Interfacial entrapment of noble metal nanoparticles and nanorods capped with amphiphilic multiblock copolymer at a selective liquid–liquid interface
Binyang
Du
*,
Xiujuan
Chen
,
Bin
Zhao
,
Aixiong
Mei
,
Qi
Wang
,
Junting
Xu
and
Zhiqiang
Fan
MOE Key Laboratory of Macromolecular Synthesis and Functionalization, Department of Polymer Science & Engineering, Zhejiang University, Hangzhou, 310027, China. E-mail: duby@zju.edu.cn; Fax: +860571087952400; Tel: +860571087953164
First published on 5th August 2010
Abstract
An amphiphilic multiblock copolymer, [poly(4-vinylpyridine)-b-polystyrene-b-poly(4-vinylpyridine)]n (P4VP-PS-P4VP)n bearing trithiocarbonate moieties (–S–CS–S–), was used as capping agent to fabricate the functional copolymer-capped Au nanoparticles. Due to the amphiphilic character of the multiblock copolymer, the (P4VP-PS-P4VP)n-capped Au nanoparticles were able to be entrapped and hence form a stable monolayer thin film at the (DMF–H2O)/diethyl ether interface. Note that DMF–H2O is a good mixed solvent for P4VP and a nonsolvent for PS, whereas diethyl ether is a good solvent for PS and a nonsolvent for P4VP. A similar procedure can be applied to prepare monolayer thin film of the (P4VP-PS-P4VP)n-capped Ag nanoparticles at the (DMF–H2O)/diethyl ether interface, which exhibits enhanced surface Raman signal. We further demonstrate that the stable monolayer of (P4VP-PS-P4VP)n-capped Au nanorods can also be fabricated by using the ligand-exchange method.
Introduction
Extensive attention and effort have been paid to assembling nanoparticles into two or three-dimensional (2D or 3D) nanostructures, which have wide applications in optical and electronic nanodevices.1,2 Various methods, such as controlled evaporation of solvents,3,4 Langmuir-Blodgett technique5,6 and self-assembly at the interface of immiscible liquids,7–13 have been employed to fabricate 2D or 3D nanostructures. Among them, the liquid–liquid interface serves as a flexible soft substrate for the rapid and spontaneous self-assembly of nanoparticles. Moreover, the rapid diffusion of nanoparticles and reagents in liquids can lead to the very efficient modification of interfacial chemistry of the nanoparticles,7 which allows the tuning of surface properties of the as-constructed nanostructures.The interfacial entrapment and self-assembly of nanoparticles at the liquid–liquid interface can be realized by appropriate surface coating.10 Most reagents used for mediating the self-assembly of nanoparticles were small organic molecules. For example, thiols containing alkyl chain with 2-bromopropionate terminal groups, crown ether, and C60 were used to induce the self-assembly of noble metal nanoparticles at the water–oil interface.10,14–16 The surface properties of these 2D self-assemblies were mainly dominated by the small organic molecules. Another class of capping agent, namely copolymer, may offer more versatile choices, which has been extensively applied for controlling the surface chemistry and functionality of the nanoparticles.17–23 However, the usage of copolymer for achieving self-assembly of nanoparticles at the liquid–liquid interface has not yet been reported. The diverse functionalities of copolymers may provide the self-assembled thin films of copolymer-capped nanoparticles with more versatile properties.
Recently, the interfacial behavior of amphiphilic block copolymers at the strongly selective penetrable interfaces has attracted strong interest from the aspects of the fundamental physics of polymers and the practical applications in material science and engineering.24,25 Note that the so-called selectively penetrable interface is made up of two immiscible liquids (or solvents). Each liquid is the good solvent of one block and the bad solvent of another block, respectively. When the amphiphilic block copolymers were in the environment of two such immiscible liquids, the copolymer chains will be readily entrapped at the liquid–liquid interface, as the hydrophilic and hydrophobic blocks of the copolymer chains tend to stay in the different liquids (sides) of the interface in order to minimize the free energy of the system. This special character of amphiphilic block copolymer at the liquid–liquid interface is thus thought to be usable for the interfacial assembly and entrapment of inorganic nanoparticles.
In the present work, an amphiphilic multiblock copolymer [poly(4-vinylpyridine)-b-polystyrene-b-poly(4-vinylpyridine)]n, (P4VP-PS-P4VP)n, containing trithiocarbonate moieties (–S–CS–S–),26–28 was used as capping agent to prepare Au and Ag nanoparticles as well as Au nanorods. Scheme 1 shows the chemical structure of the (P4VP-PS-P4VP)n multiblock copolymer. The number-average molecular weight Mn and polydispersity index PDI of (P4VP-PS-P4VP)n were 11.0 × 104 and 1.89, respectively.27,28 The fraction of 4VP was about 61.2 mol%. The value of n was about 4. These (P4VP-PS-P4VP)n-capped noble metal nanoparticles and nanorods can be spontaneously entrapped and hence form stable monolayer thin films at the (DMF–H2O)/diethyl ether interface without adding any other small molecules. Note that DMF–H2O is a good mixed solvent for P4VP and a nonsolvent for PS, whereas diethyl ether is a good solvent for PS and a nonsolvent for P4VP.29,30
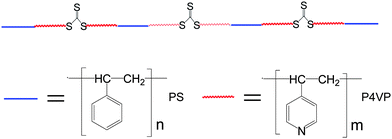 | ||
| Scheme 1 Chemical structure of (P4VP-PS-P4VP)n multiblock copolymer. | ||
Experimental
Chemical and materials
Hydrogen tetrachloroaurate (III) trihydrate (HAuCl4·3H2O), silver nitrate (AgNO3, 99%), hexadecyltrimethylammoniumbromide (CTAB, 99%) and L(+)-ascorbic acid (99%) were purchased from Acros. Sodium borohydride (NaBH4, 99%) was obtained from Aldrich. N,N-Dimethylformamide (DMF, analytical grade) was purchased from Sinopharm Chemical Reagent Co. Ltd (SCRC, China). All chemicals were used as received. The multiblock copolymer [poly(4-vinylpyridine)-b-polystyrene-b-poly(4-vinylpyridine)]n, (P4VP-PS-P4VP)n, containing trithiocarbonate moieties (–S–CS–S–) with a number-average molecular weight Mn of 11.0 × 104, polydispersity index PDI of 1.89, and 4VP fraction of 61.2 mol%, was previously synthesized.28Preparation of (P4VP-PS-P4VP)n capped Au and Ag nanoparticles in DMF
(P4VP-PS-P4VP)n-capped Au nanoparticles (Au NPs) were directly synthesized in a homogenous DMF solution in the presence of the multiblock copolymer. Typically, ∼0.566 g DMF solutions of the multiblock copolymer (4 mg ml−1, 0.1 mM) were first added under stirring into a 25 ml round-bottom flask containing 10 ml DMF solvent. ∼2.4 g of a HAuCl4 DMF solution (8 mM) was then added and stirred for 5 min. Afterwards, a NaBH4 DMF solution (16 mM, final molar ratio of NaBH4/HAuCl4 = 2.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) was added dropwise into the mixed solution under vigorous stirring for 5 min. The mixed solution was continuously vigorous stirred for 24 h. The samples were then left at least for 3 days at room temperature in order to decompose the residual NaBH4 before any further experiments.
:1) was added dropwise into the mixed solution under vigorous stirring for 5 min. The mixed solution was continuously vigorous stirred for 24 h. The samples were then left at least for 3 days at room temperature in order to decompose the residual NaBH4 before any further experiments.
A similar procedure was also used to prepare (P4VP-PS-P4VP)n-capped Ag nanoparticles (Ag NPs) in DMF. The DMF solutions of HAuCl4, AgNO3 and NaBH4 were newly prepared before use since they were unstable in air at room temperature.
Preparation of Au nanorods in DMF
The (P4VP-PS-P4VP)n-capped Au nanorods (Au NRs) were first prepared in aqueous solution according to the reported seeding method.31 The seed solutions of Au nanoparticles were freshly prepared by mixing 5.0 ml CTAB aqueous solutions (0.2 M) and 5.0 ml HAuCl4 aqueous solutions (0.5 mM) and then adding 0.6 ml ice-cold NaBH4 aqueous solutions (10 mM) under magnetic stirring for 20 min. The growth solutions were prepared by adding 5 ml HAuCl4 aqueous solutions (8 mM) into the mixtures of CTAB (32 mM, 25 ml) and AgNO3 (8mM, 0.5 ml) under magnetic stirring. Afterward, 5 ml ascorbic acid aqueous solutions (16 mM) were added as a mild reducing agent under stirring. Finally, 1 ml seed solutions were added into the growth solutions and the magnetic stirring was stopped. The growth of Au nanorods was allowed to proceed for about 24 hrs. The as-prepared CTAB-stabilized Au nanorods were then fractionalized and concentrated by centrifugation at various speeds. The as-prepared Au nanorods were first centrifuged at 9000 rpm and the precipitates were discarded. The supernatant was then centrifuged at 15000 rpm. In this case, the precipitates at 15000 rpm were collected and washed with DMF and then redispersed into the DMF solutions of the multiblock copolymer (4 mg ml−1, 0.1 mM). The multiblock copolymers readily grafted onto the Au nanorods via the coordination and complexation between the trithiocarbonate moieties (–S–CS–S–), pyridine groups and the Au atoms, stabilizing the Au nanorods. These (P4VP-PS-P4VP)n-capped Au nanorods can be stable for several months in DMF.
Self assembly of nanoparticles and nanorods at the (DMF–H2O)/diethyl ether interface
1 ml DMF solutions of nanoparticles or nanorods were added into a 25 ml clean beaker and diluted with 4 ml DMF. 1 ml of distilled water were then added and the mixtures were gently shaken. Afterward, 5 ml of diethyl ether were gently added along the wall of beaker, leading to the formation of a biphase. Finally, the biphasic mixtures were strongly stirred with a glass rod for 1 min. The nanoparticles or nanorods were slowly assembled at the (DMF–H2O)/diethyl ether interface. The mixtures with the assembled thin films of nanoparticles or nanorods were cast into a Petri dish. The assembled thin films of nanoparticles or nanorods were then floated onto carbon-coated copper grids and clean glass pieces for further characterization.Characterization of nanoparticles, nanorods, and their assembled thin films
The optical absorptions of the samples were recorded by using a Cary 100 Bio UV-vis spectrophotometer. The size and morphology of the samples were investigated by using transmission electron microscopy (TEM). FTIR spectra were recorded on a Bruker Vecter 22 FTIR spectrometer. Raman spectra were obtained on an Almega dispersive Raman microscope (Thermo Nicolet) with an argon laser. The wavelength of the argon laser was 532 nm. The TEM measurements were performed on a JEOL JEM-1200 electron microscope operating at an acceleration voltage of 60 KV. The surface morphologies of the assembled thin films were investigated by using an atomic force microscope (AFM, Seiko SPI3800N station) operated in tapping mode at room temperature in air. Silicon tips (NSG10, NT-MDT, Russia) with a resonance frequency of ca. 300 kHz were used.Results and discussion
The Au nanoparticles (Au NPs) were directly synthesized in DMF in the presence of (P4VP-PS-P4VP)n (4 mg ml−1) by the reduction of HAuCl4 with sodium borohydride. The Fourier transform infra-red (FTIR) spectrum indicates that the Au nanoparticles were capped with (P4VP-PS-P4VP)n multiblock copolymer. Fig. 1 shows the FTIR spectrum of (P4VP-PS-P4VP)n-capped Au nanoparticles. The absorption peaks at 1651 cm−1, 1630 cm−1, and 1007 cm−1 were attributed to the stretching modes of pyridine ring. It is known that the pyridine ring of P4VP has a distinct absorption peak at 1597 cm−1 and this peak will shift to a higher wavenumber due to the interaction with other atoms. It will shift to ca. 1640 cm−1 due to protonation of the nitrogen atom of pyridine ring and to ca. 1620 cm−1 due to the metal coordination.32–34 Hence, the peaks at 1651 cm−1 and 1630 cm−1may suggest that parts of the pyridine rings of P4VP blocks were coordinated with Au nanoparticles. Furthermore, the trithiocarbonate groups and sulfur atoms are well known to complex with Au and can be used as protecting agents for Au nanoparticles. However, due to the small amount of the trithiocarbonate groups contained in the multiblock copolymer, these groups can not be detected by FTIR. The peaks at 3100–3000 cm−1 and 703 cm−1 can be assigned to the aromatic rings, indicating the presence of polystyrene blocks. The properties of (P4VP-PS-P4VP)n-capped Au nanoparticles had been systematically investigated and previously reported.28 Here, we only focused on the interfacial assembly of (P4VP-PS-P4VP)n-capped Au nanoparticles at the liquid–liquid interface.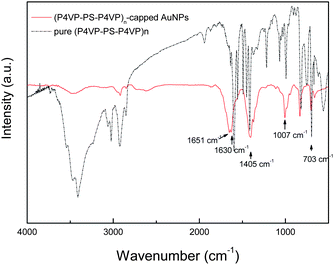 | ||
| Fig. 1 The FTIR spectra of (P4VP-PS-P4VP)n-capped Au nanoparticles and pure (P4VP-PS-P4VP)n multiblock copolymer. | ||
Typically, 1 ml DMF solutions of (P4VP-PS-P4VP)n-capped Au nanoparticles were first diluted with 4 ml DMF in a clean beaker and gently mixed with 1 ml distilled water. Note that DMF and water are fully mixable. After adding another 5 ml diethyl ether, the biphasic mixture was formed, as shown in Fig. 2A. The lower part was the DMF–H2O (5/1) phase with a wine-red color, whereas the upper part was the colorless diethyl ether phase because of the lower density of diethyl ether. The biphasic mixture was then strongly stirred with a glass rod for several tens of seconds. An interesting phenomenon was observed after the stirring was stopped. The (P4VP-PS-P4VP)n-capped Au nanoparticles were slowly assembled at the (DMF–H2O)/diethyl ether interface, as shown in Fig. 2B. Almost all of the Au NPs were entrapped at the liquid–liquid interface. No Au nanoparticle was detected in the diethyl ether phase. It is worth noting that the addition of distilled water was crucial for the interfacial entrapment and assembly. Without adding distilled water, the diethyl ether can mix with DMF to form a stable mixture of Au (P4VP-PS-P4VP)n-capped Au nanoparticles. Furthermore, the optimal amount of distilled water was found here to be 1 ml for 5 ml DMF solutions of (P4VP-PS-P4VP)n-capped Au nanoparticles. Too little or too much distilled water will not lead to self-assembly. Although the exact mechanism of distilled water is still unknown, it was thought that the addition of distilled water evoked the amphiphilic nature of (P4VP-PS-P4VP)n multiblock copolymer and changed the conformation of (P4VP-PS-P4VP)n multiblock copolymer chains in the solvents. Note that the PS block is hydrophobic, while the P4VP block is soluble in water with pH < 5.29 Because of the amphiphilic characteristic of the block copolymer, the (P4VP-PS-P4VP)n-capped Au nanoparticles can be stably dispersed in DMF and aqueous solution with pH < 5, respectively.28 Here, DMF–H2O is a good mixed solvent for P4VP and a nonsolvent for PS, whereas diethyl ether is a good solvent for PS and a nonsolvent for P4VP.29,30 The process of interfacial entrapment of the nanoparticles was mainly dominated by the surface wettability of the nanoparticles. The DMF–H2O mixture and diethyl ether hence formed a selective liquid–liquid interface for the molecular chains of (P4VP-PS-P4VP)n multiblock copolymer. The P4VP blocks tended to stay in the DMF–H2O phase, whereas PS blocks tended to penetrate into the diethyl ether phase. The (P4VP-PS-P4VP)n multiblock copolymer will form micelles with PS blocks as cores and P4VP blocks as shells in the DMF–H2O mixed solvent. Alternatively, the (P4VP-PS-P4VP)n multiblock copolymer will form inverse miclles with P4VP blocks as cores and PS blocks as shells in the DMF–diethyl ether mixed solvent. We had investigated the micelle formation of the (P4VP-PS-P4VP)n multiblock copolymers when adding water or toluene into their DMF solutions and found that the (P4VP-PS-P4VP)n multiblock copolymers formed spherical micelles in acidic water and the micelle morphologies of (P4VP-PS-P4VP)n multiblock copolymers in DMF–toluene mixed solvent depended on the block lengths of P4VP.35 Here, in the mixed solvents of DMF–H2O–diethyl ether, the molecular chains of the (P4VP-PS-P4VP)n multiblock copolymer preferred to stay at the DMF–H2O–diethyl ether interface in order to mininize the free energy of the system,24,25 leading to the interfacial entrapment of (P4VP-PS-P4VP)n-capped Au nanoparticles.
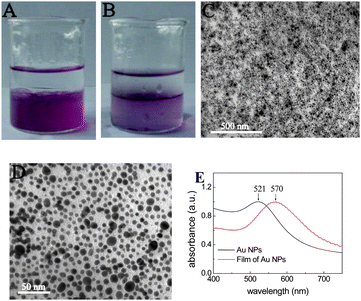 | ||
| Fig. 2 (A) The biphasic mixture with a colorless diethyl ether phase on top and a wine-red (DMF–H2O) phase containing (P4VP-PS-P4VP)n-capped Au nanoparticles below. (B) The (P4VP-PS-P4VP)n-capped Au nanoparticles were interfacially entrapped at the (DMF–H2O)/diethyl ether interface after stirring, forming a stable thin film. (C) TEM image of the thin film of (P4VP-PS-P4VP)n-capped Au nanoparticles at low magnification. (D) TEM image of the thin film of (P4VP-PS-P4VP)n-capped Au nanoparticles at higher magnification. (E) The UV-vis spectra of the thin film of (P4VP-PS-P4VP)n-capped Au nanoparticles on glass (red line) and the as-prepared Au nanoparticles in DMF solution (black line). | ||
It was also thought that the multiblock chain structure of the copolymer may play roles in the interfacial entrapment of the (P4VP-PS-P4VP)n-capped nanoparticles at the liquid–liquid interface. For the multiblock copolymer, not only the hydrophobic blocks but also the hydrophilic blocks have to form loops in the selective solvent and are hence in the confined environment.36–38 Nevertheless, Fig. 2 demonstrates that the amphiphilic multiblock copolymer can be used to tune the surface wettability of the Au nanoparticles, leading to the assembly of the nanoparticles at the liquid–liquid interface. Scheme 2 schematizes the possible mechanism of this interfacial assembly of Au nanoparticles at the selective liquid–liquid interface induced by the amphiphilic multiblock copolymer. In the common good solvent, DMF, both PS and P4VP blocks were soluble and had a random coil-like conformation. However, after adding H2O, the DMF–water mixture was the non-solvent for PS so that the PS blocks were forced to accumulate at the surface of the nanoparticles, while the P4VP block chains adopted the random coil conformation. When diethyl ether was further added into the mixed solvent, two phase structures were formed with a diethyl ether phase and a DMF–water phase. Because diethyl ether was the nonsolvent for P4VP blocks, the P4VP chains were forced to accumulate at the surface of the nanoparticles, whereas the PS blocks adopted the random coil conformation in the diethyl ether phase.
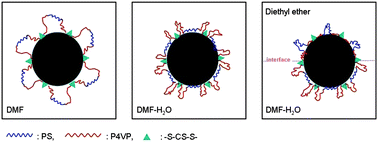 | ||
| Scheme 2 Possible mechanism of the interfacial assembly of Au nanoparticles at the (DMF–H2O)/diethyl ether interface induced by the amphiphilic multiblock copolymer | ||
The interfacial assembled thin film of (P4VP-PS-P4VP)n-capped Au NPs was transferred onto solid substrates by casting the mixture into a Petri dish. Fig. 2C and 2D show typical TEM images of the thin films of (P4VP-PS-P4VP)n-capped Au NPs. The (P4VP-PS-P4VP)n-capped Au NPs had a polydisperse size distribution with sizes of 8.8 ± 2.4 nm so that the thin films did not exhibit long-range ordering (Fig. 2D). Voids of various sizes were also observed for the thin films of (P4VP-PS-P4VP)n-capped Au NPs, which were in good agreement with those reported by Reincke et al.39 and Duan et al.10 The UV-vis spectra show a red-shift of surface plasmon resonance from 521 nm for as-prepared Au NPs in DMF solution to 570 nm for the thin film of (P4VP-PS-P4VP)n-capped Au NPs, as shown in Fig. 2E. The red-shift of surface plasmon resonance (SPR) resulted from the close contacts of Au NPs in the thin film. Lee et al. also observed the large red shift of SPR from the self-assembly of Au and Ag nanoparticles capped with functional organic molecules (crown ether or C60) at the water/oil interface.14–16 The small red shift of SPR observed here may possibly be due to the lower surface coverage of the glass slide by the thin film of (P4VP-PS-P4VP)n-capped Au NPs during the transferring process from the liquid phase to the glass substrates and to the rather poor long-range order of the nanoparticle film.
Fig. 3 shows the typical AFM images of the assembled thin film of (P4VP-PS-P4VP)n-capped Au NPs. The AFM images show that the AuNPs' thin film was rather smooth. From the phase image (Fig. 3B), it can also be clearly seen that the Au nanoparticles were indeed surrounded by the soft polymer coatings. The root-mean-square roughness of the (P4VP-PS-P4VP)n-capped Au NPs' thin film was about 2.4 nm for the measured surface area of 1 μm × 1 μm.
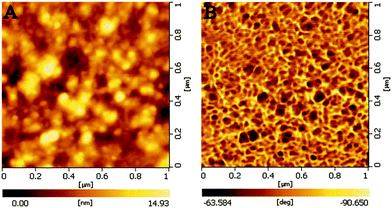 | ||
| Fig. 3 Typical AFM images of the thin film of (P4VP-PS-P4VP)n-capped Au NPs. (A) morphology image and (B) phase image. | ||
A similar procedure was also used for Ag nanoparticles (Fig. 4). The (P4VP-PS-P4VP)n-capped Ag NPs were directly prepared by the reduction of silver nitrate in DMF in the presence of (P4VP-PS-P4VP)n multiblock copolymer. However, the size and size distribution of Ag NPs in DMF can not be well controlled. The obtained (P4VP-PS-P4VP)n-capped Ag NPs had mean particle sizes of 24.9 ± 4.1 nm (Fig. 4C and 4D). Again, the (P4VP-PS-P4VP)n-capped Ag NPs can be interfacially entrapped at the (DMF–H2O)/diethyl ether interface. Interestingly, the speed of interfacial assembly was slower for (P4VP-PS-P4VP)n-capped Ag NPs than that for (P4VP-PS-P4VP)n-capped Au NPs. Due to the broad size distribution of Ag NPs, the thin film of (P4VP-PS-P4VP)n-capped Au NPs again did not exhibit long-range ordering (Fig. 4C and 4D). The interfacial assembly of (P4VP-PS-P4VP)n-capped Ag nanoparticles also exhibited enhanced surface Raman signals. Fig. 5 shows the surface-enhanced Raman scattering (SERS) spectrum of the interfacial assembly of (P4VP-PS-P4VP)n-capped Ag nanoparticles. The strong absorption peaks at 1010 cm−1, 1600 cm−1 and 3049 cm−1 indicate the presence of polystyrene and poly(4-vinylpyridine) blocks on the Ag nanoparticles.40–42 The characteristic absorption bands of aromatic and pyridine rings may overlap with each other. Fig. 6 shows the typical AFM images of the thin film of (P4VP-PS-P4VP)n-capped Ag NPs. Similarly, it can be clearly seen that the Ag nanoparticles were surrounded by soft polymer coatings from the phase image (Fig. 6B). The root-mean-square roughness of the (P4VP-PS-P4VP)n-capped Ag NPs' thin film was about 8.6 nm for the measured surface area of 1 μm × 1 μm, which was much larger than that of (P4VP-PS-P4VP)n-capped Au NPs' thin film. It is reasonable because the sizes and polydispersity of the Ag nanoparticles were much larger than those of the Au nanoparticles.
 | ||
| Fig. 4 (A) The biphasic mixture with a colorless diethyl ether phase on top and a brown (DMF–H2O) phase containing (P4VP-PS-P4VP)n-capped Ag NPs below. (B) A thin film of (P4VP-PS-P4VP)n-capped Ag NPs formed at the (DMF–H2O)/diethyl ether interface after stirring. (C) TEM image of the thin film of (P4VP-PS-P4VP)n-capped Ag NPs at low magnification. (D) TEM image of the thin film of (P4VP-PS-P4VP)n-capped Ag NPs at higher magnification. | ||
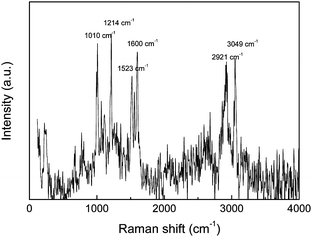 | ||
| Fig. 5 The surface-enhanced Raman scattering spectrum of interfacial assembly of (P4VP-PS-P4VP)n-capped Ag nanoparticles. | ||
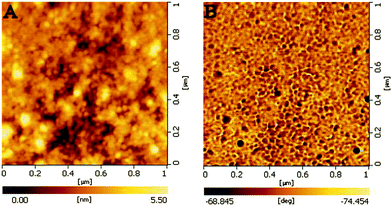 | ||
| Fig. 6 Typical AFM images of the thin film of (P4VP-PS-P4VP)n-capped Ag NPs. (A) morphology image and (B) phase image. | ||
In order to further demonstrate the universality of such a strategy and the unique capability of the multiblock copolymer, efforts were also made toward the interfacial assembly of Au nanorods capped with (P4VP-PS-P4VP)n at the (DMF–H2O)/diethyl ether interface. The attempt at the direct preparation of Au nanorods in (P4VP-PS-P4VP)n DMF solution failed. Alternatively, Au nanorods were first prepared in aqueous solution with hexadecyltrimethylammoniumbromide (CTAB) as capping agent, by using the standard protocol in the literature,31 and then transferred into the (P4VP-PS-P4VP)n DMF solution by ligand exchange. The (P4VP-PS-P4VP)n-capped Au nanorods were thus obtained. The multiblock copolymers readily grafted onto the Au nanorods via the coordination and complexation of trithiocarbonate moieties (–S–CS–S–) and pyridine groups, stabilizing the Au nanorods. These (P4VP-PS-P4VP)n-capped Au nanorods can be stable for several months in DMF. Note that the Au nanorods were unstable and precipitated in DMF without adding (P4VP-PS-P4VP)n. The aspect ratio of the Au nanorods was about 3 with length of 39.5 ± 3.6 nm and thickness of 12.5 ± 1.0 nm. Similarly, the (P4VP-PS-P4VP)n-capped Au nanorods exhibited the capability of forming interfacial assembly at the (DMF–H2O)/diethyl ether interface, as shown in Fig. 7. Due to the various orientations of Au nanorods and the coexistence of Au nanoparticles, large voids were observed for the thin film of the (P4VP-PS-P4VP)n-capped Au nanorods (Fig. 7C and 7D).
 | ||
| Fig. 7 (A) The biphasic mixture with colorless diethyl ether phase on top and the violet (DMF–H2O) phase containing (P4VP-PS-P4VP)n-capped Au nanorods below. (B) A thin film of (P4VP-PS-P4VP)n-capped Au nanorods formed at the (DMF–H2O)/diethyl ether interface after stirring. (C) TEM image of the thin film of (P4VP-PS-P4VP)n-capped Au nanorods at low magnification. (D) TEM image of the thin film of (P4VP-PS-P4VP)n-capped Au nanorods at higher magnification. | ||
Conclusion
The amphiphilic multiblock copolymer, [poly(4-vinylpyridine)-b-polystyrene-b-poly(4-vinylpyridine)]n (P4VP-PS-P4VP)n bearing trithiocarbonate moieties (–S–CS–S–), can be used as a capping agent for fabricating functional copolymer-capped noble metal nanoparticles and nanorods. When these functional nanoparticles and nanorods were introduced into a mixture of two immiscible and strongly selective solvents for (P4VP-PS-P4VP)n, namely (DMF–H2O)/diethyl ether, the (P4VP-PS-P4VP)n-capped nanoparticles and nanorods can be interfacially entrapped and hence form stable monolayer thin films at the (DMF–H2O)/diethyl ether interface. The assembled thin film of (P4VP-PS-P4VP)n-capped Ag nanoparticles also exhibited enhanced surface Raman signal. These experimental results indicated that the amphiphilic multiblock copolymers are promising for the interfacial assembly and entrapment of inorganic nanoparticles at the proper liquid–liquid interface. With the various types and diverse functionalities of monomers, the usage of copolymers may provide the self-assembled films of nanoparticles/nanorods with more versatile properties. This provides a new possibility for the fabrication of self-assembled 2D nanostructures with designable functionalities.Acknowledgements
The authors thank the National Natural Science Foundation of China (No. 20604022, 20874087, 20774079) and 863 project (No. 2009AA04Z125) for financial support.Notes and references
- A. P. Alivisatos, Science, 1996, 271, 933 CrossRef CAS.
- C. B. Murray, C. R. Kagan and M. G. Bawendi, Annu. Rev. Mater. Sci., 2000, 30, 545 CrossRef CAS.
- F. X. Redl, K. S. Cho, C. B. Murray and S. O'Brien, Nature, 2003, 423, 968 CrossRef CAS.
- E. V. Shevchenko, D. V. Talapin, A. L. Rogach, A. Kornowski, M. Haase and H. Weller, J. Am. Chem. Soc., 2002, 124, 11480 CrossRef CAS.
- J. H. Fendler, Curr. Opin. Colloid Interface Sci., 1996, 1, 202 CAS.
- J. Shan, H. Chen, M. Nuopponen, T. Viitala, H. Jiang, J. Peltonen, E. Kauppinen and H. Tenhu, Langmuir, 2006, 22, 794 CrossRef CAS.
- Y. Lin, H. Skaff, T. Emrich, A. D. Dinsmore and T. P. Russell, Science, 2003, 299, 226 CrossRef CAS.
- L. L. Dai, R. Sharma and C. Wu, Langmuir, 2005, 21, 2641 CrossRef CAS.
- A. Kumar, S. Mandal, S. P. Mathew, P. R. Selvakannan, A. B. Mandale, R. V. Chaudhari and M. Sastry, Langmuir, 2002, 18, 6478 CrossRef CAS.
- H. Duan, D. Wang, D. G. Kurth and H. Möhwald, Angew. Chem., Int. Ed., 2004, 43, 5639 CrossRef CAS.
- K. Y. Lee, M. Kim, J. Hahn, J. S. Suh, I. Lee, K. Kim and S. W. Han, Langmuir, 2006, 22, 1817 CrossRef CAS.
- H. Xia and D. Wang, Adv. Mater., 2008, 20, 4253 CrossRef CAS.
- Y.-K. Park and S. Park, Chem. Mater., 2008, 20, 2388 CrossRef CAS.
- K. Y. Lee, G. W. Cheong and S. W. Han, Colloids Surf., A, 2006, 275, 79 CrossRef CAS.
- K. Y. Lee, Y. Bae, M. Kim, G. W. Cheong, J. Kim, S. S. Lee and S. W. Han, Thin Solid Films, 2006, 515, 2049 CrossRef CAS.
- K. Y. Lee, M. Kim, S. S. Kwon and S. W. Han, Mater. Lett., 2006, 60, 1622 CrossRef CAS.
- T. Sakai and P. Alexandridis, J. Phys. Chem. B, 2005, 109, 7766 CrossRef CAS.
- K. H. Bae, S. H. Choi, S. Y. Park, Y. Lee and T. G. Park, Langmuir, 2006, 22, 6380 CrossRef CAS.
- J. Raula, J. Shan, M. Nuopponen, A. Niskanen, H. Jiang, E. I. Kauppinen and H. Tenhu, Langmuir, 2003, 19, 3499 CrossRef CAS.
- K. Ohno, K. Koh, Y. Tsujii and T. Fukuda, Angew. Chem., Int. Ed., 2003, 42, 2751 CrossRef CAS.
- L. Guo, J. Nie, B. Du, Z. Peng, B. Tesche and K. Kleinermanns, J. Colloid Interface Sci., 2008, 319, 175 CrossRef CAS.
- A. B. Lowe, B. S. Sumerlin, M. S. Donovan and C. L. McCormick, J. Am. Chem. Soc., 2002, 124, 11562 CrossRef CAS.
- M. Q. Zhu, L. Q. Wang, G. J. Exarhos and A. D. Q. Li, J. Am. Chem. Soc., 2004, 126, 2656 CrossRef CAS.
- A. Corsi, A. Milchev, V. G. Rostiashvili and T. A. Vilgis, Macromolecules, 2006, 39, 1234 CrossRef CAS.
- A. Corsi, A. Milchev, V. G. Rostiashvili and T. A. Vilgis, J. Chem. Phys., 2005, 122, 094907 CrossRef.
- R. T. A. Mayadunne, E. Rizzardo, J. Chiefari, J. Krstina, G. Moad, A. Postma and S. H. Thang, Macromolecules, 2000, 33, 243 CrossRef CAS.
- L. Zhang, Q. Wang, P. Lei, X. Wang, C. Wang and L. Cai, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 2617 CrossRef CAS.
- B. Du, B. Zhao, P. Tao, K. Yin, P. Lei and Q. Wang, Colloids Surf., A, 2008, 317, 194 CrossRef CAS.
- H. Shen, L. Zhang and A. Eisenberg, J. Am. Chem. Soc., 1999, 121, 2728 CrossRef CAS.
- S. L. N. Seung and R. N. Young, Polym. Bull., 1979, 1, 481 CAS.
- B. Nikoobakht and M. A. El-Sayed, Chem. Mater., 2003, 15, 1957 CrossRef CAS.
- J. Ruokolainen, G. ten Brinke, O. Ikkala, M. Torkkeli and R. Serimaa, Macromolecules, 1996, 29, 3409 CrossRef CAS.
- J. Ruokolainen, J. Tanner, G. ten Brinke, O. Ikkala, M. Torkkeli and R. Serimaa, Macromolecules, 1995, 28, 7779 CrossRef CAS.
- J. Ruokolainen, M. Torkkeli, R. Serimaa, S. Vahvaselka, M. Saariaho, G. ten Brinke and O. Ikkala, Macromolecules, 1996, 29, 6621 CrossRef CAS.
- B. Du, A. Mei, Y. Yang, Q. Wang and J. Xu, Acta Polym. Sin., 2009, 9, 723 Search PubMed.
- A. Halperin, Macromolecules, 1991, 24, 1418 CrossRef CAS.
- N. Sommerdijk, S. J. Holder, R. C. Hiorns, R. G. Jones and R. J. M. Nolte, Macromolecules, 2000, 33, 8289 CrossRef CAS.
- V. Hugouvieux, M. A. V. Axelos and M. Kolb, Macromolecules, 2009, 42, 392 CrossRef CAS.
- F. Reincke, S. G. Hickey, W. K. Kegel and D. Wanmaekelbergh, Angew. Chem., Int. Ed., 2004, 43, 458 CrossRef CAS.
- A. Palm, J. Phys. Chem., 1951, 55, 1320 CrossRef CAS.
- J. Zhang, Y. Gao, R. A. Alvarez-Puebla, J. M. Buriak and H. Fenniri, Adv. Mater., 2006, 18, 3233 CrossRef CAS.
- J. R. Anema, A. G. Brolo, A. Felten and C. Bittencourt, J. Raman Spectrosc., 2009, 41, 745.
| This journal is © The Royal Society of Chemistry 2010 |