Converting carbon nanofibers to carbon nanoneedles: catalyst splitting and reverse motion
Jia
Yun
a,
Rui
Wang
b,
M. H.
Hong
b,
J. T. L.
Thong
ab,
Y. L.
Foo
c,
C. V.
Thompson
ad and
W. K.
Choi
*ab
aAdvanced Materials for Micro- and Nano-Systems Programme, Singapore–MIT Alliance, Singapore 117576, Singapore. E-mail: elechoi@nus.edu.sg; Fax: +65-6779-1103; Tel: +65-6516-6473
bDepartment of Electrical and Computer Engineering, National University of Singapore, Singapore 117576, Singapore
cInstitute of Materials Research and Engineering (IMRE), Agency of Science Technology and Research (A*STAR), Singapore 117602, Singapore
dDepartment of Materials Science and Engineering, Massachusetts Institute of Technology, Massachusetts 02139, USA
First published on 10th August 2010
Abstract
Carbon nanoneedles (CNNs) were grown using a plasma-enhanced chemical vapor deposition process in which the source gas (C2H2) was turned off 10 min before the NH3 flow and plasma were turned off. It is demonstrated that tubular carbon nanofibers (CNFs) grow while the source gas is on. However, once the source gas is turned off, the Ni catalyst at the top of each CNF splits to form a small catalyst that remains at the top of the tube and a larger catalyst that travels down the interior of the tube. We postulate that the motion of the bottom (larger) catalyst is driven by etching of the graphitic walls and ‘cups’ inside the CNF. This process, combined with slowing growth of the CNFs and etching of the material above the bottom catalyst, converts the carbon nanofibers to the final nanoneedle shape.
1. Introduction
Carbon nanotubes (CNTs) are promising candidates for a wide range of applications due to their good electrical,1 thermal2 and mechanical properties.3 Chemical vapor deposition (CVD) is commonly used for synthesis of CNTs. Of the various CVD methods available, plasma-enhanced chemical vapor deposition (PECVD) is widely used to grow aligned CNTs. It should be noted, however, that a different type of carbon nanostructure, carbon nanofibers (CNFs), with unique structures and properties can also be synthesized using this method.4,5 The growth of carbon nanotubes is catalyzed by particles of materials such as Fe and Ni, and the size and location of these catalyst particles control the size and location of the resulting tubes. The most common technique for creating metallic nanoparticle catalysts over a wide area is to simply allow dewetting of thin solid metal films on a flat substrate.4 Inevitably, the nanotubes that are subsequently grown exhibit a very wide distribution of size and spacing, due to the poor control of the size and spacing of the catalysts resulting from the simple dewetting method. To exercise good control over the size and location of metal catalysts for the growth of CNTs, various patterning techniques have been used, including: electron beam lithography,6 nanosphere lithography,7 nanoimprint lithography8 and the use of porous anodized aluminum oxide templates.9,10 Among these techniques, some provide very good control of the catalyst geometry and location, but are costly and provide only small-area coverage (e.g. electron beam lithography). Others provide relatively large-area coverage, but suffer from restrictions on the catalyst geometry and location (e.g. nanosphere lithography and anodized aluminum oxide templates) or they involve relatively complicated processes to change the geometry parameters (e.g. new masks for different spacings in the case of nanoimprint lithography). On the other hand, there is research focussed on the preparation of nanostructure arrays using masks obtained from lithographic techniques and catalytic etching,11,12 self-organized masks and without any masks, via plasma-controlled self-organization.13,14We have recently15 developed a patterning technique that combines ‘top-down’ interference lithography (IL) and ‘bottom-up’ dewetting of Au or Ni particles to control particle location and to tune the diameter of the particles. We have used this technique to grow arrays of carbon nanofiber (CNF) needles with locations, diameters, and aspect ratios that are precisely controlled on silicon substrates. We were able to carry out a systematic study of the field-emission properties of the CNFs as a function of the geometric characteristics of the CNFs themselves and of the CNF array.16 While synthesizing the CNFs for the field-emission study, we noticed various intriguing phenomena that we believe have not been reported in the literature. These observations include: a reproducible method to synthesize either normal (round-topped tubular) CNFs or sharp-tipped carbon nanoneedles (CNNs), and an interesting movement of the Ni catalyst that is unique to our method of synthesis. In this paper we present the results of a detailed study of the growth mechanism of CNNs. We examine the influence of: the plasma power, the composition of the C2H2 and NH3 in our PECVD system, and variations in the presence of both the growth gases and the plasma on the geometry of the CNFs. We also show that catalyst splitting and reverse motion controllably produces CNNs from CNFs. From these results, we develop a mechanistic understanding that provides guidance on how to synthesize normal or sharp-tipped CNFs using PECVD.
2. Experimental methods
To pattern the catalyst we used a technique reported earlier that combines top-down and bottom-up approaches to provide precise in situ production and placement of metallic nanoparticles on a silicon surface.15 The top-down component of this method uses interference lithography17 (IL), while the bottom-up process involves agglomeration (dewetting) of solid metallic thin films18 (AF). This method, denoted here as IL-AF, allows positioning of metal nanoparticles at predetermined locations on a silicon surface and, further, allows accurate control the size of the nanoparticles. In the present work, we adopted this approach with slight improvements.
Fig. 1 outlines the procedure for synthesis of large-area CNF arrays on Si substrates using the IL-AF method and PECVD growth with Ni catalysts. First, a 200 nm layer of photoresist (Ultra-i 123) was spin-coated on an n-type Si (100) wafer and baked at 90 °C for 90 s. The photoresist was then patterned using a Lloyd's mirror type IL set-up with a He-Cd laser source (λ = 325 nm). The creation of periodic square patterns in the photoresist was achieved using two perpendicular exposures of 200 s each. The unexposed photoresist was etched away using Shipley Microposit MF CD-26 developer, leaving behind an array of circular-shaped photoresist dots on the Si wafer surface (see Fig. 1(a)). Next, a 20 nm thick Cr layer was evaporated onto the entire sample using an e-beam evaporator. The Cr layer on top of the photoresist was removed by lifting-off the photoresist. The photoresist lift-off and KOH anisotropic etching of the Si were achieved simultaneously by dipping the samples in a 30 wt% KOH solution at 50 °C for around 300 s (see Fig. 1(c)). The Cr served as an etching hard mask, so that beneath the holes in the Cr, arrays of 4-fold symmetric inverted pyramids were produced. We then used the Cr as a deposition mask to evaporate a 10 nm silicon oxide layer and a 30 nm Ni film (at a base pressure of 10−6 Torr) onto the patterned sample. This resulted in an oxide layer that was sandwiched between the silicon substrate and the Ni film in the pits (Fig. 1(d)). The oxide layer serves as a diffusion barrier to prevent nickel silicide formation during the growth of CNFs. Finally, the Cr, oxide, and Ni layers above were lifted off using Chromium Cermet Etchant, revealing Ni in the centers of the inverted silicon pyramids. The period of the inverted pyramid pattern was 500 nm and the Ni in the pits was around 200 nm in size. These samples were then used for the growth of large-area arrays of isolated vertically aligned CNFs synthesized using the PECVD technique. The CNF growth conditions were fixed at 700 °C at a pressure of 7.5 mbar in a 5![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) :1 mixture of NH3 and C2H2 (NH3: 50 sccm and C2H2: 10 sccm) with Ar as the plasma source and the dc voltage fixed at 462 V. We varied the plasma current such that a half and full plasma power were obtained with the plasma current fixed at 37.5 and 75 mA, respectively. We controlled the length of the CNFs by varying the time of the flow of the NH3 and C2H2 gases into the PECVD system. This technique enabled growth of single CNFs in most pits.
:1 mixture of NH3 and C2H2 (NH3: 50 sccm and C2H2: 10 sccm) with Ar as the plasma source and the dc voltage fixed at 462 V. We varied the plasma current such that a half and full plasma power were obtained with the plasma current fixed at 37.5 and 75 mA, respectively. We controlled the length of the CNFs by varying the time of the flow of the NH3 and C2H2 gases into the PECVD system. This technique enabled growth of single CNFs in most pits.
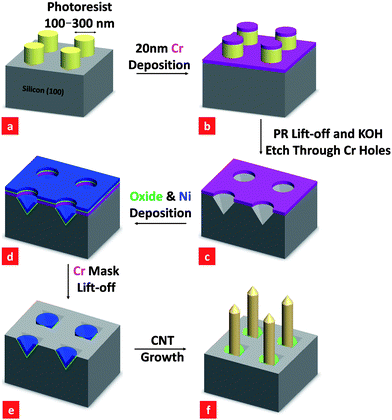 | ||
| Fig. 1 Process flow for the synthesis of arrays of carbon nanofibers (CNFs) using Ni nanoparticles as catalysts. Appropriate scanning electron micrographs are shown in Fig. 2. | ||
3. Results and discussion
Fig. 2(a) shows a scanning electron micrograph of a large-area array of isolated CNFs synthesized using the process described above. As pointed out in the previous section, with our patterning technique, even though the catalysts are initially different from the usual spherical-shaped catalysts commonly reported in the literature, we still obtained normal tubular-shaped CNFs with spherical-shaped Ni catalysts at the tip of the CNFs, as shown in the Fig 2(a). A high-resolution transmission electron micrograph of an individual tubular CNF (see, Fig 2(b)) exhibits a ‘cup-in-cup’ feature that is indicative of graphitic cups and walls. Fig. 2(d), (e) and (f) show EDX results obtained at the bottom, the top, and far away from a CNF, as shown in Fig. 2(c). These data show no Ni at the bottom of the CNF, a strong Ni signal at the top of the CNF, and only a carbon signal far away from the CNF. This is in agreement with the normal CNT tip growth mechanism commonly reported in the literature.4,6,9,10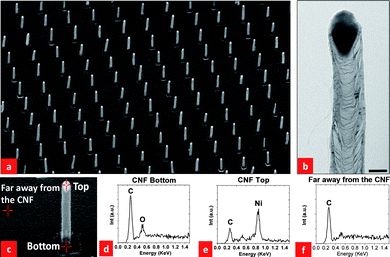 | ||
| Fig. 2 (a) An SEM image of a CNF array grown at 700 °C at a pressure of 7.5 mbar in a 5:1 mixture of NH3 and C2H2 gases (NH3: 50 sccm and C2H2: 10 sccm). Note that the SEM image was taken at a 45° tilted angle. The scale bar is 1 µm; (b) HRTEM image of a CNF, the scale bar is 50 nm; (c) locations at which EDX analyses were carried out; (d) EDX signal at the CNF bottom; (e) EDX signal at the CNF top; (f) EDX signal far away from the CNF. | ||
Fig. 3 shows SEM images of CNNs synthesized in the same PECVD system as that used to produce the CNFs in Fig. 2. The growth conditions were generally the same as those for Fig. 2, except that the CNNs were synthesized by keeping the heater and dc plasma on for another 10 min after shutting off the C2H2 supply.
 | ||
| Fig. 3 A large-area ordered array of carbon nanoneedles. The diameter of each CNN at the base was ∼200 nm, the length was ∼1 µm (45° tilt). The inset is a magnified view of a portion of the CNF array, with a scale bar of 500 nm. | ||
Fig. 4(a) and (b) show TEM images of one CNN. Lattice fringes can be seen in the image of the Ni catalyst in Fig. 4(b), but fringes cannot be seen in the CNN. This indicates that the CNN was amorphous near the tip. We have also carried out EDX measurements focused at the bottom of the CNNs, as shown in Fig. 4(c). The EDX experiments show a strong signal for Ni at the bottom of the CNNs, indicating the presence of a Ni catalyst. This is in contrast with the EDX results for the tubular CNFs in Fig. 2, for which no Ni signal was found at the bottom of the CNFs. This indicates that the growth mechanism of CNNs is different from that of tubular CNFs, and is distinct from the common observation of either a tip or base catalytic growth. It should also be noted that the limited further growth and the formation of the sharp CNN tips may be related to the decreasing supply of C2H2 after it is shut off in the last 10 min of the CNN growth process. We have also carried out EELS experiments to probe the chemical composition of the CNNs. As the CNNs were formed with only the supply of C2H2 eliminated, we expected species containing nitrogen to exist in the environment during growth and to be incorporated into the CNNs. We have, in fact, detected nitrogen in some of the CNNs. It should be noted, however, that nitrogen and nitrogen compounds would be volatile during growth and this might explain the fact that we did not manage to detect nitrogen in some other CNNs.
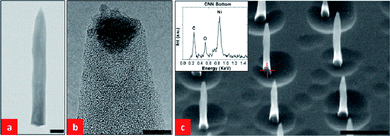 | ||
| Fig. 4 (a) TEM image of a CNN. The scale bar is 100 nm; (b) HRTEM image of the tip of the CNN shown in (a), the scale bar is 5 nm. (c) SEM image (45° tilt) of part of a CNN array with CNN lengths of 1 µm and base diameters of 100 nm, the scale bar is 1 µm. The inset is an EDX signal obtained from the bottom of one of the CNNs. | ||
We next experimented with a sample of tubular CNFs which were subjected to a treatment in NH3 with the plasma turned on (plasma current fixed at 75 mA) at 700 °C for 5 min. Fig. 5(a) and (b) show SEM images of samples before and after this treatment. Fig. 5 (c) and (d) show SEM images of CNFs that were subjected to a treatment in NH3 at 700 °C without the dc plasma turned on. Comparing the SEM images of Fig. 5(b) and (d), we note that with the plasma turned on, the CNFs have been converted to shorter CNNs. This shows that the plasma has a pronounced effect on the sharpening of the tips of the CNFs. Another interesting observation is that the original top Ni catalysts (in Fig. 5(a)) seem to have moved downwards inside the CNNs. It can be seen from Fig. 5(c) and (d) that without the plasma turned on, there appears to be no evolution of the CNFs in NH3 at 700 °C. Also, in this case, all the Ni catalysts remain at the top of the CNFs.
 | ||
| Fig. 5 (a) and (c) SEM images of as-grown tubular CNFs, (b) the same tubular CNFs heated in NH3 at 700 °C for 5 min with the plasma turned on at a current flow of 75 mA, and (d) the same tubular CNFs as in (c) heated in NH3 at 700 °C for 5 min without the plasma turned on. The scale bars in (a) and (b) are 600 nm and in (c) and (d) are 300 nm. | ||
It is important to point out the key difference in the growth process that leads to the CNNs with sharp tips in Figs. 4(b) and 5(b). In Fig. 4(b), the formation of sharp-tipped CNNs could be due to: (i) the dwindling supply of C2H2, and, (ii) etching of the CNFs in the presence of NH3 with the plasma turned on. As the CNNs in Fig. 4(b) continue to grow, albeit with shaper tips, the etching process must be less efficient than the growth at the top of the CNNs. The growth environment in the case shown in Fig. 5(b) was completely devoid of C2H2, and therefore only etching of the CNFs took place.
We will discuss the mechanism by which the top Ni catalysts move down into the CNNs in Fig. 5(b) by investigating the effect of plasma power on CNN formation. Fig. 6 shows HRTEM images of a CNN that had been subjected to a plasma treatment with the plasma current fixed at 75 mA in NH3 for 3 min. Fig. 6(a) shows that the top catalyst splits into two parts, with one remaining at the tip and the other travelling downward inside the CNN (we will call this the “body catalyst”). As the body catalyst moves downwards inside the CNN, two distinct regions develop in the CNN above and below it. As shown in Fig. 6(b), the region below the body catalyst retains the cup-in-cup structure of the original CNF. However, there is a striking reduction in the number of cup-shaped features in the region above the body catalyst, with only an amorphous sheath remaining to define the CNN wall. Fig. 6(c) shows a tail on the top Ni catalyst. This suggests that a pinch-off process is occurring, and that a process like this might have led to the formation of the larger body catalyst, presumably as a result of the splitting of the original top catalyst into two parts.
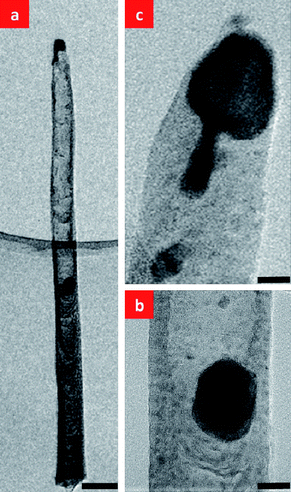 | ||
| Fig. 6 (a) HRTEM image of a CNF that was subjected to a plasma treatment (plasma current fixed at 75 mA) in NH3 for 3 min after the C2H2 was turned off, showing that the top Ni catalyst splits into two parts, with one remaining at the tip of the CNF and the other travelling down into the CNF. Note the hollow interior of the upper part of the CNF through which the bottom catalyst has passed, (b) magnified image of the body catalyst, and (c) magnified image of the top catalyst. The scale bars are (a) 100 nm, (b) 25 nm, and (c) 25 nm, respectively. | ||
Carbon nanoneedles similar in appearance and structure to those we have observed have been observed previously.19 However, catalyst splitting has not been observed previously, so an associated mechanism has not been presented. A model for the mechanism of formation of single-crystal carbon cones composed of planar stacked graphene sheets has been discussed by Denysenko et al.,20 who considered the effects of ion-assisted precursor dissociation and surface diffusion. However, the mechanism we have observed is clearly different, in that our fibers have cup structures, and cone formation occurs when only the plasma is on but no precursor gas is present. However, there may be similar roles for plasma-assisted etching and diffusion in our case.
Helveg et al. carried out TEM experiments in which in situ observations of CNF growth were made.21 They observed a liquid-like oscillation in the shape of the Ni catalysts at the CNF tips, and found that this oscillation was associated with the formation of cup-in-cup structures. They demonstrated that these shape oscillations occurred even though the Ni remained crystalline, and argued that the Ni catalysts elongated as they wet the tube at both the top cap and the cup. The energy of the Ni/graphene surface held them in contact until the energy cost of the increased interface area exceeded the cost of breaking the contact between the Ni and the bottom cap. We suggest that when the C2H2 is turned off, but the NH3 and plasma are left on, the Ni particle etches the cup at the bottom, and that this etching drives the continued extension of the Ni catalyst until it pinches-off to form two particles (see the schematic illustration in Fig.7). A Ni particle (usually a smaller one) remains at the tip to continue to wet the top cap.
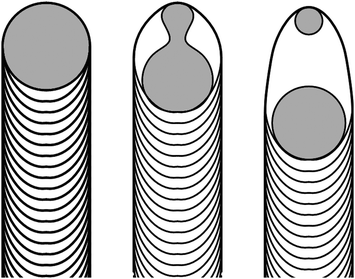 | ||
| Fig. 7 Schematic diagram of the mechanism responsible for the Ni catalyst splitting and the reverse motion observed in CNNs. | ||
In this mechanism, once the C2H2 flow is eliminated (but the NH3 and plasma remain on), the Ni catalyzes etching of the graphitic walls inside the tube structure. The mechanism for etching would be similar to that discussed in refs. 22 and 23, in which the Ni catalyzes the reaction of NH3 with the graphene to form volatile hydrocarbon products.
We further prepared three samples with tubular CNFs and subjected them to different plasma powers and durations at 700 °C in ambient NH3. Fig. 8 shows SEM images of the as-grown CNFs and the three sets of CNNs subjected to the different plasma powers and durations.
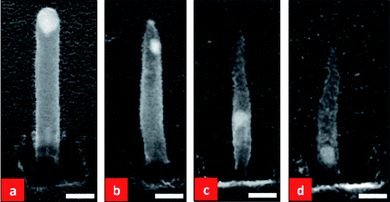 | ||
| Fig. 8 SEM images of: (a) an as-grown round-topped tubular CNF that has been annealed at 700 °C in ambient NH3 without plasma treatment. CNFs annealed under the same conditions but with (b) the plasma power set at half the normal power (plasma current fixed at 37.5 mA) for 3 min, (c) full plasma power (plasma current fixed at 75 mA) for 3 min and (d) full plasma power for 5 min. The scale bars are 100 nm for all images. | ||
If one compares Fig. 8(b) and (c), as is reasonable to expect, more significant etching of the CNNs has occurred at the higher plasma powers in (c) than (b) for a fixed duration of 3 min. With a fixed plasma power (fixing the plasma current a 75 mA), an increase in the time of the plasma from 3 to 5 min also resulted in more etching of the CNFs, as shown in Fig. 7 (c) and (d). It is also clear from Fig 8 that when the plasma power was at half and the treatment time was 3 min (Fig. 8(b)), the catalyst broke into two particles, with one particle being relatively small and remaining at the top of the CNN (the top catalyst) and the other one being bigger and starting to migrate downward into the body of the CNN (the body catalyst). By increasing the plasma to full power for 3 min (Fig. 8(c)), the top catalyst still remained at the top while the body catalyst reached the middle of the CNN. Keeping the plasma power at full power (i.e. keeping the plasma current fixed at 75 mA) and increasing the treatment time to 5 min, the body catalyst almost arrives at the bottom of the CNF (Fig. 8(d)). This is almost the situation shown in Fig. 3. Hence, we conclude that at higher plasma powers and/or for longer plasma treatment times, the top catalyst becomes smaller while the body catalyst continuously migrates downward through the CNN. Note that when the carbon source is turned off with the plasma on, carbon can be etched and re-deposited on the CNN24 resulting in the shape change in Fig. 8. With a higher plasma power for a longer duration, the etching of carbon becomes increasingly pronounced. Therefore, the CNNs become shorter and thinner and the tips becomes sharper, as a result of prolonged plasma etching. Sharp tips are crucial for optimal field emission. It is also clear from Fig. 8 that the sizes of the top and body catalysts of a CNN are closely related to the plasma power and duration used for the growth process. Typically, the top catalyst is less than 10 nm and the body catalyst depends on the initial catalyst size and is of the order of tens of nm.
We have carried out a set of experiments with CNNs that have body catalysts embedded approximately midway down the CNNs. We placed these samples in the PECVD system and re-started the growth process at the original CNF growth conditions (i.e. at 700 °C at a pressure of 7.5 mbar in a 5:1 mixture of NH3 and C2H2 (NH3: 50 sccm and C2H2: 10 sccm)). Fig. 9(a) and (b) show representative SEM images of the samples before and after the second growth. It is interesting to note that under our standard growth conditions, the body catalyst manages to “break through” the side wall of the CNN and branch out to form a second CNF, with the body catalyst now being the top catalyst of the new branching CNF. The original CNN remains the same except that it becomes slightly thicker due to carbon deposition on its side wall. This branching was observed in almost all the CNNs. Branching, instead of reversible growth, might be the result of the needle-like shape of the tube above the body catalyst.
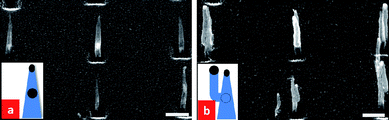 | ||
| Fig. 9 SEM images of: (a) as-grown CNNs, and (b) the same CNNs subjected to a second growth which causes branching to form a second tube. The second growth was carried out at 700 °C at a pressure of 7.5 mbar in a 5:1 mixture of NH3 and C2H2 (NH3: 50 sccm and C2H2: 10 sccm). | ||
It should be noted that, in a number of the cases described above, the CNFs or the CNNs were exposed to an ambient atmosphere (e.g. for SEM examination) and subsequently placed back in the PECVD system for further processing. We have carried out a set of experiments to confirm that the conversion of CNFs to CNNs is reproducible and is not due to extraneous phenomena resulting from exposure to air. We carried out the normal CNF growth for 3 min under our standard growth conditions, after which we maintained the flow of NH3 but purged the remaining C2H2 in the PECVD system and let the system cool down to room temperature. The PECVD system was then again set at 700 °C with the plasma turned on with a constant supply of NH3 throughout. We obtained very similar CNNs to those shown in Fig. 3. This established that the conversion of CNFs to CNNs is not related to exposure to air.
There exists a wide range of potential applications for the CNN and CNF arrays discussed in this work. For example: similar structures have been used as enzymatic glucose biosensors25, they are a candidate for making sharp probes for scanning probe nanolithography, and they could be used as building blocks for field-emitting tips or photodetectors in electronic/optoelectronic applications.
4. Conclusions
In summary, we have examined the effects of plasma power, temperature and ambient gas on the formation of CNFs and CNNs during PECVD growth and plasma-enhanced etching. We find that nanoneedles form during etching when the Ni catalysts at the top of the CNFs split into two particles. One particle remains at the top of the CNF, and continues to wet the top cap, while the other, larger, particle travels down the interior of the CNF to etch graphitic walls and cups.Acknowledgements
The authors would like to acknowledge financial support from the Singapore-MIT Alliance. YJ would like to thank the Singapore–MIT Alliance for the provision of a fellowship and WR would like to thank the National University of Singapore for the provision of research scholarship.References
- S. Fan, M. G. Chapline, N. M. Franklin, T. W. Tombler, A. M. Cassell and H. Dai, Science, 1999, 283, 512 CrossRef CAS.
- Y. Xu, Y. Zhang, E. Suhir and X. W. Wang, J. Appl. Phys., 2006, 100, 074302 CrossRef.
- M. J. Treacy, T. W. Ebbesen and J. M. Gibson, Nature, 1996, 381, 678 CrossRef CAS.
- M. Chhowalla, K. B. K. Teo, C. Ducati, N. L. Rupesinghe, G. A. Amaratunga, A. C. Ferrari, D. Roy, J. Robertson and W. I. J. Milne, J. Appl. Phys., 2001, 90, 5308 CrossRef CAS.
- A. V. Melechko, V. I. Merkulov, T. E. McKnight, M. A. Guillorn, K. L. Klein, D. H. Lowndes and M. L. Simpson, J. Appl. Phys., 2005, 97, 041301 CrossRef.
- K. B. K. Teo, M. Chhowalla, G. A. Amaratunga, W. I. Milne, G. Pirio, P. Legagneux, F. Wyczisk, D. Pribat and D. G. Hasko, Appl. Phys. Lett., 2002, 80, 2011 CrossRef CAS.
- K. H. Park, S. Lee, K. H. Koh, R. Lacerda, K. B. K. Teo and W. I. J. Milne, J. Appl. Phys., 2005, 97, 024311 CrossRef.
- S. M. C. Vieira, K. B. K. Teo, W. I. Milne, O. Groning, L. Gangloff, E. Minoux and P. Legagneux, Appl. Phys. Lett., 2006, 89, 022111 CrossRef.
- J. S. Suh, K. S. Jeong, J. S. Lee and I. Han, Appl. Phys. Lett., 2002, 80, 2392 CrossRef CAS.
- S. M. Yoon, J. Chae and J. S. Suh, Appl. Phys. Lett., 2004, 84, 825 CrossRef CAS.
- W. K. Choi, T. H. Liew, M. K. Dawood, H. I. Smith, C. V. Thompson and M. H. Hong, Nano Lett., 2008, 8, 3799 CrossRef CAS.
- M. K. Dawood, T. H. Liew, P. Lianto, M. H. Hong, S. Tripathy, J. L. T. Thong and W. K. Choi, Nanotechnology, 2010, 21, 205305 CrossRef CAS.
- N. Vourdas, D. Kontziampasis, G. Kokoris, V. Constantodis, A. Goodyear, A. Tserepi, M. Cooke and E. Gogolides, Nanotechnology, 2010, 21, 085302 CrossRef CAS.
- Z. L. Tsakadze, I. Levchenko, K. Ostrikov and S. Xu, Carbon, 2007, 45, 2022 CrossRef.
- W. K. Choi, T. H. Liew, H. G. Chew, F. Zheng, C. V. Thompson, Y. Wang, X. D. Wang and J. Yun, Small, 2008, 4, 330 CrossRef CAS.
- J. Yun, R. Wang, W. K. Choi, J. T. L. Thong, C. V. Thompson, M. Zhu, Y. L. Foo and M. H. Hong, Carbon, 2010, 48, 1362 CrossRef CAS.
- M. Walsh, PhD thesis, MIT, 2004.
- A. L. Giermann and C. V. Thompson, Appl. Phys. Lett., 2005, 86, 121903 CrossRef.
- I. Levchenko, K. Ostrikov, J. D. Long and S. Xu, Appl. Phys. Lett., 2007, 91, 113115 CrossRef.
- I. Denysenko and K. Ostrikov, Appl. Phys. Lett., 2007, 90, 251501 CrossRef.
- S. Helveg, C. Lopez-Cartes, J. Sehested, P. L. Hansen, B. S. Clausen, J. R. Rostrup-Nielsen, F. Abild-Pedersen and J. K. Nørskov, Nature, 2004, 427, 426 CrossRef CAS.
- L. Ci, Z. Xu, L. Wang, W. Cao, F. Ding, K. F. Kelly, B. I. Yajobson and P. M. Ajayan, Nano Res., 2008, 1, 116 Search PubMed.
- L. C. Campos, V. R. Manfrinato, J. D. Sanchez-Yamagishi, J. Kong and P. Jarillo-Herrero, Nano Lett., 2009, 9, 2600 CrossRef CAS.
- S. Xu, I. Levchenko, S. Y. Huang and K. Ostrikov, Appl. Phys. Lett., 2009, 95, 111505 CrossRef.
- Z. Zhu, W. Song, K. Burugapalli, F. Moussy, Y. L. Li and X. H. Zhong, Nanotechnology, 2010, 21, 165501 CrossRef.
| This journal is © The Royal Society of Chemistry 2010 |