Hybrid materials based on Pd nanoparticles on carbon nanostructures for environmentally benign C–C coupling chemistry
Javier
Guerra
abc and
María Antonia
Herrero
*a
aDepartamento de Química Orgánica, Facultad de Química, Universidad de Castilla-La Mancha, 13071 Ciudad Real, Spain. E-mail: MariaAntonia.Herrero@uclm.es; Tel: +34-926-295-300 Ext. 6744
bNanoDrugs SL, Paseo de la Innovación, 1, 02006 Albacete, Spain
cCIBERNED, Unidad Asociada Neurodeath, CSIC-Universidad de Castilla-La Mancha, Departamento de Ciencias Médicas, Facultad de Medicina, 02006 Albacete, Spain
First published on 4th June 2010
Abstract
The combination of different nanomaterials such as metallic nanoparticles and carbon nanostructures in a new hybrid material should give rise to interesting properties that combine the advantages of each of the nanocomponents. This review highlights the latest advances in the synthetic design of these hybrid materials where carbon nanostructures act as supports as well as stabilizing agents for very reactive metallic nanoparticles. The striking applications of Pd nanoparticles anchored on the surface of carbon nanostructures in C–C coupling chemistry are analyzed. Special emphasis is placed on the stability of these materials, which is linked to their recyclability. Numerous examples are given that involve the use of these catalysts in Heck, Suzuki and Sonogashira coupling reactions.
 Javier Guerra | Javier Guerra obtained his MSc in Chemistry (2000) at the University of Castilla-La Mancha (UCLM) where he finished his PhD under the tutelage of Prof. Enrique Díez Barra and Dr Juan Tejeda Sojo. Dr Guerra undertook a predoctoral stay for six months at the Debye Institute (Utrecht, the Netherlands) and spent two years as postdoctoral fellow at the University of Texas at Austin with Prof. Richard M. Crooks. Dr Guerra currently works at NanoDrugs, SL, a spin-off company led by Prof. Valentín Ceña. His areas of interest concern the development and application of new nanomaterials. |
 María Antonia Herrero | María Antonia Herrero obtained her Degree in Chemistry in 2000 and her PhD in 2006, both at UCLM. During her PhD she spent time at the Universities of Oxford and Uppsala (Sweden). Her first postdoctoral stay was at the University of Graz (Austria) under the supervision of Prof. Kappe, followed by another stay at the University of Trieste (Italy) under the supervision of Prof. Prato. During this time she broadened her knowledge of nanotechnology, especially in carbon nanostructures. Currently she is working as assistant professor at UCLM. |
1 Introduction
The recovery and recyclability of metal catalysts represents a major field of research in the quest for sustainable chemistry. The use of carbon-based nanomaterials as supports for different metallic entities enables these aims to be achieved. Carbonaceous nanomaterials are ideal supports due to their chemical inertness under moderate experimental conditions and their nanosize, which implies a better degree of dispersability in spite of their heterogeneous nature.However, better contact between the catalysts and the different components in a catalytic reaction and higher solubility are desired. In fact, a lack of solubility is the main disadvantage of pristine carbon nanomaterials for most genuine applications. This drawback is currently solved by the incorporation of different pendant units that increase the solubility.1–6 The enhancement of this property provides significant advantages for these functionalized carbon nanostructures and these include (i) easy handling of the materials, (ii) the availability of different characterization techniques and (iii) the possibility of tailoring the functionalization on the surface of carbon nanomaterials.
It is also well known that heterogeneous catalysts cannot compete in terms of efficiency with their homogeneous counterparts. This problem has been addressed by the use of metallic nanoparticles as catalysts. The high surface-to-volume ratio and the low coordination number of the metallic atoms located on the surface of the nanoparticles result in catalysts with very high activity.7 However, this high activity also represents the main drawback for these materials – i.e. the lack of stability displayed by the metallic nanocomposites. Metallic nanoparticles share properties intrinsic to heterogeneous as well as homogeneous catalysis and they are also known as “soluble-heterogeneous” catalysts.8 This terminology is consistent with Schwartz's criterion9 because nanoparticles contain multiple catalytic sites. These new concepts for homogeneous and heterogeneous catalysis are widely used.10–16 Catalytic activity and selectivity in “structure-sensitive” reactions17 depend strongly on the shape and size of the particles. While it is true that a certain measure of control over catalytic activity can be achieved by regulating the size of metallic nanocrystals,18–20 a greater degree of control can be obtained when crystal morphology is also well regulated.21,22 Heterogeneous catalysis carried out on the surface of single crystals under high vacuum has been studied thoroughly with the aim of understanding the importance of the location of the catalytic atom within a specific atomic layer.23 However, studies dealing with monodisperse soluble catalysts with a known shape and size have been increasingly reported in recent years.22,24–34 El-Sayed and co-workers have contributed towards understanding the correlation between nanoparticle shapes and the catalytic activity.24 They used an electron-transfer reaction between hexacyanoferrate(III) and thiosulfate ions catalyzed by PVP [poly(vinylpyrrolidone)]–platinum nanoparticles to study the difference in the catalytic activities of tetrahedral, cubic and near spherical particles.35–37 These authors also pointed out that aggregated material is more catalytically active than individual nanoparticles.38 This finding is consistent with results reported by Finke and co-workers, who found that bulk material is a more active catalyst than nanoclusters in the presence of an excess of coordinative ligands.39–41 Apart from these studies, to our knowledge, there are only a few examples in the literature dealing with the stability of the nanoparticle during catalytic reactions. These examples involve hydrogenation,42,43 carbonylation,44 oxidation45 and electrocatalysis.46
The search for new supports that will impart stability to the metallic nanoparticles and a high degree of contact between the reactants, reagents and catalysts has led to a new type of hybrid material that consists of metallic nanoparticles attached to the surface of carbon nanostructures, mainly carbon nanotubes. Apart from the aforementioned advantages of the soluble functionalized carbon nanotubes (CNTs), these carbon nanostructures are good candidates as supports for highly active catalysts like metal nanoparticles because CNTs do not have pores within their carbon backbone and this avoids catalytic reactions governed by diffusion processes.47
This review is split into two different parts: firstly the synthesis and design of these novel materials, and secondly their catalytic applications. As a result of the abundance of these hybrid materials and their numerous applications, we have focused on their applications in C–C coupling chemistry, where the stability of the catalyst is crucial for the efficient recovery and recyclability of the metallic particles. These hybrid catalysts are reported to be highly active with a high capability of being recycled.
2 Incorporation of the metallic particles onto carbon nanostructures
Of the different possible categorizations, we have found it very useful to classify these novel materials depending on the attachment procedure used to incorporate the metallic nanoparticles into the structure. This is not intended to be an exhaustive review of the different methods as other complementary reviews have been published.47–49 We have noted six types of attachment: electroless plating, electrodeposition, chemical reduction in the presence of a stabilizing agent, reduction in supercritical CO2, thermal decomposition and direct assembly of pre-formed metal nanoparticles.2.1 Electroless plating
The electroless plating technique involves deposition of the metal onto the carbon nanotube surface without the application of an electrical current. This approach has been widely used for the decoration of carbon nanostructures with metallic particles. In this technique, sodium hypophosphite or dimethylamine borane50 have been exploited as reducing agents on the metallic ions. In this way Pd/Sn and Pd particles have been deposited onto the surface.51 The interior of the carbon nanotubes can also be decorated with metallic particles, e.g. Ni or Co.50 Control of the particle size is limited and it is usual to find nanocrystallites that contain phosphorus or boron depending on the choice of reducing agent. More striking is the fact that certain inorganic salts can spontaneously be reduced on the carbon nanotube surface to render metallic particles. The nanotube/Au3+ and nanotube/Pt2+ systems can be categorized as thermodynamically favorable redox pairs in order to rationalize this spontaneous process.52 Other metals with lower redox potentials (Ag+, Cu2+, Ni2+) do not undergo this reduction process.47 The presence of oxygen functionalities on the CNT surface has also been claimed as part of the redox process that favors the reduction of a Pt salt and leads to deposition of Pt particles.53 Corma and co-workers also reported that the spontaneous reduction of a Pd salt and an organometallic palladium derivative (a carbapalladacycle oxime) are the Pd(0) nanoparticle precursors. The reduction peak potential for Pd2+ in THF (versus a standard hydrogen electrode) is 0.466 V for Pd(OAc)2 and 0.509 V for the carbapalladacycle oxime. These reported values are favorable for the reduction when compared with the work function of single-walled carbon nanotubes (SWNTs) (−5.0 V in a vacuum).54 These SWNTs will act as supports for the Pd(0) nanoparticles that are formed on the carbon surface.Recently, a new methodology that does not involve the use of reducing agents or electrical currents has been developed.55,56 This technique consists of ball-milling the carbon nanostructures with different metallic salts under an inert atmosphere at high temperatures (Fig. 1)55 or even under ambient conditions.56 This technique is based on mechanochemistry and is also available for other thermally conductive nanomaterials that can act as supports for the metallic nanoparticles.56 The limitation of this methodology with carbon nanomaterials is the compromise between nanoparticle formation and the structural integrity of the supports as ball-milling is also used to shorten CNTs.57–60
 | ||
| Fig. 1 SEM image of an Ag nanoparticle-decorated MWCNT sample with ∼1 mol% Ag content prepared by a ball-milling process. Reproduced with permission from ref. 55. Copyright (2009) American Chemical Society. | ||
2.2 Electrodeposition
In this approach the application of an electrical current in an electrochemical cell is responsible for the metal ion reduction and the subsequent deposition onto the surface of the nanotubes. The carbon nanostructures will act as a support and stabilize the metallic nanoparticles.61–63 Recently, McPherson et al. designed a microcapillary technique within a microelectrochemical cell that allows them to control the factors responsible for nucleation and the growth of metallic nanoparticles on the surface of pristine multi-walled carbon nanotubes (MWNTs).64 Pd and Pt can be deposited by this technique and the electrochemical parameters were identified that control nanoparticle number density, distribution and size.62,64,65 The use of controlled methodologies has enabled the introduction of Pt particles on well-aligned CNT arrays.61 Interestingly, the nanotube sidewalls serve both as the electrodeposition template and also as the wire that electrically connects the deposited nanoparticles.The differences between the nucleation and growth mechanisms for silver and platinum have been studied. It was found that silver causes a rapid nucleation that renders a high density of particles while a slower nucleation process is common for Pt particles and this gives rise to large aggregates.62 However, the main drawback of these electrochemical methods concerns the difficulties in scaling-up the process to obtain gram quantities of these metal-functionalized carbon nanostructures.48
2.3 Chemical reduction in the presence of a stabilizing agent
Under this category, we include those methodologies that require the addition of a reducing agent to form the metallic particles. Stabilizers are usually added to avoid aggregation and coalescence of the particles.7Ionic liquids have also been employed as stabilizers for Pt and Pt/Ru particles that were synthesized by the polyol method.68 Ayyappan et al. reported that nanoparticles of Ag, Au, Pd and Cu could be obtained by reduction of the corresponding metal salts using ethanol as the reducing agent under reflux.69 Polymers such as poly(vinylpyrrolidone) (PVP) provide control over the formation of the metal colloids and these macromolecules will stabilize the particles, thus avoiding aggregation and coalescence processes.
An interesting approach is the use of microwave irradiation combined with the polyol method to render monodisperse gold particles uniformly distributed on the CNT surface (Fig. 2). It is worth mentioning that these CNTs were previously modified through the introduction of organic groups such as carboxyl, carbonyl, hydroxyl and allyl terminal groups – also using the microwave technique. The organic functionalization is generated by irradiation of the CNTs with microwaves in boiling water.70
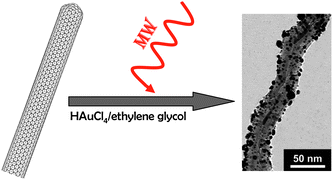 | ||
| Fig. 2 Au nanoparticles synthesized by the polyol method on the surface of CNTs. TEM image reproduced with permission from ref. 70. Copyright (2006) American Chemical Society. | ||
The use of sodium dodecyl sulfate (SDS) is quite common in this methodology when the combination of metallic nanoparticles and nanotubes is desired. SDS plays different roles as follows: (i) it is a well-known surfactant that will enhance the dispersion of the nanotubes, (ii) its presence will serve as a perfect interface between the carbon material and the metallic particles, (iii) SDS thermally decomposes in water to give 1-dodecanol (this alcohol will serve as a reducing agent as in the polyol method) and (iv) SDS, along with the 1-dodecanoic acid that comes from the polyol process, is a good stabilizer of metallic particles. As a result of these factors, SDS has commonly been chosen as a good material for the synthesis of these hybrid materials.71–75
Different molecules have been used to stabilize Pd particles and these include ionic liquids linked to CNTs79 and polymers or other macromolecules that are claimed to be responsible for stabilization of the metallic particles.7,80,81
Dendrimers that play a dual role as stabilizers and templates for the nanoparticle formation have been used to link nanoparticles that range in size from less than 1 to few nanometres.82 The metallic particle can be encapsulated within the cavities of the dendrimer, resulting in Dendrimer Encapsulated Nanoparticles (DENs),82 or chelated by multiple surface-bound dendrimers that stabilize the nanoparticle. This latter system is known as Dendrimer-Stabilized Nanoparticles (DSNs).83 For example, Imae and co-workers attached poly(amidoamine) (PAMAM) dendrimers onto the surface of MWNTs and used them as templates to introduce metal and metal oxide nanoparticles.84,85 However, attachment of the dendrimer onto the MWNT surface before metal complexation and reduction gives rise to particles that are larger than the cavities of the PAMAM dendrimer, estimated to be around 2 nm,86 indicating the presence of DSNs rather than DENs. Following a different strategy, Vijayaraghavan and Stevenson introduced previously synthesized Pt particles templated within PAMAM dendrimers onto the surface of nitrogen-doped MWNTs.87 In this study, the attachment was thought to occur because of strong van der Waals interactions with the edge plane sites of the CNTs. On the basis of this knowledge, we also used a similar strategy to attach electrostatically Au particles (around 2 nm size) encapsulated within the cavities of sixth-generation PAMAM dendrimers onto the surface of acidic-functionalized MWNTs (Fig. 3).88
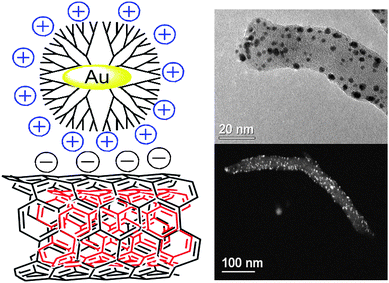 | ||
| Fig. 3 Au nanoparticles encapsulated within sixth-generation PAMAM dendrimers on the surface of MWNTs. Scheme and TEM images reproduced with permission from ref. 88. Copyright (2010) American Chemical Society. | ||
The attachment of previously synthesized PAMAM dendrimers that contain Pd(II) ions bound to their tertiary amines onto a CNT surface has recently been reported.89 In this case, the subsequent reduction renders Pd particles estimated to be around 2 nm in size. However, in this case, the deposition of the Pd particles seems to be very poor. A high level of functionalization on the carbon support is determinant for the final organic attachment of any pendant linker. In a different study, the degree of oxidation of the carbon surface proved critical to achieve a rich functionalization on the activated carbon fibers. This parameter correlates directly to the subsequent coverage of these carbon fibers with Pt DENs.90 Newkome and co-workers also synthesized CdS quantum dots encapsulated within amino-polyester dendrons and tethered them to SWNTs.91
Reduction with an aqueous solution of formaldehyde has also been used to synthesize Pt particles on MWNT surfaces and this resulted in a metal loading of 10%.92 Remarkably, it is possible to functionalize selectively the inner cavities of the nanotubes. The introduction of Pd particles within the nanotubes has been performed, with an efficient inner entrance of palladium nanoparticles that are within the range 4–6 nm. The authors postulated that the low surface tension of the solvent, water, is the reason that complete filling of the tube is achieved. A subsequent thermal process and treatment with hydrogen gas led to the formation of small and monodisperse palladium particles located in the cavities of CNTs.93,94
2.4 Reduction in supercritical CO2
The use of s-CO2 as a reducing agent has been shown to be an environmentally friendly technique to render zerovalent metal particles.95–100 High temperatures and high pressures are needed to reduce the inorganic salts that are the origin of the particles. The curvature of the nanotubes is claimed to be responsible for the final size of the particles (Fig. 4).95 The attachment of metallic particles onto the CNT surface facilitates the recovery of the catalyst and also avoids disadvantages such as discontinuous operation.100 Pt particles deposited onto the CNT surface by this method have shown remarkable efficiency as electrocatalysts in low-temperature fuel cells.101 | ||
| Fig. 4 TEM images of MWNTs with Pd particles attached on their surface after hydrogen reduction in s-CO2. From A to D, more equivalents of Pd precursor salt are added. Images reproduced with permission from ref. 95. Copyright (2003) The Royal Society of Chemistry. | ||
2.5 Thermal decomposition
Noble metal particles (Pd, Pt, Au and Ag) have been deposited onto CNT surfaces. Pd, Pt, Ag and Au nanoparticles were prepared by decomposing and reducing the metals in a solid-state mixture under a hydrogen atmosphere at temperatures that vary from 573 K to 973 K depending on the metal salt. The lack of a template renders particles that range from less than 5 nm to particles with diameters in the range of tens of nanometres.102A metal oxide particle (ZnO) has also been grown on a MWNT surface through a thermal decomposition reaction of an inorganic metallic salt [Zn(NH3)4CO3] with a polymer that contains donor groups, such as PVP, that can stabilize the final particle.103
2.6 Direct assembly of metal nanoparticles
A different strategy involves the deposition of previously synthesized particles onto CNTs. The latter materials are generally functionalized with different groups that enhance the dispersion of the tubes and also provide stabilization of the metallic particles. These groups can be linked to the CNTs electrostatically, covalently or by means of π–π stacking. An example of the latter system was described by Ou and Huang, where the agent used to detangle the carbon nanotubes and to anchor the metallic nanoparticles is bound to the nanotube surface by means of π–π interactions. These agents were 1-pyrenemethylamine, N-(1-naphthyl)ethylenediamine and phenethylamine, which can hold Au particles on the CNT surface.104 Thiol-modified DNA molecules wrapped around SWNTs provided a good dispersion and the sulfhydryl groups acted as anchor points for Au particles.105Water-in-oil microemulsions can also be used to synthesize Pd and Rh particles with hydrogen as the reductant.106 The amphiphilic surfactant bis(2-ethylhexyl) sulfosuccinate (AOT) gives rise to micelles with a polar inner environment where the metallic ions are encapsulated. Subsequent stirring with CNTs in the microemulsion solution gives rise to the linkage between the previously formed nanoparticles and the carbon nanostructures (Fig. 5).107
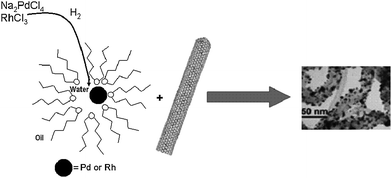 | ||
| Fig. 5 Water-in-oil microemulsions for the synthesis of metallic nanoparticles and the decoration of CNTs where the anionic surfactant is AOT, bis(2-ethylhexyl) sulfosuccinate. TEM image reproduced with permission from ref. 107. Copyright (2005) American Chemical Society. | ||
Gold particles have been stabilized using polyethyleneimine – a polymer that contains secondary amine groups and is electrostatically linked to the surface of acidic-functionalized MWNTs.108 The polymer is useful as a template for the particles and also acts as a solubilizing agent for the carbon nanotubes.
Oleylamine molecules have also been introduced by means of non-covalent interactions5 on the surface of SWNTs and this has facilitated the incorporation of pre-formed CdS quantum dots109 that are also stabilized by oleylamine (Fig. 6).110 The introduction of the metallic particles favors the dispersability, resulting in interesting optoelectronic properties and charge transfer processes.110
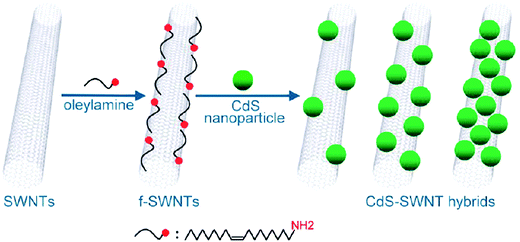 | ||
| Fig. 6 Quantum dots attached to the surface of SWNTs and stabilized by oleylamine. Scheme reproduced with permission from ref. 110. Copyright (2010) American Chemical Society. | ||
3 Application of Pd particles attached to carbon nanostructures in C–C coupling reactions
Hybrid metal/carbon materials have played an important role in C–C coupling reactions, thus highlighting the stability that the carbon supports seem to provide to the Pd nanocomposites. Numerous examples are described in the following section, with emphasis on the recyclability of the catalysts.3.1 Heck reaction
The olefination of aryl halides has been one of the preferred routes to build carbon skeletons.111–113 Homogeneous catalysts such as the Herrmann catalyst,114 palladacycles, palladium salts or palladium particles115 have shown high catalytic activity in this process.116,117 However, the reduced stability makes recovery and re-use of the catalysts difficult. Recent examples have taken advantage of the multiple possibilities provided by the functionalization of CNTs and these are discussed below.Pd particles attached to CNTs by means of a water-in-oil microemulsion and synthesized under a hydrogen atmosphere were used as a catalyst for a Heck reaction between iodobenzene and styrene (Fig. 7).107 The non-protic polar solvent N-methylpyrrolidine and a temperature of 120 °C were chosen for this reaction, which gave the trans-stilbene in 94% yield. This synthesis was completed by the hydrogenation of trans-stilbene using carbon nanotubes that also contain Pd particles on the surface as catalyst. In a different experiment, a system with Rh particles on the CNT surface also showed good catalytic activity for hydrogenation. In this work, the presence of Pd particles and Rh particles gave rise to higher efficiency in the reactions when compared with conventional heterogeneous catalysts.107 The high activity of these particles could be rationalized as being the result of the high surface area in the particles with respect to bulk material as well as the high percentage of edge and vertex sites in the nanoparticles.118,119
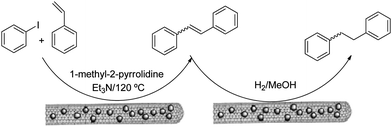 | ||
| Fig. 7 Water-in-oil microemulsions for the synthesis of metallic nanoparticles and the decoration of CNTs. Figure adapted with permission from ref. 107. Copyright (2005) American Chemical Society. | ||
Pd particles stabilized by SDS on MWNTs also showed good activity in the Heck reaction, especially when compared with a commercial palladium on carbon catalyst (Pd/C, 10 wt% from Sigma-Aldrich, Inc.).120 Simple separation of the catalyst by filtration allowed recycling of the catalytic material for further catalytic cycles. The authors reported that the efficiency of the catalytic hybrid material remained good after four cycles.
A more detailed study was performed by Corma and co-workers54 In this case, Pd nanoparticles were deposited on SWNTs. The aim was to compare different preparations of Pd-SWNT catalysts with respect to palladium on activated carbon (Pd/C). The authors pointed out that the catalytic activity will depend on the particle size, dispersion of the particles and the stabilizers used to avoid aggregation and coalescence of the particles. Three different Pd-SWNT catalysts were synthesized. Two of these examples were prepared by spontaneous reduction of Pd(OAc)2 and an oxime carbapalladacycle with the CNTs and the other one was synthesized by reduction of Pd(OAc)2 on SWNTs functionalized with dicetylmethylammonium moieties covalently linked to the SWNTs. All of the Pd-SWNTs present better activities than Pd/C. However, leaching of Pd atoms from the Pd-SWNTs catalysts was reported as a drawback. In spite of the leaching, significant decreases in the activity were not reported. This finding leads to the hypothesis that the leached Pd atoms could re-deposit onto remaining Pd particles that act as seeds for the crystal growth. This conclusion is consistent with current works, which have shown that Pd particles are precatalysts that act as a slow source of very reactive Pd atoms or tiny nanoclusters.121–129 This Pd migration is referred to as the boomerang effect. In fact, the mechanism involving the presence of Pd nanoparticles and the role of leached Pd species as real catalysts is common to all C–C coupling reactions and numerous examples will be given throughout the text. The Heck reaction has been the focus of many studies128,129 and the mechanism depicted in Fig. 8 is widely accepted for aryl iodides.
In this example a wide range of anionic species are proposed. The species depicted in bold have been detected by different techniques such as ES-MS and EXAFS.130,131 The substrate/catalyst ratio seems to be fundamental to achieve good conversions regardless of the haloarene derivative studied.128 This is because Pd nanoparticles and leached Pd species tend to aggregate and this aggregation leads to the formation of Pd black after a certain size is reached. Pd black consists of large aggregates of Pd in the micron range; these species are poorly active because of the absence of defect sites and mainly because of their small surface/volume ratio. For this reason, many authors have chosen to use stabilizers to avoid coalescence and aggregation of the Pd nanoparticles.80,123,128 Köhler and co-workers showed that even traces of Pd (10−7 mol% with respect to the haloarene) display good catalytic activity for deactivated aryl bromides and activated aryl chlorides.132
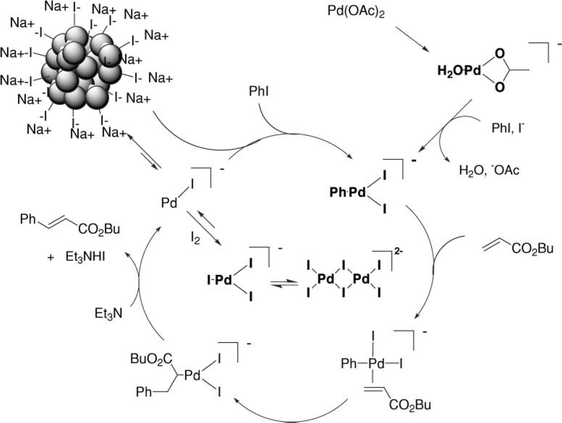 | ||
| Fig. 8 Mechanism proposed for the ligand-free Heck reaction between aryl iodide and butyl acrylate. Figure reproduced with permission from ref. 128. Copyright (2006) The Royal Society of Chemistry. | ||
One more conclusion that can be drawn from the work of Corma and co-workers54 is that Pd nanoparticles are not always the best choice and this will depend on the target reaction. If the reaction is particle-size demanding, then the presence of the nanoparticles is convenient – as is the case for C–C coupling reactions. However, when the reactions are less demanding, as in hydrogenations, Pd/C could be more effective. In any case, there is some controversy about this point because particle size and shape effects have also been found in hydrogenations.118,133
3.2 Suzuki reaction
The Suzuki reaction involves the formation of a C–C biaryl bond between aryl halogenides and arylboronic acids. Boronic acid derivatives have low toxicity in comparison to the stannane derivatives used in the Stille coupling.Carbon nanotubes can display different forms of entanglement. Aggregation in micrometre-sized carbon nanotube particles (CNTPs) is one of these patterns and this gives rise to a spherical shape (Fig. 9). Chen et al. used the polyol method in conjunction with SDS to introduce Pd particles with a size of 2.7 ± 0.7 nm.134 These Pd-CNTPs show a high catalytic activity with activated aryl iodides in ethanol under reflux. Pd-CNTPs were separated from the reaction mixture by filtration and reused several times without loss of the catalytic activity.
 | ||
| Fig. 9 SEM images reproduced with permission from ref. 134. Copyright (2008) American Chemical Society. | ||
Sodium dodecyl sulfate is a good stabilizer but it does not passivate the surface of Pd particles.134 Hence, Pd particles attached to the surface of MWNTs and using SDS as surfactant displayed good catalytic activity in reactions between iodobenzene or 4-iodoanisole and phenylboronic acid. The reaction was performed in the non-protic polar solvent dimethylformamide with heating at 110 °C. Full conversion was obtained in 2 h and the catalyst was recovered, washed and reused five times, with similar results obtained in each case. Similar conclusions were drawn for the Stille coupling with tri-n-butylphenyltin, although for this reaction heating for 12 h was necessary to achieve complete conversion.135
MWNTs have also been decorated with Pd particles formed by reduction of Pd(dba)2. The resulting material has successfully been used as a catalyst in Suzuki reactions at 90 °C using ethanol and water among other polar solvents. The catalysts could be recycled without loss of catalytic activity. Unfortunately, chloroarene derivatives were not reactive under these conditions.136 In a similar study,137 Pd particles stabilized by thiolate groups anchored on the surface of MWNTs showed reasonable catalytic activity with 4-halobenzoic acids. The Maitlis filtration test138 showed that Pd leaching occurs and, therefore, part of the catalytic process could be attributed to these leached Pd species. This situation is consistent with results reported by Köhler and co-workers with palladium on carbon and on metal oxides, where proof of leaching was shown along with a correlation with the catalytic activity in the Suzuki coupling.139
Pd particles synthesized in supercritical carbon dioxide on the MWNT surface also showed good catalytic activity in the Suzuki reaction. In this case, different aryl iodide derivatives were used and even aryl bromide derivatives. These reactions were performed in methanol under reflux and in air. Excellent yields were obtained and the catalyst was recycled six times without loss of the catalytic activity. However, after recycling, TEM experiments revealed the presence of large particles (up to 40 nm).99 This reactivity could be due to leached Pd atoms as actual catalysts for the Suzuki reaction and re-deposition onto Pd particle seeds in a boomerang effect.
Carbon nanohorns (CNHs) represent a different allotropic form of carbon. A primary CNH particle is a single graphene tube (similar in structure to single-walled carbon nanotubes) of 2–5 nm in diameter and 40–50 nm long, with a conically-closed tip. CNHs usually aggregate in assemblies that are reminiscent of dahlia flowers, with a diameter that ranges from 80 to 100 nm (Fig. 10).140
 | ||
| Fig. 10 TEM images of covalently functionalized carbon nanohorns. TEM scale bar: 100 nm. Images reproduced with permission from ref. 140. Copyright (2009) The Royal Society of Chemistry. | ||
Kaneko and co-workers used an oxidation treatment to obtain a higher surface area with respect to pristine CNHs. Pd particles (2–3 nm) were deposited on the surface of pristine CNHs and oxidized CNHs (ox-CNHs) with stabilization through the use of PVP.141 The catalytic activity of these Pd-CNHs and Pd-ox-CNHs were compared with Pd particles dispersed in activated carbon (Pd/C) as well as with Pd nanoparticles tailored pitch-based activated carbon fibers (Pd-ACFs). Pd-ox-CNHs were the most active in the Suzuki reaction between phenylboronic acid and iodobenzene, giving high yields in short time periods even at 60 °C. It is worth noting that a yield of 68% yield was obtained in only 2 h using just 10 ppm of Pd to catalyze the reaction.141 The use of tiny amounts of Pd metal in efficient catalysts for C–C coupling reactions is in agreement with results from different studies, where the minimum amount of Pd results in high conversions for these reactions.16,126,127,132,142–144 For example, Leadbeater only used 50 ppb of Pd under microwave irradiation to perform Suzuki couplings.142 However, contradictory results were reported in relation to microwave techniques applied in C–C Pd chemistry. Stepnicka and co-workers showed a rapid deactivation of Pd because the growth of Pd particles led to Pd black formation even when supported on mesoporous molecular sieves.145,146
Within the field of sustainable chemistry, Wai and co-workers synthesized tetrasubstituted olefins in supercritical CO2.99 The catalyst consisted of Pd particles (5–20 nm) and these nanocomposites were formed by reduction of PdCl2 with hydrazine in the presence of dispersed CNTs.95,96 In this case, different dibromo-substituted olefins reacted with different boronic acids to give products with a slightly higher yield in comparison to reactions catalyzed by Pd/C.
In any case, it is worth noting that other heterogeneous catalysts have resulted in good conversions with chloro-, bromo- and iodo-aryl derivatives in the Suzuki coupling. Different materials with Pd on carbon, metal oxides, non-metallic oxides and hydroxides have been used to achieve high yields.147–150 However, when homogeneous catalysts were applied under harsh conditions the formation of Pd black was observed. Different authors have shown that the Pd catalyst can be recycled by addition of iodine, with most of the original catalytic activity recovered.128,130,139
A kinetic study applied to the Suzuki reaction clearly showed a direct correlation between the leaching of Pd species and the conversion in the reaction.139 This finding was also reported for other C–C coupling reactions, e.g. the Heck coupling.13,128,144,151–154
3.3 Sonogashira reaction
MWNTs loaded with Pd particles (5–10 nm, 10 wt%) have also been used as catalysts in the alkynylation reaction.78 This cross-coupling takes place between compounds bearing terminal alkyne moieties and aryl halides. The remarkable feature in this application is that the authors aimed for the optimization of environmentally-benign conditions as follows: (i) minimize the use of toxic organic solvents using a mixture of ethanol–water, (ii) avoid the use of copper salts by carrying out the reactions in the absence of organic bases and phosphine ligands, and (iii) use microwave irradiation as the source of energy. The usefulness of this alternative way of heating has recently been recognized as it is associated with the reduction of both chemical waste and reaction times.155,156 Microwave heating has already been applied to the purification and functionalization of CNTs due to the strong characteristic absorption of microwaves by CNTs.3,157–160 In this work,78 the authors found good yields with a variety of tolanes obtained in reaction times of just 5 min. Remarkably, the authors also reported an unusual formation of enynes, which arise from the addition of a terminal alkyne to a different alkyne (Scheme 1). However, in most cases homodimerization of the terminal alkynes is favored in the Sonogashira coupling. On using Pd-MWNTs the formation of this reaction side-product is a highly regio- and stereoselective process.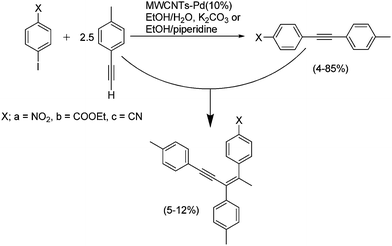 | ||
| Scheme 1 Enyne formation catalyzed by Pd nanoparticles anchored on MWNTs as a side-reaction in a Sonogashira coupling. Adapted from ref. 78 with permission of the Centre National de la Recherche Scientifique (CNRS) and the Royal Society of Chemistry. Copyright (2008). | ||
The authors pointed out the high efficiency of the Pd particles as catalysts and the good energy transfer by CNTs as possible reasons for the formation of these enyne products. Recovery of the catalyst and re-use for five additional cycles did not lead to a decrease in the catalytic activity. TEM images of the catalyst after these cycles did not show evidence of growth in the Pd particles. However, different tests aimed at quantifying leached Pd species demonstrated 1.1% leaching. Importantly, leaching processes were also evidenced in Sonogashira reactions using as catalysts Pd particles on the cavities of ordered mesoporous cubic carbon.161 In this study the authors confirmed leaching of active Pd species by the Maitlis filtration test and also by electron-dispersive spectroscopy (EDS) coupled to scanning electron microscopy (SEM). The presence of the activated carbon material as a support proved advantageous compared with unsupported Pd particles because the heterogeneous support provides a large interfacial area between the Pd particles and the reaction mixture, thus minimizing Ostwald ripening processes and slowing the Pd leaching. This situation is consistent with the mechanism previously proposed by Rothenberg and co-workers.126,127,162
4 Conclusions and outlook
The increasing number of studies concerning the attachment of metallic nanoparticles onto well-defined carbon nanostructures clearly reveals the utility of the latter as a support and stabilizer for very reactive metallic guests. However, with the exception of notorious examples, in terms of C–C coupling reactions these supports act as passive witnesses and selectivity differences are not found compared with other conventional Pd/C catalysts. Moreover, to date the real catalytic site where the reaction takes place remains unknown, although there is a lot of clear evidence for Pd leaching and therefore these highly active leached Pd species could be the real catalyst. In this situation the Pd nanoparticles would act as a slow source of active Pd species. On the other hand, one of the main advantages of well-defined carbon nanomaterials is that their functionalization has made it possible to obtain materials with greater solubility. These soluble functionalized CNTs facilitate contact between the reagents and reactants and also make it easy to recover and reuse the catalytic material.In the near future, the use of other novel carbon nanomaterials such as CNHs will be more common as materials for Pd attachment because they are better defined and traces of other metallic species are not present. In any case, better characterization of these materials is needed for further developments in this field. This aim will not be reached unless scientists can obtain more soluble materials and this has to be combined with advances in the different characterization techniques.
Acknowledgements
M. A. H. and J. G. are indebted to the Junta de Comunidades de Castilla-La Mancha (Spain) for a postdoctoral research grant. The authors acknowledge Prof. M. Prato (University of Trieste, Italy) and Prof. R. M. Crooks (The University of Texas at Austin, USA) for sharing their knowledge related to carbon nanostructures and metallic DENs, respectively. M. A. H. is grateful to DGICYT of Spain for funding through project CTQ2007-60037/BQU and to the Consejería de Educación y Ciencia (JCCM) for funding projects PBI-06-0020 and PCI08-0040. J. G. also acknowledges the MICINN (Spain)-Fondo Europeo de Desarrollo Regional (FEDER, European Union) (project CTQ2006-08871) and the Junta de Comunidades de Castilla-La Mancha (project PCI08-0033). Prof. V. Ceña is also acknowledged for financial support (NanoDrugs, SL). M. A. H. and J. G. are grateful to the Organic Chemistry Department at UCLM for support and especially to Prof. A. de la Hoz and Dr E. Vázquez for their useful comments.References
- V. Georgakilas, K. Kordatos, M. Prato, D. M. Guldi, M. Holzinger and A. Hirsch, J. Am. Chem. Soc., 2002, 124, 760–761 CrossRef CAS.
- A. Hirsch, Angew. Chem., Int. Ed., 2002, 41, 1853–1859 CrossRef CAS.
- E. Vázquez and M. Prato, ACS Nano, 2009, 3, 3819–3824 CrossRef CAS.
- M. Shim, N. W. S. Kam, R. J. Chen, Y. Li and H. Dai, Nano Lett., 2002, 2, 285–288 CrossRef CAS.
- B. Pan and B. Xing, Environ. Sci. Technol., 2008, 42, 9005–9013 CrossRef CAS.
- B. I. Kharisov, O. V. Kharissova, H. Leija Gutiérrez and U. Ortiz Méndez, Ind. Eng. Chem. Res., 2009, 48, 572–590 CrossRef CAS.
- C. A. Foss Jr. and D. L. Feldheim, Metal Nanoparticles: Synthesis Characterization & ApplicationsMarcel Dekker, New York, 2002 Search PubMed.
- J. D. Aiken III, Y. Lin and R. G. Finke, J. Mol. Catal. A: Chem., 1996, 114, 29–51 CrossRef.
- J. Schwartz, Acc. Chem. Res., 1985, 18, 302–308 CrossRef CAS.
- J. A. Widegren and R. G. Finke, J. Mol. Catal. A: Chem., 2003, 198, 317–341 CrossRef CAS.
- C. A. Jaska and I. Manners, J. Am. Chem. Soc., 2004, 126, 9776–9785 CrossRef CAS.
- S. P. Andrews, A. F. Stepan, T. Hirohisa, S. V. Ley and M. D. Smitha, Adv. Synth. Catal., 2005, 347, 647–654 CrossRef CAS.
- N. T. S. Phan, M. Van Der Sluys and C. W. Jones, Adv. Synth. Catal., 2006, 348, 609–679 CrossRef CAS.
- J. A. Widegren, M. A. Bennet and R. G. Finke, J. Am. Chem. Soc., 2003, 125, 10301–10310 CrossRef CAS.
- O. Illa, C. Rodriguez-Garcia, C. Acosta-Silva, I. Favier, D. Picurelli, A. Oliva, M. Gomez, V. Branchadell and R. M. Ortuno, Organometallics, 2007, 26, 3306–3314 CrossRef CAS.
- M. T. Reetz and E. Westermann, Angew. Chem., Int. Ed., 2000, 39, 165–168 CrossRef CAS.
- M. Boudart, Chem. Rev., 1995, 95, 661–666 CrossRef CAS.
- H. Song, R. M. Rioux, J. D. Hoefelmeyer, R. Komor, K. Niesz, M. Grass, P. Yang and G. A. Somorjai, J. Am. Chem. Soc., 2006, 128, 3027–3037 CrossRef CAS.
- R. M. Rioux, H. Song, J. D. Hoefelmeyer, P. Yang and G. A. Somorjai, J. Phys. Chem. B, 2005, 109, 2192–2202 CrossRef CAS.
- H. Ye, J. A. Crooks and R. M. Crooks, Langmuir, 2007, 23, 11901–11906 CrossRef CAS.
- R. M. Rioux, H. Song, M. Grassa, S. Habasa, K. Niesz, J. D. Hoefelmeyer, P. Yang and G. A. Somorjai, Top. Catal., 2006, 39, 167–174 CrossRef CAS.
- H. Lee, S. E. Habas, S. Kweskin, D. Butcher, G. A. Somorjai and P. Yang, Angew. Chem., Int. Ed., 2006, 45, 7824–7828 CrossRef CAS.
- G. A. Somorjai, Introduction to Surface Chemistry and Catalysis, Wiley, New York, 1994 Search PubMed.
- R. Narayanan and M. A. El-Sayed, J. Phys. Chem. B, 2005, 109, 12663–12676 CrossRef CAS.
- Y. Zhang, M. E. Grass, S. E. Habas, F. Tao, T. Zhang, P. Yang and G. A. Somorjai, J. Phys. Chem. C, 2007, 111, 12243–12453 CrossRef CAS.
- S. Panigrahi, S. Basu, S. Praharaj, S. Pande, S. Jana, A. Pal, S. K. Ghosh and T. Pal, J. Phys. Chem. C, 2007, 111, 4596–4605 CrossRef CAS.
- N. Semagina, A. Renken, D. Laub and L. Kiwi-Minsker, J. Catal., 2007, 246, 308–314 CrossRef CAS.
- R. Xu, D. Wang, J. Zhang and Y. Li, Chem.–Asian J., 2006, 1, 888–893 CrossRef CAS.
- R. J. Chimentao, F. Medina, J. E. Sueiras, J. L. Garcia Fierro, Y. Cesteros and P. Salagre, J. Mater. Sci., 2007, 42, 3307–3314 CrossRef CAS.
- T. Na, Z.-Y. Zhou, S.-G. Sun, Y. Ding and Z. L. Wang, Science, 2007, 316, 732–735 CrossRef CAS.
- F. Sen and G. Gokagac, J. Phys. Chem. C, 2007, 111, 5715–5720 CrossRef CAS.
- R. K. Sharma, P. Sharma and A. Maitra, J. Colloid Interface Sci., 2003, 265, 134–140 CrossRef CAS.
- F. J. Vidal-Iglesias, J. Solla-Gullon, P. Rodriguez, E. Herrero, V. Montiel, J. M. Feliu and A. Aldaz, Electrochem. Commun., 2004, 6, 1080–1084 CrossRef CAS.
- J. Grunes, J. Zhu, E. A. Anderson and G. A. Somorjai, J. Phys. Chem. B, 2002, 106, 11463–11468 CrossRef CAS.
- R. Narayanan and M. A. El-Sayed, J. Am. Chem. Soc., 2004, 126, 7194–7195 CrossRef CAS.
- R. Narayanan and M. A. El-Sayed, Nano Lett., 2004, 4, 1343–1348 CrossRef CAS.
- R. Narayanan and M. A. El-Sayed, J. Phys. Chem. B, 2004, 108, 5726–5733 CrossRef CAS.
- R. Narayanan and M. A. El-Sayed, J. Phys. Chem. B, 2003, 107, 12416–12424 CrossRef CAS.
- L. S. Ott, S. Campbell, K. R. Seddon and R. G. Finke, Inorg. Chem., 2007, 46, 10335–10344 CrossRef.
- C. Besson, E. E. Finney and R. G. Finke, Chem. Mater., 2005, 17, 4925–4938 CrossRef CAS.
- C. Besson, E. E. Finney and R. G. Finke, J. Am. Chem. Soc., 2005, 127, 8179–8184 CrossRef CAS.
- J. Schulz, A. Roucoux and H. Patin, Chem.–Eur. J., 2000, 6, 618–624 CrossRef CAS.
- P. J. Collier, J. A. Iggo and R. Whyman, J. Mol. Catal. A: Chem., 1999, 146, 149–157 CrossRef CAS.
- Q. Wang, H. Liu, M. Han, X. Li and D. Jiang, J. Mol. Catal. A: Chem., 1997, 118, 145–151 CrossRef.
- S. N. Sidorov, I. V. Volkov, V. A. Davankov, M. P. Tsyurupa, P. M. Valetsky, L. M. Bronstein, R. Karlinsey, J. W. Zwanziger, V. G. Matveeva, E. M. Sulman, N. V. Lakina, E. A. Wilder and R. J. Spontak, J. Am. Chem. Soc., 2001, 123, 10502–10510 CrossRef CAS.
- P. J. Ferreira, G. J. la O, Y. Shao-Horn, D. Morgan, R. Makharia, S. Kocha and H. A. Gasteiger, J. Electrochem. Soc., 2005, 152, A2256–A2271 CrossRef.
- V. Georgakilas, D. Gournis, V. Tzitzios, L. Pasquato, D. M. Guldi and M. Prato, J. Mater. Chem., 2007, 17, 2679–2694 RSC.
- G. G. Wildgoose, C. E. Banks and R. G. Compton, Small, 2006, 2, 182–193 CrossRef CAS.
- X. Peng, J. Chen, J. A. Misewich and S. S. Wong, Chem. Soc. Rev., 2009, 38, 1076–1098 RSC.
- J. Li, M. Moskovits and T. L. Haslett, Chem. Mater., 1998, 10, 1963–1967 CrossRef CAS.
- L.-M. Ang, T. S. Andy Hor, G.-Q. Xu, C.-h. Tung, S. Zhao and J. L. S. Wang, Chem. Mater., 1999, 11, 2115–2118 CrossRef CAS.
- H. C. Choi, M. Shim, S. Bangsaruntip and H. Dai, J. Am. Chem. Soc., 2002, 124, 9058–9059 CrossRef CAS.
- J. H. Chen, M. Y. Wang, B. Liu, Z. Fan, K. Z. Cui and Y. F. Kuang, J. Phys. Chem. B, 2006, 110, 11775–11779 CrossRef CAS.
- A. Corma, H. Garcia and A. Leyva, J. Mol. Catal. A: Chem., 2005, 230, 97–105 CrossRef CAS.
- Y. Lin, K. A. Watson, M. J. Fallbach, S. Ghose, J. G. Smith, Jr., D. M. Delozier, W. Cao, R. E. Crooks and J. W. Connell, ACS Nano, 2009, 3, 871–884 CrossRef CAS.
- Y. Lin, K. A. Watson, S. Ghose, J. G. Smith, Jr., T. V. Williams, R. E. Crooks, W. Cao and J. W. Connell, J. Phys. Chem. C, 2009, 113, 14858–14862 CrossRef CAS.
- H. Pan, L. Liu, Z.-X. Guo, L. Dai, F. Zhang, D. Zhu, R. Czerw and D. L. Carroll, Nano Lett., 2003, 3, 29–32 CrossRef CAS.
- F. Liu, X. Zhang, J. Cheng, J. Tu, F. Kong, W. Huang and C. Chen, Carbon, 2003, 41, 2527–2532 CrossRef CAS.
- N. Pierard, A. Fonseca, Z. Konya, I. Willems, G. V. Tendeloo and J. B. Nagy, Chem. Phys. Lett., 2001, 335, 1–8 CrossRef CAS.
- Y. B. Li, B. Q. Wei, J. Liang, Q. Yu and D. H. Wu, Carbon, 1999, 37, 493–497 CrossRef CAS.
- B. M. Quinn, C. Dekker and S. G. Lemay, J. Am. Chem. Soc., 2005, 127, 6146–6147 CrossRef CAS.
- T. M. Day, P. R. Unwin, N. R. Wilson and J. V. Macpherson, J. Am. Chem. Soc., 2005, 127, 10639–10647 CrossRef CAS.
- C. Wang, M. Waje, X. Wang, J. M. Tang, R. C. Haddon and Y. Yan, Nano Lett., 2004, 4, 345–348 CrossRef CAS.
- T. M. Day, P. R. Unwin and J. V. Macpherson, Nano Lett., 2007, 7, 51–57 CrossRef CAS.
- H. Tang, J. H. Chen, Z. P. Huang, D. Z. Wang, Z. F. Ren, L. H. Nie, Y. F. Kuang and S. Z. Yao, Carbon, 2004, 42, 191–197 CrossRef CAS.
- A. Kongkanand, K. Vinodgopal, S. Kuwabata and P. V. Kamat, J. Phys. Chem. B, 2006, 110, 16185–16188 CrossRef CAS.
- V. Lordi, N. Yao and J. Wei, Chem. Mater., 2001, 13, 733–737 CrossRef CAS.
- B. Wu, D. Hu, Y. Kuang, B. Liu, X. Zhang and J. Chen, Angew. Chem., Int. Ed., 2009, 48, 4751–4754 CrossRef CAS.
- S. Ayyappan, R. S. Gopalan, G. N. Subbanna and C. N. R. Rao, J. Mater. Res., 1997, 12, 398–401 CrossRef CAS.
- M. S. Raghuveer, S. Agrawal, N. Bishop and G. Ramanath, Chem. Mater., 2006, 18, 1390–1393 CrossRef CAS.
- M. O'Connell, S. Bachilo, C. Huffman, V. Moore, M. Strano, E. Haroz, K. Rialon, P. Boul, W. Noon, C. Kittrell, J. Ma, R. Hauge, B. Weisman and R. Smalley, Science, 2002, 297, 593–596 CrossRef CAS.
- V. C. Moore, M. S. Strano, E. H. Haroz, R. H. Hauge and R. E. Smalley, Nano Lett., 2003, 3, 1379–1382 CrossRef CAS.
- L. Jiang, L. Gao and J. Sun, J. Colloid Interface Sci., 2003, 260, 89–94 CrossRef CAS.
- C. L. Lee, C. C. Wan and Y. Y. Wang, Adv. Funct. Mater., 2001, 11, 344–347 CrossRef CAS.
- W. L. Wang, Y. Y. Wang, C. C. Wan and C. L. Lee, Colloids Surf., A, 2006, 275, 11–16 CrossRef CAS.
- M. V. Gomez, J. Guerra, A. H. Velders and R. M. Crooks, J. Am. Chem. Soc., 2009, 131, 341–350 CrossRef CAS.
- M. V. Gomez, J. Guerra, V. S. Myers, R. M. Crooks and A. H. Velders, J. Am. Chem. Soc., 2009, 131, 14634–14635 CrossRef CAS.
- J.-H. Olivier, F. Camerel, R. Ziessel, P. Retailleau, J. Amadou and C. Pham-Huu, New J. Chem., 2008, 32, 920–924 RSC.
- Y. S. Chun, J. Y. Shin, C. E. Song and S.-g. Lee, Chem. Commun., 2008, 942–944 RSC.
- L. S. Ott and R. G. Finke, Coord. Chem. Rev., 2007, 251, 1075–1100 CrossRef CAS.
- A. M. Caminade and J.-P. Majoral, Chem. Soc. Rev., 2010 10.1039/b926408f.
- R. W. J. Scott, O. M. Wilson and R. M. Crooks, J. Phys. Chem. B, 2005, 109, 692–704 CrossRef CAS.
- M. E. Garcia, L. A. Baker and R. M. Crooks, Anal. Chem., 1999, 71, 256–258 CrossRef CAS.
- X. Lu and T. Imae, J. Phys. Chem. C, 2007, 111, 2416–2420 CrossRef CAS.
- X. Lu and T. Imae, J. Phys. Chem. C, 2007, 111, 8459–8462 CrossRef CAS.
- V. Petkov, V. Parvanova, D. Tomalia, D. Swansonb, D. Bergstromb and T. Vogt, Solid State Commun., 2005, 134, 671–675 CrossRef CAS.
- G. Vijayaraghavan and K. J. Stevenson, Langmuir, 2007, 23, 5279–5282 CrossRef CAS.
- M. A. Herrero, J. Guerra, V. S. Myers, M. V. Gomez, R. M. Crooks and M. Prato, ACS Nano, 2010, 4, 905–912 CrossRef CAS.
- Y. Shen, Q. Xu, H. Gao and N. Zhu, Electrochem. Commun., 2009, 11, 1329–1332 CrossRef CAS.
- J. Ledesma-García, I. L. Escalante García, F. J. Rodríguez, T. W. Chapman and L. A. Godínez, J. Appl. Electrochem., 2008, 38, 515–522 CrossRef CAS.
- S. H. Hwang, C. N. Moorefield, P. Wang, K.-U. Jeong, S. Z. D. Cheng, K. K. Kotta and G. R. Newkome, J. Am. Chem. Soc., 2006, 128, 7505–7509 CrossRef CAS.
- W. Li, C. Liang, W. Zhou, J. Qiu, Z. Zhou, G. Sun and Q. Xin, J. Phys. Chem. B, 2003, 107, 6292–6299 CrossRef CAS.
- J.-P. Tessonnier, L. Pesant, G. Ehret, M. J. Ledoux and C. Pham-Huu, Appl. Catal., A, 2005, 288, 203–210 CrossRef CAS.
- S. Rather, R. Zacharia, S. W. Hwang, M. Naik and K. S. Nahm, Chem. Phys. Lett., 2007, 441, 261–267 CrossRef CAS.
- X. R. Ye, Y. Lin and C. M. Wai, Chem. Commun., 2003, 642–643 RSC.
- X.-R. Ye, Y. Lin, C. Wang, M. H. Engelhard, Y. Wanga and C. M. Wai, J. Mater. Chem., 2004, 14, 908–913 RSC.
- Y. Lin, X. Cui and X. Ye, Electrochem. Commun., 2005, 7, 267–274 CrossRef CAS.
- Z. Sun, Z. Liu, B. Han, S. Miao, Z. Miao and G. An, J. Colloid Interface Sci., 2006, 304, 323–328 CrossRef CAS.
- H.-B. Pan, C. H. Yen, B. Yoon, M. Sato and C. M. Wai, Synth. Commun., 2006, 36, 3473–3478 CrossRef CAS.
- L. Zhou, W. Zhang and H. Jiang, Sci. China, Ser. B: Chem., 2008, 51, 241–247 CrossRef CAS.
- Y. Lin, X. Cui, C. Yen and C. M. Wai, J. Phys. Chem. B, 2005, 109, 14410–14415 CrossRef CAS.
- B. Xue, P. Chen, Q. Hong, J. Lin, K. Lee and K. Tan, J. Mater. Chem., 2001, 11, 2378–2381 RSC.
- G. Guo, J. Guo, D. Tao, W. C. H. Choy, L. Zhao, W. Qian and Z. Wang, Appl. Phys. A: Mater. Sci. Process., 2007, 89, 525–528 CrossRef CAS.
- Y. Y. Ou and M. H. Huang, J. Phys. Chem. B, 2006, 110, 2031–2036 CrossRef CAS.
- X. Han, Y. Li and Z. Deng, Adv. Mater., 2007, 19, 1518–1522 CrossRef.
- B. Yoon, H. Kim and C. M. Wai, Chem. Commun., 2003, 1040–1041 RSC.
- B. Yoon and C. M. Wai, J. Am. Chem. Soc., 2005, 127, 17174–17175 CrossRef CAS.
- X. Hu, T. Wang, X. Qu and S. Dong, J. Phys. Chem. B, 2006, 110, 853–857 CrossRef CAS.
- K.-T. Yong, Y. Sahoo, M. T. Swihart and P. N. Prasad, J. Phys. Chem. C, 2007, 111, 2447–2458 CrossRef CAS.
- X. Li, Y. Jia and A. Cao, ACS Nano, 2010, 4, 506–512 CrossRef CAS.
- W. A. Herrmann, C.-P. Reisinger and P. Härter, in Aqueous-Phase Organometallic Catalysis: Concepts and Applications, ed. B. Cornils and W. A. Herrmann, Wiley-VCH, Weinheim, 2nd edn, 2004 Search PubMed.
- E.-I. Negishi and A. De Meijere, Handbook of Organopalladium Chemistry for Organic Synthesis, John Wiley & Sons, New York, 2002 Search PubMed.
- B. Cornils and W. A. Herrmann, Applied Homogeneous Catalysis with Organometallic Compounds: A Comprehensive Handbook in Three Volumes, Wiley-VCH, 2nd edn, Weinheim, 2002 Search PubMed.
- W. A. Herrmann, C. Brossmer, K. Öfele, C.-P. Reisinger, T. Priermeier, M. Beller and H. Fisher, Angew. Chem., Int. Ed. Engl., 1995, 34, 1844–1848 CrossRef CAS.
- L. K. Yeung and R. M. Crooks, Nano Lett., 2001, 1, 14–17 CrossRef CAS.
- S. Bräse and A. De Meijere, in Metal-Catalyzed Cross-Coupling Reactions, ed. A. De Meijere and F. Diederich, Wiley-VCH, Weinheim, 2nd edn, 2004 Search PubMed.
- I. P. Beletskaya and A. V. Cheprakov, in The Mizoroki–Heck reaction, ed. M. Oestreich, John Wiley & Sons, New York, 1st edn, 2009 Search PubMed.
- O. M. Wilson, M. R. Knecht, J. C. Garcia-Martinez and R. M. Crooks, J. Am. Chem. Soc., 2006, 128, 4510–4511 CrossRef CAS.
- T. Teranishi and M. Miyake, Chem. Mater., 1998, 10, 594–600 CrossRef CAS.
- N. Karousis, G.-E. Tsotsou, N. Ragoussis and N. Tagmatarchis, Diamond Relat. Mater., 2008, 17, 1582–1585 CrossRef CAS.
- E. V. Carino, M. R. Knecht and R. M. Crooks, Langmuir, 2009, 25, 10279–10284 CrossRef CAS.
- D. Astruc, Inorg. Chem., 2007, 46, 1884–1894 CrossRef CAS.
- D. Astruc, F. Lu and J. Ruiz Aranzaes, Angew. Chem., Int. Ed., 2005, 44, 7852–7872 CrossRef CAS.
- E. de Jesús and J. C. Flores, Ind. Eng. Chem. Res., 2008, 47, 7968–7981 CrossRef CAS.
- M. Bernechea, E. de Jesús, C. López-Mardomingo and P. Terreros, Inorg. Chem., 2009, 48, 4491–4496 CrossRef CAS.
- A. V. Gaikwad, A. Holuigue, M. B. Thathagar, J. E. ten Elshof and G. Rothenberg, Chem.–Eur. J., 2007, 13, 6908–6913 CrossRef CAS.
- M. B. Thathagar, J. E. Ten Elshof and G. Rothenberg, Angew. Chem., Int. Ed., 2006, 45, 2886–2890 CrossRef CAS.
- J. G. de Vries, Dalton Trans., 2006, 421–429 RSC.
- A. Biffis, M. Zecca and M. Basato, J. Mol. Catal. A: Chem., 2001, 173, 249–274 CrossRef CAS.
- A. H. M. de Vries, F. J. Parlevliet, L. Schmieder-van de Vondervoort, J. H. M. Mommers, H. J. W. Henderickx, M. A. N. Walet and J. G. de Vries, Adv. Synth. Catal., 2002, 344, 996–1002 CrossRef CAS.
- J. Evans, L. O'Neill, V. L. Kambhampati, G. Rayner, S. Turin, A. Genge, A. J. Dent and T. Neisius, J. Chem. Soc., Dalton Trans., 2002, 2207–2212 RSC.
- W. Kleist, S. S. Pröckl and K. Köhler, Catal. Lett., 2008, 125, 197–200 CrossRef CAS.
- J. Guerra, J. L. Burt, D. Ferrer, S. J. Mejía-Rosales and M. José-Yacamán, J. Nanopart. Res., 2009 DOI:10.1007/s11051-010-9927-0.
- X. Chen, Y. Hou, H. Wang, Y. Cao and J. He, J. Phys. Chem. C, 2008, 112, 8172–8176 CrossRef CAS.
- N. Karousis, G.-E. Tsotsou, F. Evangelista, P. Rudolf, N. Ragoussis and N. Tagmatarchis, J. Phys. Chem. C, 2008, 112, 13463–13469 CrossRef CAS.
- P.-P. Zhang, X.-X. Zhang, H.-X. Sun, R.-H. Liu, B. Wang and Y.-H. Lin, Tetrahedron Lett., 2009, 50, 4455–4458 CrossRef CAS.
- J. A. Sullivan, K. A. Flanagan and H. Hain, Catal. Today, 2009, 145, 108–113 CrossRef CAS.
- J. E. Hamlin, K. Hirai, A. Millan and P. M. Maitlis, J. Mol. Catal., 1980, 7, 543–544 CAS.
- K. Köhler, R. G. Heidenreich, S. S. Soomro and S. S. Pröckl, Adv. Synth. Catal., 2008, 350, 2930–2936 CrossRef.
- N. Rubio, M. A. Herrero, M. Meneghetti, A. Díaz-Ortiz, M. Schiavon, M. Prato and E. Vázquez, J. Mater. Chem., 2009, 19, 4407–4413 RSC.
- T. Itoh, H. Danjo, W. Sasaki, K. Urita, E. Bekyarova, M. Arai, T. Imamoto, M. Yudasaka, S. Iijima, H. Kanoha and K. Kaneko, Carbon, 2008, 46, 172–175 CrossRef CAS.
- N. E. Leadbeater, Chem. Commun., 2005, 2881–2902 RSC.
- N. E. Leadbeater and M. Marco, Org. Lett., 2002, 4, 2973–2976 CrossRef CAS.
- M. T. Reetz and J. A. G. de Vries, Chem. Commun., 2004, 1559–1563 RSC.
- J. Demel, S.-E. Park, J. Cejka and P. Stepnicka, Catal. Today, 2008, 132, 63–67 CrossRef CAS.
- J. Demel, E.-S. Sujandi, S.-E. Park, J. Cejka and P. Stepnicka, J. Mol. Catal. A: Chem., 2009, 302, 28–35 CrossRef CAS.
- W. Kleist, J.-K. Lee and K. Köhler, Eur. J. Inorg. Chem., 2009, 261–266 CrossRef CAS.
- G. Budroni, A. Corma, H. García and A. Primo, J. Catal., 2007, 251, 345–353 CrossRef CAS.
- M. Lysén and K. Köhler, Synlett, 2005, 1671–1674 CrossRef CAS.
- M. Lysén and K. Köhler, Synthesis, 2006, 692–698 CrossRef CAS.
- J. M. Richardson and C. W. Jones, Adv. Synth. Catal., 2006, 348, 1207–1216 CrossRef CAS.
- K. Köhler, S. S. Pröckl and W. Kleist, Curr. Org. Chem., 2006, 10, 1585–1601 CrossRef.
- S. S. Pröckl, W. Kleist, M. A. Gruber and K. Köhler, Angew. Chem., Int. Ed., 2004, 43, 1881–1882 CrossRef.
- M. Weck and C. W. Jones, Inorg. Chem., 2007, 46, 1865–1875 CrossRef CAS.
- A. de la Hoz, A. Diaz-Ortiz and A. Moreno, Adv. Org. Synth., 2005, 1, 119–171 Search PubMed.
- F. Langa and P. de la Cruz, in Microwaves in Organic Synthesis, ed. A. Loupy, Wiley-VCH,Weinheim, 2nd edn, 2006, vol. 1–2 Search PubMed.
- F. G. Brunetti, M. A. Herrero, J. de Munoz, S. Giordani, A. Díaz-Ortiz, S. Filippone, G. Ruaro, M. Meneghetti, M. Prato and E. Vázquez, J. Am. Chem. Soc., 2007, 129, 14580–14581 CrossRef CAS.
- E. Vázquez, V. Georgakilas and M. Prato, Chem. Commun., 2002, 2308–2309 RSC.
- K. MacKenzie, O. Dunens and A. T. Harris, Sep. Purif. Technol., 2009, 66, 209–222 CrossRef CAS.
- F. G. Brunetti, M. A. Herrero, J. de Muñoz, A. Díaz-Ortiz, J. Alfonsi, M. Meneghetti, M. Prato and E. Vázquez, J. Am. Chem. Soc., 2008, 130, 8094–8100 CrossRef CAS.
- P. Handa, K. Wikander and K. Holmberg, 2009, 117, pp. 126–135.
- M. B. Thathagar, P. J. Kooyman, R. Boerleider, E. Jansen, C. J. Elsevier and G. Rothenberg, Adv. Synth. Catal., 2005, 347, 1965–1968 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |