B) Photograph showing the emission of clusters and nanoparticles formed at various metal ion concentrations, keeping the NLf concentration the same at ∼150 μM. In visible light (above) and in UV light (below). | ||
Quantum clusters of gold were also formed by ALf and by HLf (Fig. S3 of the ESI†), irrespective of its iron content and no obvious change in the position of emission was observed. Thus Fe3+ bound to the protein does not interfere in cluster formation. The concentration at which clusters are formed is also not affected significantly in all the three proteins, NLf, ALf and HLf.
3.2 Fluorescence time resolved studies
Inset of Fig. 2A shows the emission spectra of the as-synthesized clusters. In all cases, luminescent features showed a broad emission maximum at ∼650 nm, which had two features centered at 643 and 658 nm. Multiple peak maxima in the emission are attributed to multiple cluster size distributions or the presence of different chemical environments of the metal core. The emission spectra of NLf are shown in the inset of Fig. 2B. It is clear from Fig. 1A and inset of Fig. 2B that there is a significant spectral overlap between the NLf emission and the absorption of the cluster (AuQC@NLf). From Fig. 2D we observed that when the cluster is excited at 380 nm, it shows two peaks, one at ∼450 nm, due to the weak luminescence of the protein, which was observed upon excitation of protein alone at 380 nm, thus confirming that it is from the protein,35 and another at 650 nm due to the cluster core. This spectral overlap is expected to reveal the proximity of the cluster to the tryptophan residues of the protein NLf through energy transfer from tryptophan (donor) to Au cluster (acceptor). As revealed from the inset of Fig. 2B, the overall steady state emission intensity of the donor emission (NLf) drastically decreased in the presence of the acceptor (AuQC@NLf). Also, the faster decay of the donor (NLf) in the presence of acceptor (AuQC@NLf) (Fig. 2B) as compared to the free donor confirms Förster resonance energy transfer from tryptophan residues of NLf to the Au cluster of AuQC@NLf (Table 1). FRET in AuQC conjugated to fluorophore has been reported by our group earlier.36 The calculated donor-to-acceptor energy transfer efficiency from steady-state and time-resolved studies are found to be 80 and 43%, respectively. For a well-behaved system, the ratio of the integrated areas under the time-resolved emission decays for donor and donor–acceptor complexes should correlate with the integrated areas under the steady-state emission spectra. In principle, one of the methods, namely steady-state or time-resolved, can be used to estimate energy transfer efficiency. However, there could be a disagreement between the estimated energy transfer efficiency from these two methods as explained below. FRET is a nonradiative process whereby an excited state donor (D) transfers energy to a proximal ground state acceptor (A) through long-range dipole–dipole interactions. The rate of energy transfer is highly dependent on many factors, such as the extent of spectral overlap, the relative orientation of the transition dipoles, and, most importantly, the distance between the donor and acceptor molecules. In the steady-state, there is also a possibility of quenching due to re-absorption of the donor emission by the acceptor through the radiative mechanism of energy transfer, which is not a dipole oriented process. In case of time-resolved measurement, this radiative energy transfer does not contribute to the overall lifetime of the fluorophore. Therefore, the energy transfer is overestimated in the steady-state and includes the re-absorption, thereby giving a higher efficiency value. In one of our recent studies, we have reported the potential danger of using steady-state fluorescence quenching to conclude the nature of energy transfer as Förster type and to estimate energy transfer efficiency.37 As the dimensions of the Lf protein, measured by X-ray diffraction, are 13.89 × 8.7 × 7.34 nm,38 the distance between the donor tryptophan residues and the cluster protected by the protein is likely to be less than 10 nm, which is essential for FRET to occur. To the best of our knowledge, this is the first time we report the FRET process occurring between the protein and AuQC inside the protein. It is to be noted that the measured quantum yield gives values of 6.0%, 5.9%, 1.23% for AuQC@NLf, AuQC@apoLf, AuQC@HLf (100% Fe3+ saturated), respectively. This enhancement of quantum yield of clusters synthesized in protein molecules is still under investigation and will be presented in a subsequent report.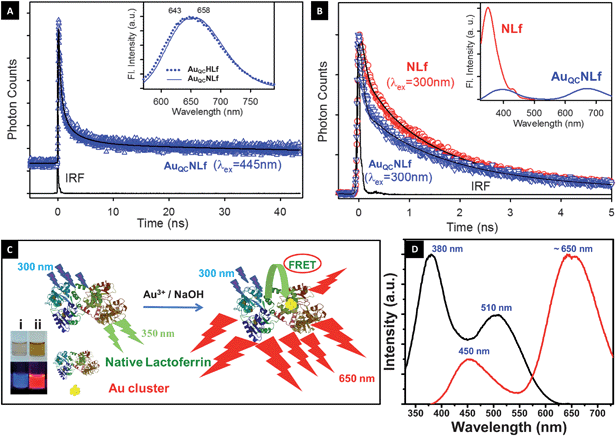 | ||
| Fig. 2 (A) Fluorescence decay profile of AuQC@NLf cluster at 650 nm (excitation at 445 nm). Inset shows emission spectra of AuQC@NLf (broken line) and AuQC@HLf (solid line), excited at 445 nm. (B) Picosecond-resolved fluorescence transients of donor (NLf) and donor–acceptor (AuQC@NLf) at 350 nm (excitation at 300 nm). Steady-state PL quenching of the tryptophan emission (donor; NLf) in the presence of acceptor Au cluster (AuQC@NLf) is shown in the inset. (C) Schematic of the occurrence of FRET between Lf and cluster. Inset shows the photographs of (i) NLf (ii) AuQC@NLf (from left to right) taken in visible light (above) and in UV light (below). (D) Photoluminescence spectra AuQC@NLf show emission at ∼650 nm (excitation wavelength 380 nm); the excitation spectra collected correspond to 650 nm emission, which shows peaks at 380 nm and 510 nm. The emission at 450 nm is due to the weak luminescence of the protein when excited at 380 nm. | ||
| Tryptophan emission (350 nm) | τ 1/ns | τ 2/ns | τ 3/ns | <τ>/ns |
|---|---|---|---|---|
| NLf | 0.11 (49%) | 1.17 (37%) | 4.11 (14%) | 1.06 |
| AuQC@NLf | 0.05 (71%) | 0.84 (16%) | 3.38 (13%) | 0.60 |
Time-resolved decay of AuQC@NLf was measured at 650 nm. Data were collected using a picosecond-resolved time correlated single photon counting (TCSPC) technique (Fig. 2A). Lifetime values were obtained by the multi-exponential fitting of the luminescence at 650 nm (an excitation wavelength of 445 nm was used to avoid exciting tryptophan). Time components are 0.18 ns (58%), 0.82 ns (22%), 3.58 ns (11%) and 110.70 ns (9%). The uplift of the decay with respect to the IRF (instrument response function) is due to the presence of a long time component of ∼111 ns (9%) which is not complete within the experimental time window of 50 ns. A fast time scale major component is seen in a number of QCs of gold. Examples include, Au25, Au22 and Au23.6 In general, these clusters exhibit a large component of a few tens of picoseconds and several slow components of nanoseconds. A similar situation is seen in the case of AuQC@BSA.7
3.3 TEM and MALDI-TOF-MS studies
TEM images have shown the presence of sub-nanometre sized clusters in AuQC@NLf (Fig. S4 of the ESI†) which are compared with the TEM image of AuNP@NLf, formed at higher gold concentrations. The clusters are shown with white circles. At lower gold concentration, a larger fraction of smaller clusters is seen, in agreement with the optical absorption spectrum. These clusters are sensitive to high energy electron beams and nanoparticles were formed due to continuous electron beam irradiation due to aggregation of the clusters (Fig. S5 of the ESI†). Therefore, the image (Fig. S4 of the ESI†) was taken immediately after exposure. The cluster core dimension could not be established accurately from the current images, but it is below 1 nm. EDAX of the sample was taken, which shows the presence of Au, S and Fe (Fig. S6 of the ESI†).Mass spectra of the protein and the cluster were measured by MALDI-TOF-MS (Fig. 3). Sinapinic acid mixed with urea was used as the matrix for the samples. The detailed method of sample preparation is explained in supplementary information 7.† The spectra were collected in the positive mode. The mass spectrum of NLf shows two distinct peaks at 41.5 and 83.0 kDa due to the di- and mono- cations. These peaks are in agreement with the previously reported values.39,40 The protein and the cluster samples did not show features beyond 100 kDa. The major peak observed for NLf was at m/z ∼83024. AuQC@NLf showed two distinct, but low intensity peaks at m/z ∼85570 and ∼87965 besides the parent protein peak. The difference between the peak at m/z 87965 and the parent protein corresponds to Au25 and we tentatively assign the peak to an Au25 core encapsulated in the protein, However, smaller clusters are also expected to be in the protein, as shown by the peak at ∼m/z 85570. The cluster cores alone were not observed, as expected as they are fragile species and are tightly bound in the protein. The overall ion intensity was significantly reduced in the cluster sample and the peaks were broadened. This spectral behavior is consistent with BSA clusters.7 As the intensity was low, a post source decay analysis could not be done to understand the nature of the cluster. The protein structure itself appears to have undergone significant changes as the dication intensity was poor. The data suggest that the cluster is fully encapsulated by a single protein molecule.
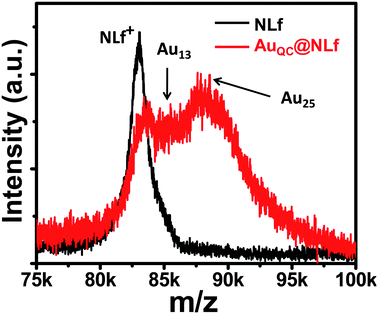 | ||
| Fig. 3 MALDI mass spectra of NLf and AuQC@NLf. Spectrum of NLf was measured at pH 12. There are two cluster features. | ||
3.4 XPS studies
Further analysis was done with X-ray photoelectron spectroscopy (XPS), for the optimized concentration of gold:NLf (17:1). All the expected elements were seen. The Au core level showed principal Au 4f7/2 and Au 4f5/2 components at 84.2 and 87.9 eV binding energy. This confirms the formation of stable Au clusters by reduction of HAuCl4. The core is close to Au(0), although a minor shift to the higher binding energy, due to reduced size is noted (Fig. 4A). The spectrum has a higher binding energy component, although weak, which may be attributed to the smaller clusters or surface atoms of a given cluster. The S 2p region suggests that the monolayer of the clusters is sensitive to the X-ray beam and as a result, two 2p3/2 features are seen at 165.8 and 168.0 eV, attributed to oxidized states of sulfur (Fig. 4B). This is seen in thiolate protected clusters upon X-ray exposure, leading to sulfite, sulfonate and sulfate species.41 Au:S atomic ratio measured from the area of Au 4f and S 2p features is 1:3.75 which implies that there is free lactoferrin in the sample even at optimized concentration. This is consistent with the MALDI-MS data.
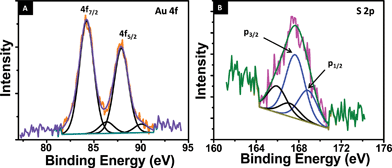 | ||
| Fig. 4 (A) Au 4f core level photoelectron spectrum of the AuQC@NLf cluster. (B) S 2p core level photoelectron spectrum of AuQC@NLf. The components are fitted. Two sets of spin–orbit split components are marked in both. | ||
3.5 FTIR and CD spectroscopic studies
CD spectroscopy and FTIR spectroscopy are excellent tools to monitor changes in the secondary structure of proteins. We have employed both these methods to study the conformational behavior of NLf before and after the formation of the cluster. Fig. 5A shows a comparison between the FTIR spectra of NLf and AuQC@NLf. The characteristic stretching and bending vibrations arising due to amide bonds which link amino acids are assigned as amide I (1600–1690 cm−1), amide II (1480–1575 cm−1), amide III (1229–1301 cm−1) and amide A (∼3300 cm−1).42 The band appearing at ∼700 cm−1 can be assigned to –NH2 and –NH wagging and that at ∼2960 cm−1 is due to C–H vibrations.43 Other bands are those at ∼1400 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching of COO−), ∼1468 cm−1 (C–H deformation of >CH2), and ∼3500 cm−1 (O–H stretching).44 As Lf is a glycoprotein, the broad structure from 900–1200 cm−1 is due to C–O, C–C stretches and C–O–H, C–O–C deformation of carbohydrates.44 In general, the cluster spectrum is broader than the protein and several features are merged. As the amide I band is most sensitive to protein secondary structures, we have studied the second derivative of FTIR spectra of NLf and AuQC@NLf in the 1600–1690 cm−1 range (Fig. 5B). Generally the band assignments in the amide I region for protein secondary structures are as follows. The bands between 1648–1660 cm−1 are assigned to α-helices and the bands between 1612–1642 cm−1 are attributed to β-sheets. Similarly the bands between 1662–1688 cm−1 are assigned to β-turns and the band at ∼1648 ± 2 cm−1 indicates the presence of random coil structures.42,45,47 Hence, in the second derivative FTIR spectrum of NLf, the intensity of the bands at 1651 and 1657 cm−1 were assigned to α helices. For β-sheets the bands assigned are from 1615–1639 cm−1. A new band appearing at 1648 cm−1 in the case of AuQC@NLf is assigned to random coil and unordered structures and the bands between 1664–1685 cm−1 are assigned to β-turns. By comparing NLf and AuQC@NLf spectra, it can be seen that after the formation of the clusters, there is substantial change in the conformation of the protein as there is decrease in intensity of α-helical and β-sheet structures in the 1600–1655 cm−1 region. Thus a large change in the protein structure is evident.
O stretching of COO−), ∼1468 cm−1 (C–H deformation of >CH2), and ∼3500 cm−1 (O–H stretching).44 As Lf is a glycoprotein, the broad structure from 900–1200 cm−1 is due to C–O, C–C stretches and C–O–H, C–O–C deformation of carbohydrates.44 In general, the cluster spectrum is broader than the protein and several features are merged. As the amide I band is most sensitive to protein secondary structures, we have studied the second derivative of FTIR spectra of NLf and AuQC@NLf in the 1600–1690 cm−1 range (Fig. 5B). Generally the band assignments in the amide I region for protein secondary structures are as follows. The bands between 1648–1660 cm−1 are assigned to α-helices and the bands between 1612–1642 cm−1 are attributed to β-sheets. Similarly the bands between 1662–1688 cm−1 are assigned to β-turns and the band at ∼1648 ± 2 cm−1 indicates the presence of random coil structures.42,45,47 Hence, in the second derivative FTIR spectrum of NLf, the intensity of the bands at 1651 and 1657 cm−1 were assigned to α helices. For β-sheets the bands assigned are from 1615–1639 cm−1. A new band appearing at 1648 cm−1 in the case of AuQC@NLf is assigned to random coil and unordered structures and the bands between 1664–1685 cm−1 are assigned to β-turns. By comparing NLf and AuQC@NLf spectra, it can be seen that after the formation of the clusters, there is substantial change in the conformation of the protein as there is decrease in intensity of α-helical and β-sheet structures in the 1600–1655 cm−1 region. Thus a large change in the protein structure is evident.
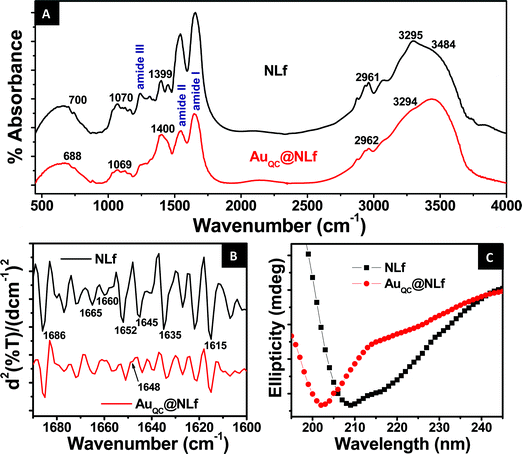 | ||
| Fig. 5 (A) Comparison of the FTIR spectra of NLf and AuQC@NLf. (B) Comparison of the second derivative FTIR spectrum in the amide I region (1600–1690 cm−1) of NLf and AuQC@NLf, showing bands appearing due to secondary structures. (C) Comparison between the CD spectra of NLf and AuQC@NLf. | ||
Fig. 5C shows a comparison of the Far-UV CD spectra of NLf and AuQC@NLf. In the case of NLf, two negative bands at 208 nm and 222 nm indicate the presence of α-helix in the protein secondary structure. However, the valley at 222 nm is shallower than usually found for other proteins containing α helical structures.45,46 This may be interpreted as the presence of β-sheets and random coil structures in this conformational state. However, after the formation of clusters, the CD spectrum shows that the negative band at 208 nm present in NLf CD spectrum is shifted to 202 nm in the case of AuQC@NLf. This implies a decrease in α-helical content and increase in β-sheet and random coil structures47 which corroborates with the FTIR result.
3.6 pH and metal ion sensitivity
The as-synthesized AuQC@NLf was tested for its sensitivity towards metal ions such as Cu2+, Ag+, Ca2+, Ni2+, Co3+, Fe3+ and Zn2+. Chlorides and nitrates of the metals were used. Final concentration of the metal ions in the 1000 ppm cluster solution was 10 ppm. After 10 min, photoluminescence spectra were taken to study the sensitivity of the cluster towards metal ions. Maximum quenching of luminescence intensity was observed for cupric ions and in the case of silver ions luminescence intensity was increased about two fold. The reason of AuQC@NLf quenching by Cu2+ at 10 ppm was found to be aggregation manifested as a decrease in absorbance in UV-Vis (Fig. S9 of the ESI†). However, ferric ions had very little effect on luminescence intensity since its concentration was very low in this case (Fig. 6A). This change can be observed visually too as shown in Fig. 6B. Studies with various Cu2+ salts showed that the anion dependence of luminescence was minimal.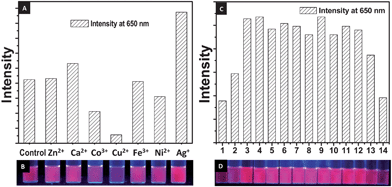 | ||
| Fig. 6 (A) Bar diagram showing changes in the luminescence intensity of clusters in presence of different metal ions and (B) corresponding photograph of the solutions in UV light. (C) Bar diagram showing the changes in luminescence intensity of clusters at various pH and (D) corresponding photograph of the solutions in UV light. | ||
The sample was tested for its stability as a function of pH. Deionised water was used for preparing the cluster solution and pH was adjusted to the desired value using NaOH and HCl by keeping the cluster concentration constant. As the cluster is encapsulated in a protein, the effect of pH on the capping must be prominent compared to its effect on the Au core. Hence while studying effect of pH on AuQC@NLf emission, samples at different pH were excited at 370 nm and emission intensity of clusters at 650 nm was studied. Fig. 6C shows the variation in intensity of emission at various pH. As the pH of the solution was changed to alkaline, a red shift was observed in the emission peak. The data are presented in the supplementary information (Fig. S8 of the ESI†). The clusters show maximum intensity near neutral pH, but the luminescence is observable even to the naked eye throughout the entire window investigated, although it decreases significantly at extremes of acidity and alkalinity.
4. Conclusions
Luminescent gold clusters have been synthesized using a therapeutically important protein, lactoferrin. The iron binding milk protein-based synthesis may be considered as green. Two distinct cluster cores have been identified by mass spectrometry and sub-nanometre cores were observed in TEM. Both Au nanoparticles and luminescent clusters are prepared by the same protein, by varying the concentrations. Cluster formation was observed in all the three forms of the protein, NLf, ALf and HLf. Products were characterized by UV-visible, photoluminescence, XPS, FT-IR and CD spectroscopies and mass spectrometry. FRET occurring between protein and cluster was characterized. The cluster showed metal ion sensitivity; maximum quenching was observed with cupric ions and not with ferric ions. Clusters were found to be stable over the entire pH range of 1–14 and a red shift was observed in the emission peak in extreme alkaline pH. As the protein is known for its diverse properties and applications, we expect this new system of gold quantum clusters in lactoferrin to be useful in drug delivery, imaging and diagnostics. Further studies will be required to get precise details about location of the cluster inside the protein. These luminescent clusters inside the protein have opened up new area at the nano–bio interface. The present study opens up many questions such as how the functionality of the protein changes upon cluster formation, whether the protein's cryptic epitopes are exposed and in turn, does it become immunogenic, whether iron binding sites remain functional or impaired by cluster formation and what is the nature of cluster formation in other forms of the protein. Detailed studies are required to answer these.Acknowledgements
Thanks are due to Tablets India Pvt Ltd, Chennai, India for their kind gift native bovine lactoferrin. We thank the department of Science and Technology, Government of India for constantly supporting our research program on nanomaterials. P. K. V. thanks CSIR, India, for a fellowship. S. K. P. thanks DST, India, for financial support (grant SR/SO/BB-15/2007).References
- X. Michalet, F. F. Pinaud, L. A. Bentolila, J. M. Tsay, S. Doose, J. J. Li, G. Sundaresan, A. M. Wu, S. S. Gambhir and S. Weiss, Science, 2005, 307, 538–544 CrossRef CAS.
- A. V. Kabashin, P. Evans, S. Pastkovsky, W. Hendren, G. A. Wurtz, R. Atkinson, R. Pollard, V. A. Podolskiy and A. V. Zayats, Nat. Mater., 2009, 8, 867–871 CrossRef CAS.
- A. E. Nel, L. mädler, D. Velegol, T. Xia, E. M. V. Hoek, P. Somasundaran, F. Klaessig, V. Castranova and M. Thompson, Nat. Mater., 2009, 8, 543–557 CrossRef CAS.
- A. M. Derfus, W. C. W. Chan and S. N. Bhatia, Nano Lett., 2004, 4, 11–18 CrossRef CAS.
- T. S. Hauck, R. E. Anderson, H. C. Fischer, S. Newbigging and W. C. W. Chan, Small, 2009, 6, 138–144.
- M. A. H. Muhammed, P. K. Verma, S. K. Pal, R. C. Arun Kumar, S. Paul, R. V. Omkumar and T. Pradeep, Chem.–Eur. J., 2009, 15, 10110–10120 CrossRef CAS.
- M. A. H. Muhammed, P. K. Verma, S. K. Pal, A. Retnakumari, M. Koyakutty, S. Nair and T. Pradeep, Chem.–Eur. J., 2010, 16, 10103–10112 CrossRef.
- A. Retnakumari, S. Setua1, D. Menon, P. Ravindran, M. A. H. Muhammed, T. Pradeep, S. Nair and M. Koyakutty, Nanotechnology, 2010, 21, 1–12.
- T. U. B. Rao and T. Pradeep, Angew. Chem., Int. Ed., 2010, 49, 3925–3929 CAS.
- J. Zheng, P. R. Nicovich and R. M. Dickson, Annu. Rev. Phys. Chem., 2008, 58, 409–431.
- Y. Negishi, K. Nobusada and T. Tsukuda, J. Am. Chem. Soc., 2005, 127, 5261–5270 CrossRef CAS.
- N. Nishida, H. Yao and K. Kimura, Langmuir, 2008, 24, 2759–2766 CrossRef CAS.
- I. Díez, M. Pusa, S. Kulmala, H. Jiang, A. Walther, A. S. Goldmann, A. H. E. Müller, O. Ikkala and R. H. A. Ras, Angew. Chem. Int. Ed., 2009, 121, 2156–2159 CrossRef.
- Z. Wu, C. Gayathri, R. Gil and R. Jin, J. Am. Chem. Soc., 2009, 131, 6535–6042 CrossRef CAS.
- J. Xie, Y. Zheng and J. Y. Ying, J. Am. Chem. Soc., 2009, 131, 888–889 CrossRef CAS.
- H. Wei, Z. Wang, L. Yang, S. Tian, C. Hou and Y. Lu, Analyst, 2010, 135, 1406–1410 RSC.
- M. B. Dickerson, K. H. Sandhage and R. R. Naik, Chem. Rev., 2008, 108, 4935–4978 CrossRef CAS.
- H. Jenssen and R. E. W. Hancock, Biochimie, 2009, 91, 19–29 CrossRef CAS.
- W. S. Kim, M. M. Rahman and K. I. Shimazaki, J. Food Saf., 2008, 28, 23–33 CrossRef CAS.
- J. M. Steijns and A. C. M. V. Hooijdonk, Br. J. Nutr., 2000, 84, S11–S17 CAS.
- Y. A. Suzuki, V. Lopez and B. Lönnerdal, Cell. Mol. Life Sci., 2005, 62, 2560–2575 CrossRef CAS.
- A. Pierce and D. Legrand, Biochimie, 2009, 91, 1–164 CrossRef CAS.
- B. E. Britigan, T. S. Lewis, M. Waldschmidt, M. L. McCormick and A. M. Krieg, J. Immunol., 2001, 167, 2921–2928 CAS.
- A. Ekins, A. G. Khan, S. R. Shouldice and A. B. Schryvers, BioMetals, 2004, 17, 235–243 Search PubMed.
- Z. M. Qian, H. Li, H. Sun and K. Ho, Pharmacol. Rev., 2002, 54, 561–587 CrossRef CAS.
- A. K. Gupta and A. S. G. Curtis, Biomaterials, 2004, 25, 3029–3040 CrossRef CAS.
- J. R. Kanwar, K. P. Palmano, X. Sun, R. K. Kanwar, R. Gupta, N. Haggarty, A. Rowan, S. Ram and G. W. Krissansen, Immunol. Cell Biol., 2008, 86, 277–288 CrossRef CAS.
- K. Hu, J. Li, Y. Shen, W. Lu, X. Gao, Q. Zhang and X. Jiang, J. Controlled Release, 2009, 134, 55–61 CrossRef CAS.
- R. Huang, W. Ke, L. Han, Y. Liu, K. Shao, L. Ye, J. Lou, C. Jiang and Y. Pei, J. Cereb. Blood Flow Metab., 2009, 29, 1914–1923 Search PubMed.
- Y. Nojima, Y. Suzuki, K. Iguchi, T. Shiga, A. Iwata, T. Fujimoto, K. Yoshida, H. Shimizu, T. Takeuchi and A. Sato, Bioconjugate Chem., 2008, 19, 2253–2259 CrossRef CAS and references therein.
- C. Constantinescua, A. Palla-Papavlua, A. Rotarua, P. Florianb, F. Chelub, M. Icriverzib, A. Nedelceaa, V. Dincaa, A. Roseanub and M. Dinescu, Appl. Surf. Sci., 2009, 255, 5491–5495 CrossRef.
- Public summary of positive opinion for orphan designation of hypothiocyanite/lactoferrin for the treatment of cystic fibrosis, Pre-authorisation Evaluation of Medicines for Human Use, European Medicines Agency, 2009-09-07, Doc.Ref.: EMEA/COMP/392984/2009 Search PubMed.
- Meveol: orphan drug status granted by the FDA for the treatment of cystic fibrosis, United States Food and Drug Administration, 2009-11-05. http://www.bioalaxia.eu/content/meveol-orphan-drug-status-granted-fda-treatment-cystic-fibrosis.
- R. R. Arnold, J. E. Russell, W. J. Champion, M. Brewer and J. J. Gauthier, Infect. Immun., 1982, 35, 792–799 CAS.
- B. L. Fletcher and A. L. Tappel, Lipids, 1971, 6, 172–177 CrossRef CAS.
- M. A. H. Muhammed, A. K. Shaw, S. K. Pal and T. Pradeep, J. Phys. Chem. C, 2008, 112, 14324–14330 CrossRef CAS.
- P. Majumder, R. Sarkar, A. K. Shaw, A. Chakraborty and S. K. Pal, J. Colloid Interface Sci., 2005, 290, 462 CrossRef CAS.
- L. Meagher and H. J. Griesser, Colloids Surf., B, 2002, 23, 125–140 CrossRef CAS.
- B. O. Keller and L. Li, J. Am. Soc. Mass Spectrom., 2006, 17, 780–785 CrossRef CAS.
- K. Sparbier, T. Wenzel and M. Kostrzewa, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2006, 840, 29–36 CrossRef CAS.
- T. Laihoa, J. A. Leiroa and J. Lukkarib, Appl. Surf. Sci., 2003, 212–213, 525–529 CrossRef.
- J. Kong and S. Yu, Acta Biochim. Biophys. Sin., 2007, 39, 549–559 CrossRef CAS.
- W. K. Surewicz, H. H. Mantsch and D. Chapman, Biochemistry, 1993, 32, 389–393 CrossRef CAS.
- Infrared and Raman Spectroscopy of Biological Materials. Practical Spectroscopy Series, ed. H. U. Gremlich and B. Yan, CRC Press, Taylor and Francis group, 2000, vol. 24, pp. 329–330 Search PubMed.
- L. Shang, Y. Wang, J. Jiang and S. Dong, Langmuir, 2007, 23, 2714–2721 CrossRef CAS.
- N. Greenfieldi and G. D. Fasman, Biochemistry, 1969, 8, 4108–4116 CrossRef CAS.
- M. Carbonaro and Nucara, Amino Acids, 2010, 38, 679–690 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional information concerning SDS-PAGE analysis of NLf, solid state luminescence, PL spectra of AuQC@ALf, AuQC@NLf and AuQC@HLf and comparison at different temperatures, TEM images, EDAX, UV-Visible spectra. See DOI: 10.1039/c0nr00377h |
| This journal is © The Royal Society of Chemistry 2010 |