Sorbents for CO2 capture from flue gas—aspects from materials and theoretical chemistry
Niklas
Hedin
*,
LiJun
Chen
and
Aatto
Laaksonen
Department of Materials and Environmental Chemistry, Berzelii Center EXSELENT on Porous Materials, Arrhenius Laboratory, Stockholm University, S-106 91, Stockholm. E-mail: niklas.hedin@mmk.su.se
First published on 2nd August 2010
Abstract
Predictions of future climate change have triggered a search for ways to reduce the release of greenhouse gases into the atmosphere. Carbon capture and storage (CCS) assists this goal by reducing carbon dioxide emissions, and CO2 adsorbents in particular can reduce the costs of CO2 capture. Here, we review the nanoscale sorbent materials that have been developed and the theoretical basis for their function in CO2 separation, particularly from N2-rich flue gases.
 Niklas Hedin | Niklas Hedin, MSc. Chemical Engineering and PhD Physical Chemistry from the Royal Institute of Technology, Stockholm, Sweden. Post doctoral research with Bradley Chmelka in University of California at Santa Barbara, US (2001–2003) and with Sebastian Reyes at ExxonMobil Corporate Research Laboratories in Annadale, US (2004–2006). Now Associate Professor in Materials Chemistry, Department of Materials and Environmental Chemistry at Stockholm University, Sweden. Main research interest is adsorbents that could allow a cost effective separation of CO2 from N2-rich gases; special focus on molecular details on solid–gas interactions. |
 LiJun Chen | LiJun Chen was born in Zhejiang, China in 1979. She obtained a Bachelor's degree in Applied Chemistry in 2002 and PhD in Physical Chemistry in 2007 at Jilin University. Since 2007 she has been working as a postdoctoral student in the Department of Materials and Environment Chemistry, Stockholm University. Her main research interests covered the development of multi-scale simulation methods bridging molecular dynamics, dissipative particle dynamics with the application in simulations of macromolecules. Now she focuses on the theoretical studies of the porous materials combining quantum chemistry, molecular dynamics and Monte Carlo approaches. |
 Aatto Laaksonen | Aatto Laaksonen is a Professor in Physical Chemistry at Stockholm University; BSc. Mathematics, Stockholm University; PhD in Physical Chemistry Stockholm University 1981; postdoctoral research with Dr Victor Saunders 1982, Daresbury Laboratory, UK; postdoctoral research with Dr Enrico Clementi 1983–1985, IBM research laboratories in Poughkeepsie and in Kingston, USA. Active in the area of computational materials science, being responsible for modelling work to design new nano- and mesoporous materials for separation and storage of gases and for heterogeneous catalysis processes inside the pores within the newly established center-of-excellence at Stockholm University. |
1. Introduction
Cost-effective large-scale separation and capture of CO2 from gas mixtures, followed by storage (and possibly recycling of the carbon and the oxygen) are of the utmost importance, due to the increasing impact of CO2, a greenhouse gas, on global warming.1,2 It is a priority to find inexpensive, effective, and robust materials and technologies that reduce emissions of CO2 and other greenhouse gases and that are suitable for installation in power plants and industries that rely on fossil fuels.3,4Three types of gas mixtures are targets for capture and separation technologies: the components of flue gases (mainly CO2/N2) and natural gases (mainly CH4/CO2), and precombustion gas mixtures that contain H2. The aim for precombustion separation is to develop materials that adsorb and separate H2, which is important for certain new CCS precombustion capture procedures. Aside from the components in flue gases, natural gases, and hydrogen, many other gases can be found in gas mixtures. For example, water is found in small amounts in most systems. Water generally leads to complications in separation applications, although under certain circumstances it may contribute constructively to the process. Surprisingly, only a few computational investigations have included water in detailed studies of its role in this regard.
The use of appropriate nanostructured materials may potentially reduce the costs associated with CCS. For example, membranes or adsorption-driven processes may be used to separate CO2 from N2-rich flue gases.5 CO2 is currently separated from N2-rich gases via absorption by aqueous solutions of, for example, simple alkanolamines or chilled NH3.2 If increased CO2 selectivity can be achieved, adsorption-mediated separation of CO2 from flue gases can potentially separate CO2 from N2 at a much lower cost than that associated with current technologies.6 Recent efforts to develop adsorbent materials have focused on zeolites, metal organic frameworks (MOFs), and hybrid systems, as well as highly alkaline adsorbents. Alkaline hybrid systems may selectively adsorb CO2 over N2. Carbon and silica materials are also of great interest in this regard. Among solution-based technologies, ionic liquids may potentially be used for CO2 capture. Here, we review the adsorbents for CCS that have been described in the literature, with an emphasis on both materials and computational investigations into the uptake and selectivity of CO2 from N2-rich gases.
The free energy of gas molecules decreases in the presence of the attractive electronic environment at an interface.7 The density of gas molecules increases close to an interface, and the average diffusion length of each molecule decreases. Development of a surface excess is called “adsorption” and is an exothermic process that arises as a consequence of attractive interactions and the associated loss of entropy.7 Adsorption is typically divided into physi- and chemisorption, in which no chemical bonds are formed during physisorption. During chemisorption, the gas and the adsorbents undergo electronic reconfiguration. Both types of adsorption are relevant to CO2 separation. Adsorbents contain nanoscale morphological features and are typically divided into three classes based on pore size: according to the International Union of Pure and Applied Chemistry (IUPAC), microporous materials have pores smaller than 2 nm, mesoporous materials have pores between 2 and 50 nm, and macroporous materials have pores larger than 50 nm. For CO2 capture, sorbents with high specific surface areas have proven to be the most promising.8–14
The main workhorses in computational studies of carbon capture and separation are molecular dynamics (MD) simulations, grand canonical Monte Carlo (GCMC) simulations,15,16 and ab initio quantum chemistry (QC) calculations at suitable levels of theory. In QC, density functional theory (DFT) is the first choice because it includes electron correlation effects. However, DFT demands care when applied to systems that contain hydrogen bonds or weak interactions. All modeling methods (MC, MD, and QC) can be used either independently or in conjunction with experiments (in complementarity) to gain information at the molecular level. The large data sets obtained from these traditional calculations may be used in newer methods, such as kinetic Monte Carlo (kMC) simulations. The kMC method requires a detailed knowledge of the system, including all stationary states on the free energy surface and the barriers that separate these states.
Transition state theory (TST) can be applied in kMC simulations, and has been used in adsorption studies reported in the literature.17 MC simulations performed in the grand canonical ensemble yield statistical and ensemble properties of the adsorption and desorption processes and predict the preferred locations of adsorption. MD simulations model the transport properties (such as diffusion) of gases as they interact with (are captured by) a sorbent material. Selective capture requires that target captured gases quickly find their way (i.e., are “channeled”) to the area of a surface designed for absorption. Optimally, other gas components encounter this surface more slowly or at a later time.
MD simulations are useful at several stages of the modeling process. After constructing the molecular structure and framework of an adsorbent substrate, it is important to submit the material to repeated simulated annealing steps to test the stability and robustness of the model substrate prior to introducing gas molecules. An accurate model for the adsorbate is also very important. For example, CO2 does not have an electric dipole moment; however, it has a large electric quadrupole moment that arises from the strong dipole moments of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds. At short distances, OCO is locally highly polar and requires an accurate description of the electrostatic component of its force field (FF) used in a simulation. This is especially important when CO2 is present in zeolites, which have a strong ionic character due to the zeolite metal atoms or additional ions hosted in the structure.
O bonds. At short distances, OCO is locally highly polar and requires an accurate description of the electrostatic component of its force field (FF) used in a simulation. This is especially important when CO2 is present in zeolites, which have a strong ionic character due to the zeolite metal atoms or additional ions hosted in the structure.
A major component of many gas mixtures is N2. Although it appears to be a simple molecule, the triple bond sometimes complicates the theoretical calculation of its molecular properties. N2 often serves as a benchmark molecule for new computational schemes. Ab initio calculations and simulations can be applied when additional levels of detail are required for modeling the structures or interactions in a simulation. QC calculations are important for distinguishing chemisorption from physisorption, because these calculations permit evaluation of the electronic rearrangements associated with a particular process. Most importantly, QC calculations aid investigations into reaction mechanisms; e.g., the mechanisms that underlie catalyzed reactions. QC calculations are routinely used to develop FFs for atomic charges or to fine-tune descriptions of bonded interactions. Empirical FFs are the most important component of a computer simulation, because the integrity of the simulation results depends directly and critically on these FFs.
During the past decade, significant progress has been made in the development of FFs that describe the adsorption of greenhouse gases. Several improved parameterizations have been suggested for zeolites, MOFs, and silicas, which will be discussed in this review. As an example of the parameterization, García-Sánchez et al.18 recently developed a complete FF that accurately reproduced the adsorption properties of CO2 in a variety of zeolites with various topologies and compositions. The FF parameters were obtained by numerical optimization using their own experimental data, and were subsequently validated by comparison with available literature data. Their model explicitly distinguished silicon from aluminium using different charge for oxygen atoms bridging two silicon atoms (qOSi) and oxygen atoms bridging one silicon and one aluminium atom (qOAl). The Lennard-Jones interactions between CO2 and the zeolite were modeled by only taking into account the interactions between carbon dioxide and O atoms and Na cations of the zeolite. An essential feature of their model was the mobility of the Na cations in the framework, which they claimed to be vital to reproduce accurately adsorption of carbon dioxide in faujasites. The FF was fully transferable between zeolite framework types and was applicable to all possible Si/Al ratios (with sodium as an extra-framework cation).
This review focuses mainly on the work performed in the first decade of this century. During this period, many novel types of nanoscale materials have been discovered and tested for potential application to separation and temporary storage technologies. We review the material development, modeling, and simulation efforts. The extensive range of related engineering literature is not reviewed here; for this, we refer the reader to other reviews.2,19,20,21
2. Sorbents for CO2 capture from flue gases
A variety of sorbents have been investigated as potential CO2 capture substrates for use with flue gases in pressure swing or temperature swing adsorption processes.4,7,22 Successful adsorbents must fulfil a range of chemical, chemical engineering, and solid mechanics criteria. As an introduction, we refer the reader to an excellent but only partly overlapping review by Choi et al., which considered adsorbents for the adsorptive capture of CO2 from large point sources.23 We review the theoretical advances in combination with aspects from material chemistry.Adsorbents can be categorized in many ways, including by composition (as in the present study) or by pore dimensions. Zeolites, carbon molecular sieves (CMSs), and MOFs are examples of microporous adsorbents with molecule-sized pore windows. These materials have been shown to separate gas molecules by equilibrium, kinetic, or molecular sieving mechanisms. Gas molecules have different effective kinetic diameters, and CO2 appears to have the smallest kinetic diameter. CO2, N2, and CH4 have effective kinetic diameters in zeolites of 0.33, 0.365, and 0.38 nm, respectively.24 The exact values are under discussion and appear to be substrate dependent.4,24,25 For simulations of gas molecules in sorbents, several well-known sorbate models, and force fields, for the sorbate–sorbate and sorbate–sorbent interactions have been developed and used. Intermolecular interactions are usually represented by a Lennard-Jones term and an electrostatic term:
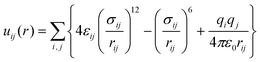
| Adsorbate | Model | σ/nm | (ε/kB)/K | q/e | Ref. |
|---|---|---|---|---|---|
| a The parameters were fitted to the experimental VLE data of bulk CO2. b The parameters were fitted to reproduce the solid CO2 structure. c The parameters were fitted to reproduce the vapor–liquid coexistence curves. d N2 was represented as a three-site model, with a positive charge (q+ = 2q−) in the middle of the N–N bond. | |||||
| C–C (CO2) | EPM247,a | 0.2785 | 28.999 | +0.6645 | 17,18,35,36,29,28 |
| O–O (CO2) | EPM247 | 0.3064 | 82.997 | −0.33225 | 17,18,35,36,29,28 |
| C–C (CO2) | Maurin et al.48,b | 0.383 | 46.601 | +0.72 | 42,46,47 |
| O–O (CO2) | Maurin et al.48 | 0.336 | 76.452 | −0.36 | 42,46,47 |
| C–C (CO2) | TraPPE49,50,c | 0.280 | 27.0 | +0.70 | 33,32,29,34 |
| O–O (CO2) | TraPPE49,50 | 0.305 | 79.0 | −0.35 | 33,32,29,34 |
| N–N (N2) | Murthy et al.51,d | 0.332 | 36.4 | −0.482 | 29,28 |
| CH4–CH4 | TraPPE49,50 | 0.373 | 148.0 | 0 | 29 |
| CH4–CH4 | Jiang et al.52 | 0.3812 | 148.2 | 0 | 32 |
| Sorbent | T/K | p CO2/kPa | n/mmol g−1 | Ref. |
|---|---|---|---|---|
| a Note: AP = n-propyl amine; PEI = polyethyleneimine; for other abbreviations see the text or the references. | ||||
| Zeolites and all-silica microporous solids | ||||
| NaX | 306; 295 | 67; 100 | 5.4; 4.5 | 8,9 |
| NaY; HY | 295; 295 | 100; 100 | 4.0; 1.1 | 9 |
| Na-ZSM-5; H-ZSM-5 | 297; 295 | 0.67; 100 | 1.8;1.9 | 8,9 |
| Silicalite | 295 | 100 | 1.4 | 8,9 |
| NaA | 298 | 93 | 4.1 | 10 |
| β-zeolite | 303 | 100 | 1.8 | 53 |
| Na-mordendite | 308 | 1000 | 2.8 | 54 |
| Herschelite | 298 | 690 | 2.0 | 55 |
| ITQ-6; ITQ-6/AP | 293; 293 | 100; 100 | 1.1; 1.2 | 56 |
| Aluminium phosphates | ||||
| ALPO4-5 | 165 K, P/P0 = 0.06 | 3.3 | 57 | |
| ALPO4-14 | 290 | 100 | 2.2 | 58 |
| SAPO4-34 | 298 | 100 | 3.4 | 59 |
| STA-7 | 303 | 2800 | 7.5 | 60 |
| Metal–organic frameworks | ||||
| MOF-177 | 298 | 3000 | 32 | 11 |
| IRMOF-1/MOF-5 | 298 | 3000 | 21 | 11 |
| IRMOF-6 | 298 | 3000 | 18 | 11 |
| IRMOF-3 | 298 | 3000 | 18 | 11 |
| ZIF-69 | 273 | 100 | 3.1 | 61 |
| ZIF-20 | 273 | 100 | 3.1 | 61 |
| ZIF-100 | 298 | 110 | 0.95 | 62 |
| MIL-53 | 304 | 1600 | 9.0 | 63 |
| MIL-100 | 303 | 6000 | 18 | 12 |
| MIL-101(Cr) | 298 | 5000 | 40 | 12 |
| Cu2(BTC)2 | 298 | 3000 | 11 | 11 |
| Imidazolate MOF | 253 | 100 | 4.9 | 64 |
| Covalent organic frameworks and porous organics | ||||
| COF-10/102/103 | 298 | 5500 | 22–27 | 13 |
| Carbons | ||||
| BF CMS | 298 | 280 | 2.6 | 65 |
| PX21 | 303 | 5000 | 15 | 14 |
| BPL | 303 | 5000 | 10 | 14 |
| CFCMS | 298 | 5700 | 11 | 66 |
| AC3000 (carbon) | 303 | 4000 | 11.7 | 67 |
| Graphene | 195 | 100 | 8.0 | 68 |
| CMK-3 | 298 | 100 | 1.7 | 69 |
| Amine modified mesoporous silica | ||||
| SBA-12/AP | 298 | N/A | max: 1.0 | 70 |
| MCM-41/PEI | 348 | N/A | 215 mg g−1-PEI | 71 |
| SBA-15/Hyperbranched amines | 298 | N/A | 3.1 | 72 |
| MCM-48/AP | 298 | 100 | 2.2 | 73 |
| Xerogel/AP | 298 | 100 | 1.2 | 73 |
| AMS | 298 | N/A | 1.25 | 74 |
| MCM-41/pore expanded/TRI | 358 | N/A | 2.7 | 75,76 |
| MCM-41/pore expanded/DEA | 298 | 100 | 3.25 | 77,78 |
| Silica monolith/TEPA | 348 | N/A | 260 mg g−1 sorbent | 79 |
| Other sorbents | ||||
| HTlc | 673 | 100 | 1.5 | 80 |
| CARiACT/PEI | 313 | N/A | 3.1 | 81 |
| Diaion/PEI | 313 | N/A | 2.5 | 82 |
| MCM-41 Silica | 298 | 2500 | 9.6 | 83,84 |
| Amine-modified hydrothermal carbon | 253 | 100 | 4.1 | 85 |
In simulation of zeolites and carbons, the Lennard-Jones and partial charges of the individual atoms are either derived from ab initio calculations28 or from “fitting” to experimental isotherms.18,29 For MOF frameworks, the Lennard-Jones parameters are often extracted from generic force fields, in particular UFF30 or DREIDING,31 that contain parameters for the whole periodic table.17,32,33,34 Salles et al.35,36 made use of the intramolecular and nonbonded parameters, for a particular organic moiety, from the Consistent Valence Force Field (cvff).37 The interactions between inorganic and organic ligands, including bond stretching, angle bending, a torsion term and nonbonded parameters, were adjusted from the DREIDING force field in order to reproduce the structural features of both large and narrow pore forms of MIL-53. The modeling captures accurately the swelling/shrinkage of the structure. The partial charges of the framework atoms were usually calculated from DFT using Hirshfeld,38 Mulliken,39 ChelpG40 and Merz–Kollman41 charge calculating methods.17,34,42 In particular, an efficient and systematic first principles parameterization of force fields based on MM343 for MOFs using a genetic algorithm approach was proposed by Tafipolsky and Schmid44
The economical separation of CO2 from point sources, using adsorbent-driven processes, imposes many demands on the sorbent. Both the uptake capacity for CO2 and the preferential CO2-over-N2 selectivity are important for economical pressure or temperature swing adsorption processes. Wiley et al. showed that increasing the CO2 selectivity of the adsorbents could radically lower the costs of separation.6 Sensitivity to water, cost, and recyclability are important economical factors. Although the engineering aspects of adsorbent-driven CO2 separation are not considered here, this field of research would benefit substantially from closer collaboration between engineers and chemists.
2.1. Zeolites and all-silica microporous solids
Zeolites are microporous aluminosilicates that occur naturally, but can also be synthesized in the laboratory. Zeolites contain a network of interconnecting channels or cages that can be used to separate gas molecules via equilibrium, kinetic, or molecular sieving mechanisms. Zeolites are typically described by the number of oxygen atoms encircling the smallest windows that form a percolating network: 8-, 10-, and 12-ring zeolites. Zeolites are described as having either cages or channels. In zeolites of the cage type, the pore window is an aperture that constricts the diffusive paths. Channel-type zeolites contain a locally tubular diffusive path. Fig. 1 shows the structures of some of the zeolites discussed in this review.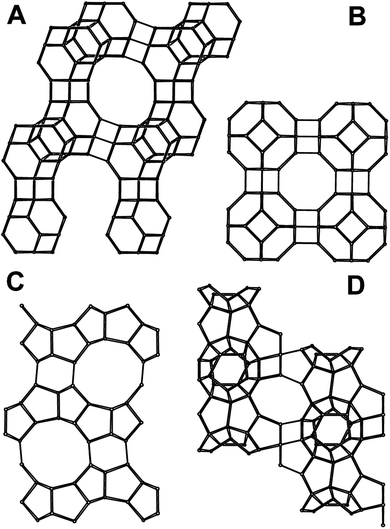 | ||
| Fig. 1 Zeolite structures: (A) zeolite X, structural code FAU; (B) zeolite A, structural code LTA; (C) ZSM-5, structural code MFI; (D) ZSM-58, structural code DDR. Redrawn from the Database of Zeolite Structures85 with permission. | ||
Zeolite X (Fig. 1a) is the most widely studied sorbent for CO2 capture. It has a caged structure (structural code FAU,85 Faujasite) and window apertures defined by 12-membered rings, which allow unhindered access for CH4, N2, CO2, and H2O. Zeolite X has a Si/Al ratio of 1.3,86 and the presence of alumina gives the framework a negative charge. The cations of zeolite X affect the heat of adsorption for CO2, such that increases in the heat of adsorption result from increased monovalent charge densities.8,87 Harlick and Tezel concluded that NaX or NaY were the best adsorbents for separation processes (Fig. 2).9 Cavenati et al. showed that NaX yielded CO2-over-N2 adsorption selectivity at pressures as high as 3.2 MPa at 298 K.88 The adsorption of CO2 was shown to be higher in NaX than in NaZSM-5, even though the heats of adsorption were similar.8 Brandani and Ruthven studied the uptake of CO2 in NaX, CaX, LiLSX, and NaLSX (zeolite LSX had a Si/Al ratio of 1) and recognized that water compromised the uptake of CO2.89 When water was adsorbed, the electric field gradients decreased and CO2, with its significant electric quadrupolar moment, was less prone to adsorb. The presence of water vapor in CO2 capture processes could be a serious problem for NaX.
Papadopoulos and Theodorou90 investigated the sorption dynamics of CH4, CO2, H2, and D2 in the frameworks of ITQ-1 and NaX zeolites using atomistic and mesoscopic computer simulations. They found that the loading dependence of self-diffusivity (Ds) was affected by the energetic inhomogeneity of the sorption sites and/or the site topology. The diffusion and adsorption of CO2 inside the pores of Li+, Na+, and K+ ion-exchanged X-type zeolites were simulated by Nakazaki and others91 using MD and MC simulation methods. CO2 was found to diffuse into the zeolite pores, collide with pore walls, and remain trapped in the supercages of the zeolite structures. CO2 was found to adsorb strongly near the 3B site of the Li+ ions. MD simulations by Jia and Murad92 studied gas separation using zeolite membranes (FAU) and two binary mixtures: O2/N2 and CO2/N2. These mixtures were found to exhibit different behavior in the presence of the membrane. For the O2/N2 mixture, adsorption and loading in the membrane were similar for both O2 and N2. The observed drop in selectivity resulted from interactions between gas molecules: O2 slowed the rate of diffusion of N2, while N2 slightly increased the rate of diffusion of O2 when passing through the pores. CO2, on the other hand, was selectively adsorbed and loaded in the zeolite, and did not leave much space for N2 adsorption. While N2 continued to have a higher diffusion rate than CO2, so few N2 molecules were present in the zeolite that the selectivity showed a significant increase. Similar to the above study, Jia and Murad93 investigated gas separation efficiencies in three aluminium-rich zeolite membranes by MD simulation, with the aim of studying the effects of pore size, pore structure, state condition, and composition on the permeation of two binary gas mixtures: O2/N2 and CO2/N2. (The three membranes consisted of zeolites with different structures and structural codes: FAU, MFI, and CHA.)
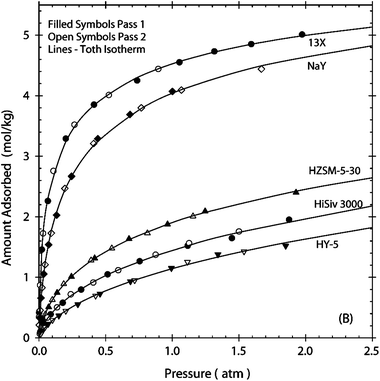 | ||
| Fig. 2 Carbon dioxide adsorption isotherms for different zeolites. From Harlick and Tezel,9 reprinted with permission. | ||
Zeolite Y has the same general structure as zeolite X, but the Si/Al ratio is 2–3.24 The prospects for using NaY to capture CO2 were investigated and found to be reasonably promising.9 The more hydrophobic NaY structure had fewer cations than did NaX, and the heat of adsorption for CO2 was lower. Galhotra et al. studied CO2 adsorption in zeolite Y, varying the cation type and water content.94 Infrared spectroscopy data showed that most CO2 molecules were physisorbed; however, some chemisorbed CO2 was detected. Harlick and Tezel measured the CO2 adsorption isotherms for NaY at different pressures [0.5–202 kPa] and temperatures [293–473 K]. The collected data were well represented as a Toth isotherm.95
Shao et al.96 studied CO2 and N2 adsorption in NaY over a wide range of temperatures, from 303 K to 473 K, and at pressures up to 100 kPa. They found that the adsorptive uptake of CO2 by the NaY was higher than in any other porous material reported, suggesting that NaY is a good adsorbent for CO2 capture at high temperatures. The intensity of the interactions between CO2 and the walls of the cavities in the zeolite were heterogeneously distributed. Maurin et al.97,98 combined GCMC simulations with adsorption measurements on LiY and NaY (FAU) zeolites at various temperatures. A new FF for the Li+-CO2 interaction was derived based on ab initio calculations. Two types of adsorption behavior were observed for NaY and LiY zeolites at 323 K and 373 K. Ghoufi et al.99 studied the adsorption of CO2, CH4, and an equimolar mixture in NaY by combining GCMC simulations with volumetric and gravimetric uptake experiments. The simulations showed a high CO2-over-CH4 selectivity in NaY across the range of pressures studied, revealing preferential adsorption sites for both adsorbates. Plant et al.100 carried out both GCMC and MD simulations to study CO2 adsorption in NaX and NaY (FAU). A new FF for the Na+–CO2 interaction was derived using quantum calculations. Two types of adsorption behavior were observed for NaY and NaX.
Plant et al.101 carried out MD simulations to study the cation rearrangement in NaX and NaY (FAU) during the process of CO2 adsorption. GCMC simulations, combined with microcalorimetry measurements, were performed by Maurin et al.98 to study the interaction of CO2 with two types of FAU surface. Plant et al.102 combined quasi-elastic neutron scattering (QENS) with MD simulations to investigate CO2 dynamics in LiY and NaY (FAU), where the transport diffusivity (DT) was shown to increase with loading, whereas self-diffusivity (Ds) decreased. In addition, the authors showed that LiY exhibited significantly slower CO2 self-diffusion due to the initial strong interactions between Li+ cations and the adsorbate molecules.
The adsorption of CO2 on HY (FAU, Si/Al = 8) was investigated by Pulido et al.103 in a combined study of variable-temperature IR spectroscopy and DFT/coupled cluster (CC) calculations. The calculations showed that weak interactions played an important role in adsorption. The calculated and experimental stretching frequencies were in good agreement. The adsorption of CO2 in the alkali-exchanged zeolite Y (Li+, Na+, K+, and Cs+) was investigated using DFT calculations (Plant et al.104). The cation–CO2 geometry was investigated as a function of the nature of the alkali cations. The calculated adsorption enthalpies showed a decrease from Li+ to Cs+ and reproduced experimental microcalorimetry results. Chatterjee and Iwasaki105 investigated the separation of gas components from a mixture of CO2, N2, CH4, C2H6, and SF6, with a focus on the selective permeation of CO2 from a mixture of CO2/N2 through a NaY membrane. Reactivity descriptors and interaction energies were calculated using DFT. Permeation, as a function of the affinity between gas molecules and the membrane wall, was analyzed to predict the optimal affinity strength (higher selectivity) for CO2. Their results predicted the experimentally observed selective permeation order of C2H6 < CH4 < SF6 < CO2 < N2.106
Zeolite A (Fig. 1b) has small interconnecting windows composed of 8-rings. The small windows render zeolite A suitable for molecular sieving. The Ca2+ form of zeolite A is called 5A or CaA, the Na+ form is called 4A or NaA, and the K+ form is called 3A or KA, where the number corresponds to the approximate molecular window size. It is worthwhile comparing these sizes with the effective kinetic diameters given in Table 3. In an early seminal study, Breck et al. showed that the capacity for CO2 adsorption varied with the Na+/K+ ratio in zeolite A (Fig. 3).10 CaA had the largest heat of adsorption for CO2 among the solid–CO2 pairs studied by Harlick and Tezel.9 The large heat is related to the very small pore size, the properties of Ca2+, and the large number of aluminium atoms present in the structure. Zeolite A was shown to have an overall capacity for CO2 sorption at 393 K that was lower than the capacity of NaX. The capacity loss was attributed to a stronger tendency toward forming chemisorbed carbonate species in zeolite A. The capacity of CaA was even lower than that of NaA.
Liu and Yang107 used GCMC to study the adsorption of supercritical CO2 on two types of zeolite with identical chemical compositions but different pore structures, NaA and NaX. Jaramillo and Chandross108 developed FFs for calculating the adsorption of NH3, CO2, and H2O on NaA by GCMC. They also studied the geometry of the adsorption sites and correlated the loading with geometry. At low pressures, the adsorption geometry of CO2 molecules was such that the longitudinal axis was directed toward the center of the supercage. At higher pressures, the two oxygen atoms were found to be equidistant from the Na+. Akten et al.109 used GCMC simulations to assess the adsorption selectivity of CO2/N2 and CO2/H2 mixtures as a function of temperature and gas composition. At room temperature, NaA showed a strong selectivity for CO2 over both N2 and H2. The selectivity decreased slightly at high pressures. Ideal adsorption solution theory (IAST) predicted the adsorption selectivity at low partial pressures of CO2 using a functional form that accurately described the isotherm for CO2. IAST was found to perform reasonably accurately in modeling these adsorbed mixtures. Izumi et al.110 precisely evaluated the window shrinkage of NaA, at a resolution of 0.01 nm, by calcination, rehydration, partial K+ exchange, and low-temperature adsorption. The oxygen selectivity in a binary O2/N2 system and the CO2 selectivity in a binary CO2/N2 were studied by MAS-NMR, single crystal X-ray diffraction (SCXD), and MD simulations. We recently showed that a high CO2-over-N2 selectivity could be achieved by carefully tuning the Na+/K+ ratio in zeolite A.111
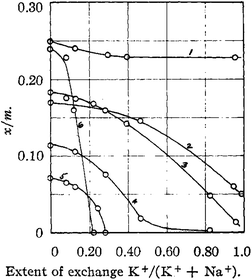 | ||
| Fig. 3 From Breck et al.,10 the uptake of different gases in NaKA: (1) water at 298 K and 0.6 kPa; (2) methanol at 298 K and 0.5 kPa; (3) CO2 at 298 K and 93 kPa; (4) ethylene at 298 K and 93 kPa; (5) ethane at 298 K and 93 kPa; (6) O2 at 90 K and 93 kPa. Reprinted with permission. | ||
All-silica microporous materials (or zeolites with very large Si/Al ratios) are hydrophobic, and hence much less sensitive to water adsorption than are aluminium-rich zeolites. This property has prompted many recent investigations into their ability to separate CO2/CH4 and CO2/N2 gas mixtures. CO2 sorption on ZSM-5 and silicalite112 (Fig. 1c) have been studied for this purpose.8,9,113–115 ZSM-5 and silicalite have the structural code MFI and have intersecting channels with 10-membered rings.85 These materials permit unhindered access to the porous network for all molecules relevant to CCS processes. It is instructive to see the apertures of the windows with the kinetic diameters listed in Table 3. The silicalite material contains no cations, and the heat of adsorption for CO2 was determined to be small (27 kJ mol−1).113
Previous studies measured the binary adsorption isotherms of CO2, N2, and methane on a ZSM-5 sorbent with SiO2/Al2O3 ratios of 30 and 280.114,115 Hirotani et al.116 studied the adsorption of CO2 in silicalite and NaZSM-5 using GCMC and found good agreement between the calculated adsorption isotherms and the experimental results collected by Yamasaki et al.117 and Geiger et al.118 The adsorption energy for NaZSM-5 was attributed to the electric field of the sodium cations. Zukal et al.119 measured the adsorption isotherms of CO2 in cation-exchanged (Li+, Na+, K+, Cs+) MCM-22, with varying molar ratios of Si/Al and at different temperatures. They calculated the isosteric heats of adsorption to gain insight into the interaction of CO2 with alkali metal cations. GCMC simulations were carried out by Leyssale et al.120 to study the thermodynamic properties of CO2 and CH4 adsorbed on the siliceous forms of MCM-22 and ITQ-1 (MWW), with their two independent pore systems. ITQ-1 was found to be CO2-selective in CO2/CH4 mixtures, with a maximum in selectivity observed at low temperatures, high pressures, and CH4-rich gas compositions. Yue and Yang121 studied the adsorption and diffusion of binary mixtures of supercritical CO2 and benzene on silicalite using GCMC and MD simulations. Their simulation results suggested that supercritical CO2 fluid could be used to efficiently desorb larger aromatics in the zeolitic materials. MD simulations revealed that the large adsorbed benzene molecule had a pronounced effect on the diffusion of CO2. Papadopoulos et al.122 carried out coherent QENS experiments and MD simulations to study the concentration dependence of N2 and CO2 transport diffusion in silicalite-1. Sorbate–sorbate interactions were found to be much more attractive in CO2/silicalite-1 than in N2/silicalite-1. GCMC simulations of the adsorption of CO2 and N2 were carried out by Himeno et al.123 on all-silica DDR (Fig. 1d) and MFI (Fig. 1c). The simulated sorption capacities, isosteric heats of adsorption, and Henry's constants, for all-silica DDR and MFI, agreed well with the experimental data.
García-Pérez et al.124 studied the adsorption properties of CO2, N2, and CH4 in all-silica microporous materials using molecular simulations. They computed the adsorption isotherms at a wide range of pressures and temperatures, and for pure (single-component), binary, and ternary component mixtures with varying bulk compositions.
A shortcoming of membranes is that they typically cannot possess both high adsorption and high diffusion selectivity. However, there are exceptions: for example, DDR membranes. Jee and Sholl125 used MD simulations to study the diffusion of CO2, and a transition state theory-based kinetic Monte Carlo scheme to accurately describe the extremely slow diffusion of CH4 (less than 10−7 cm2 s−1, which is beyond the reach of MD) inside all-silica DDR (Si120O240) applying an improved FF, yielding results that agreed well with experiments.126,127 They observed that the characteristics of CO2/CH4 diffusion in DDR were different from the characteristics of diffusion in nanoporous materials. In DDR, the diffusion rates of CO2 were only weakly affected by the presence of the much slower-diffusing CH4. They claimed that this unusual phenomenon related to different adsorption sites and diffusion mechanisms of the two species. In DDR, the 8-membered rings (8MR, 0.36 × 0.44 nm) and the 19-hedra cages (∼0.6 nm) are the structural features that are relevant for molecular transport and adsorption. The 8MR are the most energetically favorable adsorption sites for CO2; CO2 adsorbs in the 19-hedra cages only after the 8MR are occupied. However, CH4 can only occupy the larger 19-hedra cages. The adsorption of CO2 in the 8MR hinders the hopping of CH4 through the 8MR windows into the 19-hedra cages. Competitive adsorption of CO2 and CH4 occurs only in the 19-hedra cages. The large pore size ensures that CO2 diffusion will not be impeded significantly by CH4. The diffusive transport of CO2 is only weakly affected by the presence of CH4, while the more rapidly diffusing CO2 molecules retard the slowly diffusing CH4 molecules. Accordingly, they suggested a modified IAST, which describes the adsorption of mixtures of CH4 and CO2 in the 19-hedra cages of DDR and predicts the total adsorbed amount of CO2 by adding the adsorbed CO2 in the 8MR windows directly from the single-component data. It proved to perform better than the conventional IAST for this gas mixture. The combination of rapidly diffusing CO2 and slowly diffusing CH4 in DDR makes tit attractive for membrane-based or kinetically driven adsorption separations. The difference in diffusivities can enhance the adsorption-based selectivity of DDR for CO2 relative to CH4.
Krishna and van Baten128 used GCMC and MD simulations to screen 12 microporous zeolitic structures to determine which membrane structure yielded the best selectivity for CO2 separation from CH4. They found that CHA and DDR, which have cages separated by narrow windows, provided the best selectivity with respect to permeation. Selassie et al.129 performed atomistic MD simulations of the diffusion behavior of CO2 and N2, as both single components and as binary mixtures, in three all-silica microporous structures that contained variations in the pore structure: ITQ-3 (ITE; 8-ring), silicalite (MFI; 10-ring), and ITQ-7 (ISV; 12-ring). CO2 consistently diffused more slowly than did N2; however, the behavior within ITQ-7 and silicalite was found to differ from that within ITQ-3.
Krishna and van Baten130 studied the separation of CO2 from gaseous mixtures containing CH4, N2, or Ar in cage-type all-silica microporous solids (DDR, CHA, LTA, and ERI). All of these microporous solids contained 8-ring structures separated by narrow windows, and the selectivity for CO2 separation was found to be dictated by both the adsorption and diffusion characteristics. Their GCMC simulations showed that a much higher proportion of CO2 was present in the window regions of cage-type structures than was present within the cages themselves. MD simulations of self-diffusion in binary mixtures showed that CO2 slowed the diffusion of the partner molecules to a much greater degree than that predicted by Maxwell–Stefan (MS) diffusion theory, parameterized by pure component data. GCMC and MD simulation results suggested that DDR and CHA should yield high permeation selectivities for membrane-based separation in CO2/CH4, CO2/N2, and CO2/Ar mixtures. For N2/CH4 separation, DDR and FRI were found to be good choices.
Krishna and van Baten reported the results of GCMC simulations131 for the adsorption of CO2/CH4, CH4/N2, and CO2/Ar mixtures in DDR structures, and observed that the window regions contained essentially no CH4 or Ar. These molecules were predominantly adsorbed within the cages, whereas CO2 and N2 molecules were adsorbed both within the cages and in the window regions. MD simulations showed that those CO2 molecules adsorbed strongly at the windows, which hindered inter-cage diffusion of other components in the mixtures. MS theory did not describe this effect.
Krishna et al.132 carried out MD simulations to estimate the dependence of MS CH4 and CO2 diffusivity on loading, within three structural topologies characterized by: (i) intersecting channels, (ii) one-dimensional channels, and (iii) cages separated by windows. Krishna et al.133 performed MD simulations to determine DS for CH4 and CO2, for both pure components and in 50–50 mixtures, over a range of molar loadings in MFI, CHA, and DDR structures. They found that the inter-cage hopping events of molecules in CHA and DDR structures, in which the cages were separated by narrow windows, were practically independent of one another; consequently, the diffusivities of pure components were the same as those in the mixture. However, in MFI, which contained intersecting channels, species that are more mobile diffused significantly slower in the mixture. Van den Bergh et al.134 introduced a new model, the “relevant site model”, to describe the dependence of diffusion in zeolites on loading. This model assumed that segregated adsorption in cage-like zeolites, within the MS framework for mass transport, described diffusivity data for N2 and CO2 in DDR135 (8-ring and cage-like all-silica structure) very well up to saturation. They also successfully extended the model to non-isothermal diffusivity data from CO2 and N2 in the DDR all-silica structure. Ohta et al.136 performed dynamic Monte Carlo (DMC) simulations to estimate the perm-selectivity of binary mixtures (CO2/N2) in zeolite-like porous membranes using several hypothesized porous structures. The rates of hopping between different adsorption sites, estimated using an empirical atomistic FF, were used to parameterize the DMC simulations. The simulation times permitted by DMC were much longer than those permitted by conventional MD simulations were. Goj et al.137 studied the adsorption of CO2 and N2, as both single components and as binary mixtures, in three all-silica structures with different pore structures (silicalite, ITQ-3, and ITQ-7) using atomistic MC simulations. All of the all-silica materials were found to preferentially adsorb CO2 over N2 in single-component and in mixture adsorption studies. The observed CO2-over-N2 selectivity varied strongly with changes in the crystal structure. The highest selectivity was found for ITQ-3.
Many zeolites occur in nature, some of which have been tested for CO2 adsorption. Clinoptilolites are examples of such natural zeolites. They are typically relatively hydrophobic, have a high Si/Al ratio, and are known to be good SO2 sorbents.138 Aguilar-Armenta et al. studied the equilibrium and kinetic uptake of CO2, N2, and CH4 in clinoptilolites.139 CO2 sorption and selectivity in Mordendite (MOR) was studied at high pressures. The selectivity for CO2-over-N2 adsorption was found to be higher in the protonated than in the sodium form, thought to reflect the weaker affinity for N2 adsorption in the protonated form.54 Siriwardane et al. showed that most CO2 was physisorbed, and that high sodium content promoted a high uptake of CO2.140
2.2 Aluminium phosphates
Microporous aluminophosphates (ALPO4) and silicoaluminophosphates (SAPO4), with structures and properties similar to those of zeolites, were developed by Flanigen and coworkers.141,142 The overall framework of the ALPO4 materials was neutral; however, the variations in the electric field gradients were significant, and the interaction between this material and CO2 was strong and exothermic. CO2 adsorption in ALPO4-5 (AFI) was measured by Martin et al.57 The measured adsorption capacity for CO2 was large for ALPO4-14 (AFN).58 The SAPO4 framework was negatively charged and required cations for overall charge balance. After exchanging the cations with strontium, SAPO4-34 (CHA) showed significantly enhanced adsorption properties for CO2 at pressures that were low relative to the structure containing Na+ and Ag+ ions.59 SAPO-34 is a candidate material for membranes or kinetic adsorbents for CO2/CH4 separation.Deroche et al.60 investigated the adsorption properties of CO2 in SAPO4 STA-7 (SAV) in a combined GCMC and microcalorimetry study. Newly derived interatomic potentials were used to describe the interaction between CO2 and the Brønsted acid sites, yielding good agreement with experimental data. One method for enhancing CO2 adsorption in ALPO4 and SAPO4 materials achieved additional alkalinity by introducing nitrogen atoms. The number of basic sites in the modified SAPO4-34 was found to be correlated with the number of nitrogen atoms present.143 We anticipate continued experimental and theoretical treatment of CO2 adsorption and selection in ALPO4 microporous solids. These materials are less hydrophilic than zeolites, may be synthesized with a variety of structures, and have yet to be extensively investigated for this purpose.
2.3 MOFs
MOFs are porous structures with very large pore sizes, composed of both inorganic and organic building blocks. MOFs are crystalline materials that commonly feature interconnected pores and are composed of a metal ion coordinated by a relatively rigid organic linker. Two representative MOFs are presented in Fig. 4. After their discovery, these materials generated considerable attention in the literature. Several excellent reviews provide introductions to these materials.144–148 Düren et al.149 gave a useful, short tutorial review describing the application of molecular simulations to predictions of the adsorption behavior of MOFs. They described the molecular-level insights that may be gained from characterizing the adsorption properties of metal–organic frameworks.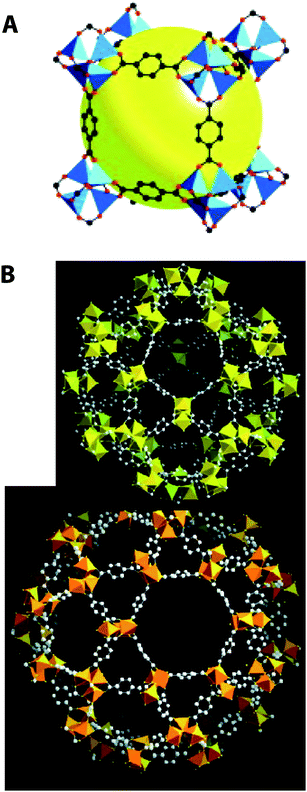 | ||
| Fig. 4 Two representative MOFs. (A) IRMOF-1 or MOF-5;150 (B) MIL-101 with its large and small pores.12 Redrawn with permission. | ||
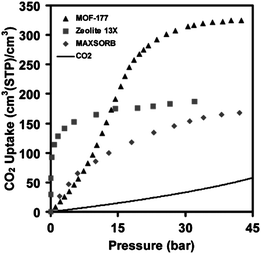 | ||
| Fig. 5 Uptake of CO2 on MOF-177 as compared with zeolite X, porous carbon (MAXSORB), and pressurized CO2.11 Reprinted with permission. | ||
Walton et al.33 demonstrated that the shapes of the adsorption isotherms of CO2 in IRMOF-1 could be predicted by molecular simulations using a rigid crystal structure. They claimed that the sorbate–sorbate electrostatic interactions were essential for predicting the inflections and steps of the adsorption isotherms. The adsorption equilibrium and diffusion of CO2 on microporous metal–organic framework crystals (MOF-5, or IRMOF-1) were studied by Zhao et al.,153 the Freundlich adsorption isotherm equation can fit well the CO2 adsorption, and MOF-5 (Fig. 4a) was found to be an attractive adsorbent for separation of CO2 from flue gas.
Yang et al.154 performed GCMC simulations of CO2/H2 mixtures to study gas separation in three pairs of isoreticular metal–organic frameworks (IRMOFs), with and without catenation, at room temperature. They found that CO2 selectivity in the catenated MOFs with multi-porous frameworks was much higher than that in the non-catenated MOFs. The electrostatic interactions appeared to be important for selectivity, even qualitatively changing the adsorption behavior and playing a dominant role, particularly at low pressures. Liu et al.155 performed a systematic molecular simulation of three pairs of IRMOFs to compare the adsorption separation selectivity of these MOFs in the presence of CH4/H2 mixtures. They showed that the CH4 selectivity in the interpenetrated IRMOFs was greatly enhanced relative to the noninterpenetrated IRMOFs, due to the formation of additional small pores and adsorption sites in the interpenetrating frameworks. The authors showed that IAST was likely to be applicable, even to interpenetrated MOFs with complex structures.
Bastin et al.156 examined a microporous MOF Zn(BDC)(4,4′-Bipy)0.5 (MOF-508b, BDC = 1,4-benzenedicarboxylate, 4,4′-Bipy = 4,4′-bipyridine) for the separation and removal of CO2 from binary CO2/N2 and CO2/CH4, and ternary CO2/CH4/N2 mixtures, providing the first reported use of microporous MOFs for the separation and removal of CO2 from binary and ternary mixtures using fixed-bed adsorption. Barcia et al.157 studied the adsorption of CO2, N2, and CH4 on crystals of MOF-508b, at temperatures in the range 303–343 K and at partial pressures up to 450 kPa. MOF-508b was found to be very selective for CO2, and the loadings of CH4 and N2 were practically temperature-independent. The Langmuir isotherm model provided a good representation of the equilibrium data. A dynamic model based on the linear driving force (LDF) approximation for mass transfer was used to describe the adsorption kinetics of single, binary, and ternary breakthrough curves with good accuracy. The LDF model has been successfully tested (in previous studies158,159) in simulations of the breakthrough curves for alkanes in zeolitic materials. The set of equations was solved numerically using the orthogonal collocation method. The intra-crystalline diffusivity of CO2 was found to be an order of magnitude faster than the intra-crystalline diffusivities of CH4 or N2.
Zeolitic imidazole frameworks (ZIFs) have structures formed by heterocyclic and nitrogen-containing linkers with topologies very similar to those of zeolites. Metals play a similar role on ZIFs to that of Si and Al atoms on zeolites, by mainly contributing with electrostatic interactions, the vdW contributions can be ignored. This fact makes ZIFs substantially different from other MOFs.
Using high-throughput experimental techniques, Yaghi and coworkers identified a range of candidate materials with a high capacity for CO2 adsorption.61,62,152 Liu et al.160 developed a FF to describe the framework atoms of two typical ZIFs, ZIF-68 and ZIF-69. They used this FF in a combined GCMC and MD simulation study to investigate the adsorption and diffusion behavior of CO2 in ZIFs. Their results showed that the small pores in ZIF-68 and ZIF-69 provided preferential adsorption sites for CO2 molecules. Rankin et al.161 computed the adsorption and diffusion properties of CO2, N2, CH4, and H2 in ZIF-68 and ZIF-70 using atomistic simulations. The simulated adsorption and diffusion of the quadrupolar molecules depended dramatically on the atomic charges used. The agreement between simulations and experiments152 for the N2 adsorption isotherms in ZIF-68 and -70 was very good when charge–quadrupolar interaction terms were included, whereas simulations over-predicted the amount of CO2 adsorbed at 298 K if these terms were dropped.
Babarao et al. reported an MC/MD simulation study for upgrading natural gas (CO2/CH4, CO2/N2 mixtures) in rho zeolite-like metal–organic frameworks (rho-ZMOF).162 CO2 was preferentially adsorbed relative to CH4, N2 due to the strong electrostatic interactions of CO2 with the ionic framework and Na+ ions. At ambient temperature and pressure, the CO2 selectivities were 80 for the CO2/CH4 mixture, and 500 for the CO2/N2 mixture. In particular, they described the effects of water for the separation of CO2/CH4 mixture on this material.163 They found that the selectivity decreased by one order of magnitude with a trace amount of H2O added into CO2/CH4 mixture.
Debatin et al. synthesized another microporous zinc–organic framework rescently by in situ synthesis of an imidazolate-4-amide-5-imidate ligand; this sorbent was shown to have a very high CO2 uptake.64
In particular, MIL-53 shows “breathing” phenomenon upon temperature change or host–guest interactions. Such flexible and dynamic frameworks are interesting as they open potential applications for high-performance molecular recognition and high selectivity for guest inclusion and release.
Based on the partial charges of the framework derived from DFT calculations,42 Ramsahye et al. performed a GCMC simulation to compare the CO2 adsorption mechanisms at work in two members of the MIL-n family of hybrid metal–organic framework materials, MIL-53 (Al) and MIL-47 (V).46,47 A structural transition between large-pore and narrow-pore forms was observed in MIL-53 (Al) around 600 kPa, although this transition was not seen for MIL-47 (V). They derived a “composite” absolute isotherm from the calculated CO2 adsorption isotherms by applying the X-ray diffraction structures of MIL-53np (Al) (narrow pore) and MIL-53lp (Al) (large pore) respectively. They found it to be comparable to experimental data collected by microcalorimetry. The “composite” differential enthalpy of adsorption agreed reasonably well with experimental results. At low CO2 loadings, snapshots from GCMC showed typical pore-bridging double interaction (Fig. 6), OCO2–Hμ2-OH distances of 0.198 and 0.176 nm, which is only possible in the narrow pore MIL-53 (Al). At high loadings, the reach of the saturation capacity of the MIL-53np (Al) leads to an increased level of intermolecular interactions between CO2 molecules and break the pore-bridging geometry and weaker adsorbate–adsorbent interaction geometries are formed. They argued that this was likely to be ultimately responsible for a transition from the narrow pore structure to the large pore version. Those adsorption geometries were confirmed in a comprehensive DFT study by the same group.164 The lack of μ2-OH groups within the pore of MIL-47 (V) makes it a homogeneous adsorbent for CO2, thus no “breathing” effect is observed.
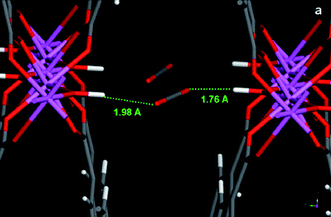 | ||
| Fig. 6 From Ramsahye et al.46 The double interaction, the most prevalent for the Al-containing structure at a CO2 loading of 2 molecules per unit cell (u.c.). | ||
Salles et al.35 used MD simulations to study the thermal activation and guest-induced structural transformations of MIL-53 (Cr).165 Capture of the two-step structural switching, induced by CO2 adsorption in the Cr-containing framework at finite temperatures, was shown to be successful. They found that the proper parameterization of the bonded interaction within the framework was important in reproducing the breathing process. The inorganic node was described by a Cr–O bonded intramolecular term and nonbonded Lennard-Jones (LJ) interactions. An additional torsion term (Cr–Ocarboxyl–Ccarboxyl–Ocarboxyl) was also included for interactions between the inorganic and organic parts. With energy minimization, they confirmed bistable behavior of the large-pore and narrow-pore structures observed by experiment, with only 5 kJ mol−1 per formula unit in favor of the large-pore structure. The simulated vibrational frequencies for the MIL-53 (Cr) framework were in good agreement with infrared data. They also calculated that a transition from the narrow-to-large-pore structure could be thermally activated at a temperature above 600 K. Starting with the bare large pore structure from XRPD data, with loading of different number of CO2 molecules per cell, they performed a series of NσT ensemble MD simulations at 300 K, and derived the unit cell volumes evolution with MD simulation time. From the final equilibrated cell volumes, the evolution of the unit cell volume of MIL-53 (Cr) as a function of the CO2 loading was derived. Their calculations predict the predominance of the large-pore form at very low loading and above 5.2 CO2 per u.c., whereas the narrow-pore version was present in the intermediate domain of loading, within the same range as that obtained from in situ XRPD and manometry experiments. From the MD snapshots, within the narrow-pore channels, the CO2 molecules were aligned along the direction of the tunnel, parallel to each other, leading to a double interaction [OCO2–Hμ2-OH and CCO2–Oμ2-OH] with the μ2-OH groups present at the pore wall. The geometries were in good agreement with the in situ XRPD analysis165 and the DFT calculations164 (Fig. 7). By using the modified FF, they simulated the transport diffusivity of CO2 in MIL-53 (Cr).36 It was the first time to combine QENS experiments and MD simulations to follow the transport diffusivity of a guest molecule confined in a highly flexible MOF-type material characterized by a spectacular phase transition between two distinct structural forms.
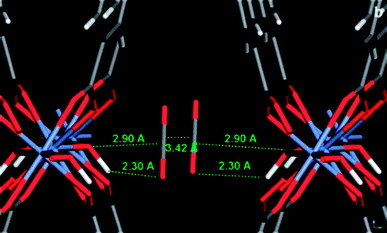 | ||
| Fig. 7 From Ramsahye et al.46 Arrangement of 2 CO2 molecules in a MIL-53np (Cr) calculated from DFT. | ||
Coombes et al.166 carried out DFT and FF-based calculations to model the “breathing” of MIL-53(Cr) in both its large- and narrow-pore forms. They found that the sorbate-free large-pore structure appeared to be the global minimum. Their calculations, in which water molecules were introduced into the structures, illustrated the physisorption-driven pore breathing process, in which water molecules in the narrow-pore form were more strongly stabilized. Hammon et al. studied binary adsorption of CO2 and CH4 in MIL-53(Cr), and discussed the possibility of using this MOFs for the pressure swing-driven separation of CH4 and CO2.167
Based on studies using a combined computational methodology, MD and GCMC simulations, DFT calculations, and TST, Watanabe et al.17 predicted that the MOF Cu(hfipbb)(H2hfipbb)0.5 would have a very high selectivity in the kinetic membrane-based separation of CO2/CH4 mixtures. It contains cages (∼5.1 × 5.1 Å) connected by small windows (∼3.5 × 3.2 Å). This structure feature makes this microporous MOF an equally promising material as DDR125 and SAPO-3459 for CO2 -over- CH4 separations.
Atomistic GCMC simulations were carried out by Babarao et al.173 to study the adsorption capacities for pure CO2, pure CH4, and their binary mixtures (in silicalite, C-168 schwarzite, and IRMOF-1) at room temperature. Although IRMOF-1 had a significantly higher adsorption capacity than did either silicalite or C-168 schwarzite, the adsorption selectivity of CO2 over CH4 was found to be similar in all three adsorbents. A dual-site Langmuir–Freundlich equation was used to satisfactorily describe the isotherms. They also studied174 the self-diffusion, corrected diffusion, and transport diffusion of CO2 and CH4 in silicalite, IRMOF-1, and C-168 schwarzite by MD simulations, and evaluated the activation energies at infinite dilution. An Arrhenius expression was employed to evaluate the energy from the observed diffusivities at various temperatures. The Maxwell–Stefan model predictions for self-diffusion, corrected diffusion, and transport diffusion for pure CO2 and CH4 agreed well with the simulation results. Based on the adsorption and self-diffusivity in the CO2/CH4 mixture, the permselectivity was found to be marginal in IRMOF-1, slightly enhanced in MFI, and greatest in C-168 schwarzite. Although IRMOF-1 had the largest storage capacity for CH4 and CO2, its selectivity was not satisfactory.
Babarao et al.175,176 studied the adsorption and separation of CO2/CH4 mixtures using molecular simulations in a series of MOFs with unique characteristics, such as exposed metals (Cu-BTC, PCN-6′, and PCN-6), catenation (IRMOF-13 and PCN-6), and extra framework ions (soc-MOF). The exposed metals and catenation were found to slightly enhance the selectivity of CO2 over CH4. The extraframework ions NO3− in charged soc-MOF act as additional adsorption sites for quadrupolar CO2 molecules, and the selectivity in soc-MOF was 1 order of magnitude higher than in IRMOFs and PCNs and the highest among various MOFs reported to date.
Farrusseng et al.177 systematically studied the heats of adsorption of many adsorbates, including CO2, CH4, N2 on a series of MOFs: IRMOF-1, IRMOF-3, and Cu-BTC, combining experiment and simulation. Simulations predict a large temperature dependence of the heat of adsorption in Cu-BTC, which is reduced significantly when the small pockets are blocked. Martin-Calvo et al.178 used MC simulations to study the adsorption and separation of natural gas components in IRMOF-1 and Cu-BTC metal–organic frameworks. They estimated the adsorption isotherms of pure components, binary, and five-component mixtures. Their simulations indicated that though IRMOF-1 had a significantly higher adsorption capacity than Cu-BTC, the adsorption selectivity of CO2 over CH4 and N2 is found to be higher in the latter, proving that the separation efficiency was largely affected by the shape, the atomic composition and the type of linkers of the structure.
Yang et al.179 reported a systematic computational study of the effects of organic linker, pore size, pore topology, and electrostatic fields on the adsorption and diffusion behavior of CO2 in nine typical metal–organic frameworks (MOFs). They showed that the high CO2 adsorption capacity could be described as the complex interplay of these properties. The MOFs in this study showed higher CO2 adsorption capacities than did either zeolites or carbon materials under practical conditions, and the most suitable pore size was found to be 1.0–2.0 nm. In addition, the DS values for CO2 in the MOFs were comparable to those observed in zeolites.
Keskin and Sholl180 introduced an efficient approximate method for screening MOFs based on atomistic models that sped the computational time associated with models of membrane applications. They validated the model via comparison with detailed calculations of the permeation of CH4/H2, CO2/CH4, and CO2/H2 mixtures at room temperature through IRMOF-1 and Cu-BTC membranes. The model was then applied to six additional MOFs (IRMOF-8, -9, -10, and -14, Zn(bdc)(ted)(0.5), and COF-102) to estimate the effects of chemical diversity and interpenetration in MOF membranes on the separation of light gases.
Yazaydin et al.34 screened 14 MOFs for CO2 capture from flue gas at an operation pressure below 1 bar using a combined experimental and modeling approach (Fig. 8). They found that MOFs with a large adsorption capacity for CO2 at high pressures often do not perform well at low pressures. IRMOF-1 and MOF-177 are among the lowest performing materials. Below 1 bar, CO2 uptake correlates well with the heat of adsorption, thus MOFs having a high density of open metal sites are promising. They found that changing the metal from Zn in M\DOBDC (DOBDC = dioxybenzenedicarboxylate) to Mg, Co, or Ni provides large changes in CO2 uptake. M\DOBDC have open metal sites that can interact with adsorbate molecules, and Mg\DOBDC performs particularly well. HKUST-1 (also known as Cu(BTC)), UMCM-150, and UMCM-150(N)2 have lower density of open metal sites than M\DOBDC and perform not as well as M\DOBDC. The LJ parameters in their force field for the MOF atoms were taken from DREIDING or UFF force field. Partial charges were fitted from DFT cluster calculations. In spite of bad performance for strong interactions between open metal sites and CO2, their model correctly predicted the top 5 MOFs: Pd(2-pymo)2, Mg\DOBDC, Ni\DOBDC, Zn\DOBDC, and Co\DOBDC in agreement with the experiments.
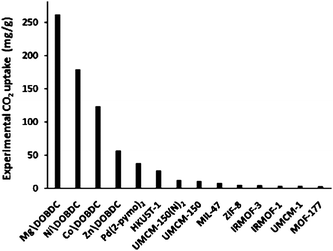 | ||
| Fig. 8 From Yazaydin et al.34 Experimental CO2 uptake in screened MOFs at 10 kPa. Data obtained at 293–298 K. | ||
The force field parameters of molecular mechanics for MOFs are often not available, because of the wide range of possible inorganic fragments involved. The generic force field like UFF and DREIDING are derived by a number of established rules from atomic parameters are usually used to describe the MOF framework. These parameters are usually not very accurate and the framework is usually kept frozen at the experimentally determined structure. It limits the applicability of molecular in screening and design of MOFs for with specific gas adsorption behaviors, especially when MOFs have flexible frameworks. High quality force field parameters are required to describe precisely the “breathing” mechanism involved in flexible MOFs.
Tafipolsky and Schmid44 proposed an efficient, systematic strategy to parameterize a force field of molecular mechanics from first principles reference data by optimizing a novel objective function with a genetic algorithm. Due to the efficiency of this approach, it is possible to abandon the need for transferability of the parameters. It is a “bonded” force field, i.e., bond stretching, angle bending, torsion and stretch–stretch, stretch–bend cross terms are also considered in the metal ion interacting with its surrounding atoms. Using this approach, the authors claimed that a database of force field parameters, suitable for molecular simulations of coordination polymers, and considering the framework flexibility, could be parameterized from different types of secondary building units and linkers. Applying this scheme to MOF-5, in a “building block” approach, parameters are derived for the two model systems, zinc formate (Zn4O(O2CH)6), and dilithium terephthalate. Reference data were obtained from density functional theory. The resulting potential gives excellent agreement with the structure, vibrational frequencies, thermal behavior, as well as elastic constants for the periodic MOF-5. Because no experimental data were used in the parameterization, the method could possibly also be used for not yet synthesized systems and allow for screening of MOFs.
Aiming at the design of linker molecules, which could form parts of new MOFs with enhanced affinity for CO2 adsorption at low pressure, Torrisi et al. studied interaction of CO2 with functionalized benzenes using density functional theory. These moieties contained methyl groups, halogen substituents,185 polar side groups substituents,186 including –NO2, –NH2, –OH, –SO3H, and –COOH. Halogen substituents have an electron-withdrawing effect on the aromatic ring, which destabilizes the π–quadrupole interaction between benzene and CO2, thus have negative effect on CO2 adsorption. Methyl groups, on the other hand, have a positive inductive effect, which strengthens the CO2-aromatic interaction. They found that the best substituents were –NH2, –SO3H, and –COOH. Such polar groups can lead to a whole range of favorable configurations, where a variety of host–guest interactions are at play, including lone-pair electron donation, H-bond like interactions. In addition, they pointed out that in a nanoporous material, additional effects restrict the freedom for the gas molecules to diffuse away from sorption sites. Molecular confinement, such as binding to more than one ligand, CO2–CO2 interactions, etc., could further serve to enhance the affinity for CO2.
2.4 COFs and porous polymers
MOFs and zeolites are typically hydrophilic, and their applications toward CO2 separation from flue gases necessitate a drying stage. To circumvent such drying, significant efforts have been devoted to identifying materials that do not contain hydrophilic cation sites. A variety of organic porous materials has been produced without metal ions, with good prospects for CO2 separation. Choi et al. studied CO2 sorption in a three-dimensional (3D) polymeric network.187 NASA uses a hybrid material that contains polyethylenimine (PEI) bonded to a high-surface-area polymethylmethacrylate mixed with polyethylene glycol (PEG) for CO2 capture during space travel.188 Budd et al. described microporous materials made from soluble polymers,189 and Ritter et al. studied the limits of microporosity for CO2 sorption in certain polyimides.190Covalent organic frameworks (COFs) are crystalline organic porous materials without metal ions. Furukawa et al. demonstrated high capacities for CO2 adsorption in COFs, studying H2, CO2, and CH4 adsorption in seven COFs over a range of pressures and temperatures. The studied COFs were COF-1, COF-5, COF-6, COF-8, COF-10, COF-102, and COF-103. COFs-102 and -103 showed very high CO2-uptake capacities.13
CO2 adsorption in COFs containing three-dimensional (3D) (COF-102, COF-103, COF-105, and COF-108), two-dimensional (2D) (COF-6, COF-8, COF-10), and one-dimensional (1D) (COF_NT) structures were estimated by Babarao and Jiang191 using computer simulations. The dimensionalities of the COFs refer to the dimension of the channel systems of these sorbents. In this earlier work, COF-105 and COF-108 appeared to have exceptionally high adsorption capacities, surpassing even the capacity of MOF-177. The authors found molecular-based structure–function correlations useful for predicting capacity and for screening COFs for CO2 adsorption. Yang and Zhong192 carried out GCMC simulations to investigate the adsorption properties of CO2, CH4, and H2 in 2D COFs with varying pore sizes. They predicted a stepped behavior common in gas adsorption, in which multilayer formation was the underlying mechanism. In general, they observed that temperature, pore size, the strength of interactions between adsorbates, and the strength of interactions between adsorbates and adsorbents affected the properties of the stepped mechanism.
Barbarao and Jiang193 reported a systematic molecular simulation study of CO2 adsorption in a series of MOFs (IRMOF1, Mg-IRMOF1, Be-IRMOF1, IRMOF1-(NH2)4, IRMOF10, IRMOF13, and IRMOF14), as well as UMCM-1, a fluorous MOF (F-MOF1), and a covalent–organic framework (COF102). The authors concluded that the affinity of these adsorbents for CO2 could be enhanced by the addition of functional groups. The pore size was also observed to constrict, via interpenetration of the framework, which simultaneously increased the isosteric heat and Henry's constant, yielding stronger adsorption at low pressures. The authors observed that the organic linkers played a critical role in determining the free volume and accessible surface area of the material, and the organic linkers largely determined the estimated CO2 adsorption at high pressures. COF-102 was found to be a promising CO2 adsorption candidate with a high adsorption capacity at very low pressures.
2.5 Carbons
Porous carbons have been investigated as potential CO2 sorbents, revealing a distinct advantage over zeolites in terms of hydrophobicity. Still, the uptake of CO2 was reduced by competitive adsorption of water onto the carbons.194 Many types of porous carbons have been developed: activated carbons, carbon molecular sieves (CMS), carbon nanotubes (CNT), and more exotic constructs such as NanoBuds and graphene.Montoya et al. presented an experimental and theoretical study that examined the mechanisms underlying the sorption process on carbon surfaces, characterizing two surface regions in which adsorption took place. In the low-coverage region, the heat of adsorption decreased rapidly for increased adsorbate concentrations, which was interpreted as a characteristic of a broad spectrum of binding sites.200 At high loadings, the heat was found to be nearly independent of the extent of loading. Levesque and Lamari201 calculated the isosteric heat of CO2 adsorption on activated carbon using GCMC simulations. The authors discussed the possibility of estimating the isosteric heat of a macroscopic sample from adsorption isotherms computed for a distribution of slit pores of a given size.
Tenney and Lastoskie202 performed GCMC simulations to investigate the influence of surface heterogeneity on the predicted adsorption behavior in activated carbons and coal. Isotherms were calculated for CO2 adsorption inside slit-shaped pores characterized by several levels of chemical heterogeneity, such as oxygen and hydrogen content, pore width, and surface functional group orientation. The heterogeneities were present on the scale of ∼10 nm2. The computed adsorption capacity was observed to increase in regions containing an excess of surface oxygen content, although exceptions to this trend were observed. Electrostatic adsorbate–adsorbent interactions significantly influenced adsorption on the model surfaces.
The preparation of mesoporous carbon materials with a narrow PSD and crystallographically organized pores has been described previously.203 For a detailed review of the preparation of these materials and their related silica materials, the reader is referred to Zhao et al.204 These preparations typically involved a multistep procedure in which a mesoporous silica mold was produced with the help of amphiphilic molecules. The mold was then filled with carbon-containing moieties that were subsequently carbonized. Finally, the silica mold was removed by chemical means. CO2 adsorption on these ordered mesoporous carbons has been studied previously.45 Peng et al.205 carried out GCMC simulations to investigate the adsorption of CH4 and CO2 mixtures on the ordered carbon material CMK-1, to study the effects of temperature, pressure, pore width, and bulk composition on adsorption capacity, the local density profile, snapshots, and the solid–fluid potential curves. In this context, a snapshot means the spatial distribution of CO2 and CH4 at a certain moment in time. The electrical and thermal properties of carbon render it a good adsorbent for electrically induced temperature swing adsorption processes.
Liu et al.206 developed an improved non-linear DFT (NLDFT) technique and combined this approach with PSD analysis of adsorbent activated carbon materials. They predicted the adsorption equilibria of high-pressure gas mixtures onto activated carbon. For two gas mixtures, CH4/N2 and CO2/N2, the authors improved the predictability of the adsorption equilibrium in the gas mixtures under high-pressure conditions, particularly the predictability of the weakly adsorbed species. CMC simulations were carried out by Cao and Wu207 to investigate the separation of H2 and CO2via adsorption in activated carbons using slit-pore models. At room temperature, the CO2-over-H2 selectivity reached approximately 90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, indicating that H2 and CO2 could be efficiently separated. Heuchel et al.208 predicted the adsorption properties of pure single-component gases and binary mixtures of CH4 and CO2 on a specific activated carbon, A35/4, using GCMC simulations. The PSD for the carbon was determined from the CH4 and CO2 isotherms at 293 K. Using the PSD and simulated adsorption densities in single pores, it was possible to predict, in good agreement with experiment data, (i) the adsorption ratios of binary mixtures containing CO2 and CH4, and (ii) the adsorption of both pure components at higher temperatures.
:1, indicating that H2 and CO2 could be efficiently separated. Heuchel et al.208 predicted the adsorption properties of pure single-component gases and binary mixtures of CH4 and CO2 on a specific activated carbon, A35/4, using GCMC simulations. The PSD for the carbon was determined from the CH4 and CO2 isotherms at 293 K. Using the PSD and simulated adsorption densities in single pores, it was possible to predict, in good agreement with experiment data, (i) the adsorption ratios of binary mixtures containing CO2 and CH4, and (ii) the adsorption of both pure components at higher temperatures.
The adsorption capacity and sorption kinetics of CO2 and N2 were studied by Vyas et al. on CMSs with varying amounts of deposited coke. Large amounts of deposited coke were correlated with a high capacity for CO2 sorption.210 Ahmad and coworkers studied CO2 uptake by CMSs prepared from palm shells. The most suitable samples for CO2-over-N2 selection were those prepared at an intermediate carbonation temperature of 1273 K, with a deposition time longer than 20 min.211–213 Carrot et al. prepared CMSs from polyester fibers and CVD of benzene at 1073 K, obtaining the best sorbents after 10 min of coke deposition. Still, these sorbents had a smaller capacity for CO2 than the commercial CMS Takeda 3A sorbent.214 CO2 sorption on Takeda 3A was studied in detail by Rutherford et al., who also studied CO2 uptake in CMS-5A.215–217 Yang et al. studied the kinetic separation of CH4/CO2 on CMSs,65 and Nabais et al. prepared monolithic CMS materials by carbonization and activation, and observed excellent CO2-over-N2 selectivities.218 Jayaraman et al. analyzed the utility of two commercial CMSs, Bergbau-Forschung and Takada 3A, in two process cycles, and focused on enhancing the separation by taking advantage of the differential diffusion rates of CO2 and CH4 in these commercial CMSs.219 Campo et al.220 studied transport mechanisms by comparing a CMS membrane with a commercial CMS adsorbent. The adsorption equilibrium isotherms of N2, Ar, CO2, and O2 were determined.
Lafyatis et al. synthesized and studied the uptake of CO2 in a series of CMSs. They derived CMSs from poly(furfuryl alcohol) and studied how the carbonization temperature and time affected the microporosity. High capacities were observed for the uptake of CO2: nearly 8 wt% at a relative pressure of 0.015 (P/P0). The diffusivity of CO2 was shown to decrease for CMSs with smaller pores.221 Solid-state nuclear magnetic resonance (13C NMR) spectroscopy has been used to show the presence of furanic rings in samples pyrolyzed at relatively low temperatures. The presence of these moieties was correlated with CO2 diffusivities in these materials.222 Nguyen and Bhatia223 studied the accessibility of Ar, N2, CH4, and CO2 in disordered microporous carbons, using TST, MD simulations, and reverse Monte Carlo (RMC) simulations with realistic carbon models, in an effort to understand the kinetic restrictions imposed on adsorbate molecules by the narrow pore mouths of coals and molecular sieve carbons.
Fomkin224 experimentally examined the CO2 uptake in a range of adsorbents, including the microporous carbon AUK, at various temperatures and pressures. Pantatosaki et al.225 used GCMC simulations and experimental adsorption isotherms to characterize microporous carbon and to obtain the PSD. They obtained PSDs under the assumption of slit and cylindrical pores at temperatures of 298 and 308 K. Steriotis et al.226 and Samios et al.227 used GCMC simulations to study the structural configurations of CO2 molecules adsorbed in microporous carbons. The authors discussed the local density profiles and angular distributions of the axes of the adsorbed molecules within the pores. These calculations addressed the densification process and molecular packing in the micropores.
Samios et al.228 used GCMC simulations and CO2 experimental isotherm data at low and high temperatures to characterize the microporous carbon materials. They studied the PSD, the densification process in the micropores, and the structure of CO2 molecular packing within the individual pores, addressing the effects on the local density of temperature, pore size, electric field gradient–electric quadrupole interactions, and molecular elongation of the adsorbates. Vishnyakov et al.229 studied CO2 adsorption in slit-shaped carbon micropores at 273 K using GCMC simulations and NLDFT. For pore widths in the range 0.3–1.5 nm, NLDFT estimations of the CO2 adsorption isotherms were generally in agreement with the GCMC estimations. Samios et al.230 developed a method to determine PSDs in the micropore regime, based on GCMC simulations and measured isotherms.
Ravikovitch et al.237 presented a unified approach to pore size characterization in microporous carbonaceous materials, such as activated carbon and carbon fibers, using N2, Ar, and CO2 adsorption measurements based on NLDFT and GCMC methods. Huang et al.238 performed GCMC simulations to investigate the adsorption behavior of an equimolar CO2/CH4 mixture in the presence of CNTs. The authors performed simulations to model the adsorption of the gas mixture onto five armchair CNTs [(6, 6), (7, 7), (8, 8), (9, 9), and (10, 10)],† with diameters varying from 0.678 to 1.356 nm, at seven temperatures (283, 293, 303, 313, 323, 333, and 343 K) and under seven pressures (1, 5, 10, 15, 20, 25, and 30 MPa), to characterize the effects of temperature, pressure, and pore size on the adsorption behavior. The authors found that CO2 was preferentially adsorbed onto the CNT surfaces under all conditions investigated. For each type of CNT, the adsorption capacity for CO2 was estimated to be much higher than that of CH4. CO2 adsorption in CNTs appeared to increase dramatically with increasing CNT diameter. The selectivity of CNTs for CO2 was no higher than that of activated carbons, zeolites, and MOFs reported in the literature.
Konstantakou et al.239 performed GCMC simulations, in combination with experimental data, to characterize adsorption in microporous carbons (AX-21 in particular), with the goal of determining the optimal PSD for adsorption. Adsorption isotherms were calculated from the GCMC simulations for several pore widths up to 3.0 nm and for the adsorption of H2 at 77 K. Quantum corrections were introduced by applying the Feynman–Hibbs effective potential. Skoulidas et al.240 used atomistic MD simulations to examine the adsorption and transport diffusion of CO2 and N2 in SWNTs at room temperature as a function of nanotube diameter. The results were consistent with previous predictions that transport diffusivity of molecules inside carbon nanotubes is extremely rapid relative to transport in other porous materials. Sinnott et al.241 carried out MD simulations to study molecular motion and the separation of molecular mixtures in carbon nanotube systems, for mixtures of CH4, C2H6, n-C4H10, i-C4H10, and CO2. Not surprisingly, they found that molecules (at 300 K) diffused from areas of high density to areas of low density throughout the nanotubes.
Xu et al.242,243 performed non-equilibrium MD (NEMD) simulations of transport and separation characteristics of binary and ternary gas mixtures consisting of CO2, CH4, and H2 through a carbon nanopore in the presence of an external chemical potential gradient. The authors addressed the effects of temperature, feed composition, and pore size on transport properties, investigating in detail the adsorption and separation characteristics. Müller244 performed GCMC simulations on the adsorption of N2, CO2, and C2F6 (three quadrupolar molecules) inside SWCNTs and predicted a tilted ordering not previously reported, which was rationalized as resulting from a combination of steric effects and an anisotropic attraction pattern.
Su and Lua245 determined the theoretical upper limit of the permeation rate of gases (with different masses, for activation energies of 0 kJ mol−1) by the Knudsen diffusion mechanism. The distribution and magnitude of the potential energy of interaction between gas molecules and the carbon pore wall was strongly dependent on the pore size in the modeled membrane. MC simulations were performed by Jia et al.246 to investigate the separation behavior of gas mixtures composed of CO2 and N2, using a model of a carbon membrane under various conditions. These calculations indicated that CO2 was strongly adsorbed on the surface of the membrane. Yang and Zhong247 carried out extensive GCMC simulations to study the adsorption behavior and orientational structure of CO2 confined in slit graphite pores.
With the goal of designing new carbon materials, Terzyk et al.249 performed GCMC simulations to test the ability of fullerene-intercalated graphene nanocontainers (NanoBuds) to adsorb CH4 and CO2. By combining quantum mechanics and molecular simulations, Jiang and Sandler28 investigated the adsorption of pure CO2 and N2, and their mixtures at room temperature in C168 schwarzite, as a model for nanoporous carbons. Schwarzites are theoretical bicontinuous porous structures analogous to fullerenes. The inclusion of the adsorbates' electric quadrupole moment in the simulation did not affect N2 adsorption, although it did affect CO2 adsorption at high coverage. The selective adsorption of CO2 over N2 by C-168 schwarzite, using a model flue gas, was predicted to be significantly higher when ab initio potentials were used than when the Steele potential was used, illustrating the importance of an accurate adsorbate–adsorbent interaction potential in determining gas adsorption. Nanohorns are small, open nanoscaled carbon objects. Urita et al. studied the semiconducting behavior of nanohorns under CO2 and O2 adsorption, reporting increased conductivity when CO2 adsorbed to single-walled carbon nanohorns (SWNH), whereas the oxidized SWNH showed reduced conductivity upon CO2 adsorption.250 These findings may be relevant to electric swing adsorption.
2.6 Other porous oxides
γ-Alumina (γ-Al2O3) is commonly used as a porous support for a variety of catalytic applications. Dewaele et al. measured a high energy of desorption for CO2 from a γ-alumina support with a surface area of 153 m2 g−1, as expected from the Lewis base character of the substrate.251 Two commercial alumina substrates were studied by Rodrigues et al., and one of the sorbents had a high capacity for CO2 of 3.5 μmol m−2.252 Pokrovski et al. studied the adsorption of CO2 on m-ZrO2 (monoclinic) and t-ZrO2 (tetragonal), and found a much higher CO2 sorption capacity on m-ZrO2 than on t-ZrO2.253 This observation was ascribed to the surface basicity of the monoclinic form. Knöfel et al. studied the uptake of CO2 on mesoporous titania and found a high heat of CO2 adsorption.254Belmabkhout et al.82,83 reported high equilibrium adsorption capacities and good separation capabilities for CO2, CH4, N2, H2, and O2 in periodic mesoporous MCM-41 silica. MCM-41 is a hexagonally structured mesoporous material, and MCM-48 is a cubic structured mesoporous material in the class of M41S solids developed by Mobil in the late 1980s.26 IAST was validated and used for the prediction of CO2/N2, CO2/CH4, and CO2/H-2 binary mixture adsorption equilibria. MCM-41 showed preferential CO2 adsorption over the other gases. Schumacher et al.255 proposed a methodology with which to design hybrid organic–inorganic adsorbents, based on periodic mesoporous silicas using kMC simulations to generate realistic model adsorbents. The authors carried out GCMC simulations of adsorption in these model materials. The capabilities of the method were demonstrated experimentally.
He and Seaton256 reported the adsorption isotherms and the isosteric heats of adsorption of pure methane, ethane, and CO2, and for mixtures of methane and CO2, in the periodic mesoporous silica MCM-41. The energies of adsorption, for pure CO2 and for CO2 from a CO2/methane mixture, were determined to be heterogeneously distributed, reflecting electrostatic interactions between CO2 and the adsorbent. Yoshioka et al.257 used NEMD simulations to study the mechanisms involved in pressure-driven gas permeation through a micropore on vitreous SiO2 membranes. The study was performed to investigate the dependencies of the permeance of helium and CO2 molecules on temperature and pore size. Yang et al.258 carried out MD simulations of dense CO2 on amorphous dehydroxylated silica surfaces. Permeability through molecular sieving membranes was investigated by Takaba et al.,259 who employed GCMD to investigate the temperature dependence of H2, Ne, Ar, O2, N2, and CO2 adsorption. Activated transport was observed when the pore size of the membrane was smaller than 1.2 times the molecular diameter. Finally, Yoshioka et al.260 used a particle-generating NEMD method to simulate He and CO2 gas permeation at various temperatures and pressures through cylindrical pores that mimicked microporous silica.
2.7 Amine-modified mesoporous silica
Many studies have examined CO2 adsorption in modified porous silica sorbents containing amine groups. The amine groups could be introduced by chemical surface modification or simply by coating or filling the pores. Previous studies have also investigated silica materials chemically modified by n-propylamine moieties.72,75,261–268 Leal et al. showed that such amine-modified materials adsorbed significant amounts of CO2 and that CO2 was chemisorbed on the material.261 Angeletti et al. showed that the same modifications produced excellent sites for Knoevenagel condensation.268 Huang et al. studied CO2 uptake under dry and moist conditions on MCM-48 modified with propylamine functional groups and found that the presence of water doubled the amount of CO2 adsorbed.75 Chaffee et al. observed a similar uptake from “wet CO2”, although the rate of uptake was lower than that under dry conditions.263Sayari et al. expanded the pores of MCM-48 and modified them with pendant n-propylamine moieties. The authors concluded that the mechanisms describing the interaction between CO2 and the amine functionalities were related to the chemistry present in amine solutions. Bicarbonate appeared to form only when large amounts of water were present; i.e. at conditions under which capillary condensation of water took place. Under dry conditions, propylammonium + propylcarbamate ion pairs formed.266 Che et al. used a new chemistry to synthesize n-propylamine-functionalized silica materials (the AMS class269) in a highly controlled manner, although this chemistry does not introduce a large number of amine groups. Kim et al. studied the CO2 uptake on such sorbents.74
Mesoporous silicas modified with amine or amine-like functionalities other than propylammonium have also been studied.70,75–79,264,270 Kim et al. modified MCM-48 substrates with n-propylamine, polymeric n-propylamine, pyrrolidinepropyl, and polyethyleneimine (PEI) and observed that the highest capacity for CO2 sorption was achieved by the n-propylamine-modified substrate.264 Zelenak et al. showed that the n-aminopropyl-modified material had the highest capacity among three modifications studied: n-propylamine, 3-(methylamino)propyl, and 3-(phenylamino)propyl. A high basicity produced high levels of CO2 sorption. Weak bases showed rapid regeneration, which may be beneficial in practical applications.70 Zhao et al. showed that n-propylamine modifications yielded the highest capacities for CO2 uptake among the modifications studied (n-propylamine, bis-ethanol amine, and amidine).270 Ionic liquids tethered to silica and quaternary amines catalytically produced cyclic carbonates from adsorbed CO2.271
Harlick and Sayari studied the performance of CO2 adsorption on triamine-modified MCM-41.75–78 The material, with expanded pores, showed significant advantages relative to the non-expanded material, and was found to outperform zeolite X in humid environments. Covalently tethered polyethyleneimine272 on mesoporous silica has been shown to effectively capture CO2 in a reversible manner.72,273 The high amine loading, as well as the chemical anchoring of the alkaline moieties to the silica surface, made these materials highly useful for CO2 capture.
Previous studies have examined CO2 sorption in mesoporous silica, in which the pores have been physically filled with amines.73,83,274–277 The CO2-uptake capacities of these materials are large, but their tolerance for recycling processes, without leaching the filler material, remains under investigation.74 Xu et al. studied the CO2 uptake in MCM-41 materials filled with PEI and observed high capacities with an atypical temperature dependence. The materials adsorbed more CO2 at higher temperatures than at low temperatures.73,274–277 An uptake as high as 24.6 wt% CO2/PEI was observed, which is higher than the uptake of pure PEI.275 Chen et al. studied CO2 absorption in monolithic silica with hierarchically textured pores impregnated with amines, for tetraethylenepentamine (TEPA) a high uptake was observed.79
CO2 has been shown to chemisorb on many solids.278–280 Chemisorption of CO2 on porous silica materials, modified with n-propylamine, has been studied by infrared spectroscopy.73,254,280–282 The chemistries of CO2 and amines are well understood. Thermally unstable ammonium carbamate salts have been shown to form in the absence of water, releasing CO2 upon heating.283 Battjes et al. showed that alkylammonium–carbamate ion pairs were the product of a reaction between primary or secondary amines and CO2.284 At low temperatures (273 K), the dimeric form of carbamic acid was observed as the chemisorption product.285,286 CO2 was chemisorbed on n-propylamine-modified silica via two different mechanism in the presence or absence of water.77,266,274,275,280,287,288 The carbamate ion pairs were shown to react with CO2 and H2O to form bicarbonate groups in the following ratios: one mole of amines chemisorbed one mole CO2 in the presence of water; in the absence of water, two amines were required to chemisorb one mole of CO2. One of the authors of this review recently detected both chemisorbed and physisorbed CO2 on mesocaged silica adsorbents tethered by n-propylamines. Sorbents that were post-synthetically modified with n-propylamines took up more CO2 at high temperatures than at low temperatures, indicating the presence of a kinetic barrier. A high degree of heterogeneity in the coating was required to promote the formation of propylammonium-propylcarbamate ion pairs.289
The activation of sorbents using amine groups has attracted interest because it provides a method for achieving higher selectivity of CO2 adsorption from flue gases. Gray et al. determined, somewhat surprisingly, that amine-enriched carbon sorbents had much lower capacities for CO2 adsorption than did commercially available carbon sorbents.290 Lu et al. prepared composite materials in which 3-aminopropyltriethoxysilane was condensed onto zeolites, activated carbons, and CNTs. The highest uptake was observed for the amine–CNT composite.291 Dillon et al. covalently attached PEI to fluorinated SWNTs and studied adsorption on these materials using a variety of techniques. They found a high CO2 uptake (9.2% w/w) and discussed various potential applications of the PEI-SWNTs composites.292 One of the authors of this review has studied CO2 uptake in porous carbons derived from hydrothermally treated glucose to which were attached chemically tethered amine functional groups; these materials showed a high level of CO2 uptake.84
Only a few molecular simulation studies can be found for these systems. Chaffee293 prepared a series of silica models with mesoporous dimensions, 3-D periodicity, varying pore diameter (22–33 Å), and with a varying density of silanol functional groups (2–9 OH per nm2) on the internal surfaces. Models of inorganic–organic hybrid material were prepared by attaching grafted aminopropylhydroxysilyl groups at the locations providing the greatest (calculated) energy relief. The gas–solid molecular behavior at the modified interface were analyzed and visualized. Chen et al.294 prepared an atomistic slit model to represent the propylamine-grafted mesoporous amorphous silica pore surface. Applying GCMC, they simulated CO2, CO2/N2, CO2/H2O mixtures adsorption isotherms, studied the effects of temperature, calculated the selectivity of CO2 over N2. They predicted high CO2/N2 selectivity upon the amine modification of this amorphous silica material. In combination with calculations using quantum mechanics, the influence of H2O on the CO2 uptake were studied. The diffusion of CO2 and H2O was estimated by MD simulations.
2.8 Hydrotalcite and other sorbents
Many studies have investigated chemisorbents for CO2, mainly Hydrotalcites (HTlc) and calcium oxide-based sorbents. For a detailed review, we refer the reader to Choi et al.24 Here, we include some selected references to highlight the importance of these sorbents for the capture of CO2 from point sources. HTlc was studied as a selective adsorbent for CO2. HTlcs are alkaline clays (layered double hydroxides).295–300 Yong et al. studied Pural™ MG50 and MG70 and showed CO2 sorption (at 573 K and 100 kPa) exceeding 0.3 mmol g−1.295 Ritter et al. showed that potassium-exchanged HTlcs adsorbed CO2 reversibly at high temperatures.296–299 Yavuz et al. studied Ga3+-substituted HTlc, which, together with K+, proved to be a robust and promising material for sorption of CO2.300 Lee and Sircar proposed a temperature swing adsorption process using Na2O on an alumina substrate,301 and Wu et al. studied a calcium-based sorbent for CO2 capture.302 Notably, this sorbent had the potential for use in precombustion CO2 capture. Solieman et al. studied Li2ZrO3, BaO, and CaO sorbents, with CaO found to be the most suitable sorbent for reforming methane.303Dolomite is an inexpensive sedimentary MgCaCO3 material and is a weak Lewis base. Duffy et al. studied the adsorption properties of dolomite towards acidic gases.304 The surface area was increased in a process in which some MgO was produced, but the CO2 capacity was not very high. Rajabbeigi et al.305,306 developed models for nanoporous materials and inorganic membranes, in which interconnected pores of irregular shapes, sizes, and connectivity were used to model adsorption in three silicon carbide (carborundum) membranes. Non-equilibrium MD methods were used to study the transport and separation properties of this membrane in the presence of two binary gaseous mixtures, H2/CO2 and H2/CH4. Bulnes et al.307 studied the adsorption of binary mixtures on solid heterogeneous substrates using MC simulation of a lattice gas model. The adsorption process was monitored via total and partial isotherms, and via the difference in heats of adsorption for the species in the mixture. The uptake of acidic gases by calcite (CaCO3) was studied by Santschi and Rossi, who reported that CO2 interacted specifically with calcite and formed bicarbonates with OH groups on the surface.308 Mömming et al. studied the sorption of CO2 using a frustrated Lewis acid–base pair composed of an organic borane and an organic phosphine.309,310 A similar approach may potentially be used on porous substrates.
3. Conclusions
In this review, we attempted to provide an overview of the significant results related to CO2 sorbent development, from the perspective of materials and theoretical chemistry. Such developments would benefit from collaborations between experimentalist and theoreticians. We hope that there will be more truly interdisciplinary studies on CO2 sorbents, where chemical, physical, and engineering aspects are treated in an integrated manner.We discussed the materials in eight groups: zeolites and microporous silicates, aluminium phosphates, MOFs, COFs, carbons, other porous oxides, amine-modified mesoporous silicas, hydrotalcite and other sorbents. In screening and designing of MOFs, better force fields are needed and searches for such are ongoing. Many of these sorbents show great potential as CO2 sorbents. Amine-modified porous solids have been studied in detail experimentally, but only few theoretical studies of these complex solids have been performed. These amine-containing solids show high CO2-over-N2 selectivity, high operational efficiency, and are robust towards water. They could potentially be used; however, a fair amount of engineering studies need to be performed.
Additional developments in developing CO2 sorbents are forthcoming, and we are certain that combined experimental and theoretical approaches will enable the development of CO2-selective sorbents, without the energetic penalties associated with strong chemisorbents. Zeolites are typically hydrophilic and render them difficult to use for CO2 capture from flue gases. Hydrophobic microporous solids are more robust towards the presence of water vapor. Many different sorbents (zeolites, CMS, ALPO4, silicates, MOFs, COFs) could potentially enable molecular sieving or kinetic selection of CO2-over-N2. In our opinion, only a few studies have seriously considered the prospects for developing kinetically active or molecular sieving as a means of separating CO2 from N2-rich flue gas.
References
- S. Rackley, Carbon Capture and Storage, Butterworth-Heinemann, Cambridge, 2009 Search PubMed.
- D. Aaron and C. Tsouris, Sep. Sci. Technol., 2005, 40, 321–348 CrossRef CAS.
- R. M. A. Roque-Malherbe, Adsorption and Diffusion in Nanoporous Materials, CRC Press, Baton Rouge, 2007 Search PubMed.
- R. T. Yang, Gas Separation by Adsorption Processes, Imperical College Press, London, 1997 Search PubMed.
- J. Baxter, Z. Bian, G. Chen, D. Danielson, M. S. Dresselhaus, A. G. Fedorov, T. S. Fisher, C. W. Jones, E. Maginn, U. Kortshagen, A. Manthiram, A. Nozik, D. R. Rolison, T. Sands, L. Shi, D. Sholl and Y. Wu, Energy Environ. Sci., 2009, 2, 559–588 RSC.
- M. T. Ho, G. W. Allinson and D. E. Wiley, Ind. Eng. Chem. Res., 2008, 47, 4883–4890 CrossRef CAS.
- D. M. Ruthven, Principles of Adsorption and Adsorption Processes, John Wiley & Sons, Inc., New York, 1984 Search PubMed.
- J. A. Dunne, M. Rao, S. Sircar, R. J. Gorte and A. L. Myers, Langmuir, 1996, 12, 5896–5904 CrossRef CAS.
- P. J. E. Harlick and F. H. Tezel, Microporous Mesoporous Mater., 2004, 76, 71–79 CrossRef CAS.
- D. W. Breck, W. G. Eversole, R. M. Milton, T. B. Reed and T. L. Thomas, J. Am. Chem. Soc., 1956, 78, 5963–5971 CrossRef CAS.
- A. R. Millward and O. M. Yaghi, J. Am. Chem. Soc., 2005, 127, 17998–17999 CrossRef CAS.
- P. L. Llewellyn, S. Bourrelly, C. Serre, A. Vimont, M. Daturi, L. Hamon, G. De Weireld, J. Chang, D. Hong, Y. K. Hwang, S. H. Jhung and G. Ferey, Langmuir, 2008, 24, 7245–7250 CrossRef.
- H. Furukawa and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 8875–8883 CrossRef CAS.
- S. Sircar, T. C. Golden and M. B. Rao, Carbon, 1996, 34, 1–12 CrossRef CAS.
- D. Frenkel and B. Smit, Understanding of Molecular Simulation: From Algorithms to Applications, Academic Press, San Diego, 2002 Search PubMed.
- D. Nicholson and N. G. Parsonage, Computer Simulation and the Statistical Mechanics of Adsorption, Academic Press, San Diego, 1982 Search PubMed.
- T. Watanabe, S. Keskin, S. Nair and D. S. Sholl, Phys. Chem. Chem. Phys., 2009, 11, 11389–11394 RSC.
- A. García-Sánchez, C. O. Ania, J. B. Parra, D. Dubbeldam, T. J. H. Vlugt, R. Krishna and S. Calero, J. Phys. Chem. C, 2009, 113, 8814–8820 CrossRef CAS.
- Z. Yong, V. Mata and A. E. Rodrigues, Sep. Purif. Technol., 2002, 26, 195–205 CrossRef CAS.
- K. B. Lee, M. G. Beaver, H. S. Caram and S. Sircar, Ind. Eng. Chem. Res., 2008, 47, 8048–8062 CrossRef CAS.
- C. M. White, B. R. Strazisar, E. J. Granite, J. S. Hoffman and H. W. Pennline, J. Air Waste Manage. Assoc., 2003, 53, 645–715 CAS.
- D. M. Ruthven, F. Shamsuzzaman and K. S. Knaebel, Pressure Swing Adsorption, John Wiley and Sons, Inc, New York, 1994 Search PubMed.
- S. Choi, J. H. Drese and C. W. Jones, ChemSusChem, 2009, 2, 796–854 CrossRef CAS.
- D. W. Breck, Zeolite Molecular Sieves, Robert E. Krieger Pub. Co., Malabar, 1984 Search PubMed.
- R. C. Reid, J. M. Prausnitz and T. K. Sherwood, The Properties of Gases and Liquids, McGraw-Hill, New York, 1977 Search PubMed.
- J. S. Beck, J. C. Vartuli, W. J. Roth, M. E. Leonowicz, C. T. Kresge, K. D. Schmitt, C. T. W. Chu, D. H. Olson, E. W. Sheppard, S. B. Mccullen, J. B. Higgins and J. L. Schlenker, J. Am. Chem. Soc., 1992, 114, 10834–10843 CrossRef CAS.
- D. Y. Zhao, J. L. Feng, Q. S. Huo, N. Melosh, G. H. Fredrickson, B. F. Chmelka and G. D. Stucky, Science, 1998, 279, 548–552 CrossRef CAS.
- J. W. Jiang and S. I. Sandler, J. Am. Chem. Soc., 2005, 127, 11989–11997 CrossRef CAS.
- B. Liu and B. Smit, Langmuir, 2009, 25, 5918–5926 CrossRef CAS.
- A. K. Rappe, C. J. Casewit, K. S. Colwell, W. A. Goddard and W. M. Skiff, J. Am. Chem. Soc., 1992, 114, 10024–10051 CrossRef CAS.
- S. L. Mayo, B. D. Olafson and W. A. Goddard, J. Phys. Chem., 1990, 94, 8897–8909 CrossRef CAS.
- S. Keskin, J. C. Liu, J. K. Johnson and D. S. Sholl, Microporous Mesoporous Mater., 2009, 125, 101–106 CrossRef CAS.
- K. S. Walton, A. R. Millward, D. Dubbeldam, H. Frost, J. J. Low, O. M. Yaghi and R. Q. Snurr, J. Am. Chem. Soc., 2008, 130, 406–407 CrossRef CAS.
- A.Ö. Yazaydin, R. Q. Snurr, T.-H. Park, K. Koh, J. Liu, M. D. LeVan, A. I. Benin, P. Jakubczak, M. Lanuza, D. B. Galloway, J. J. Low and R. R. Willis, J. Am. Chem. Soc., 2009, 131, 18198–18199 CrossRef CAS.
- F. Salles, A. Ghoufi, G. Maurin, R. G. Bell, C. Mellot-Draznieks and G. Férey, Angew. Chem., Int. Ed., 2008, 47, 8487–8491 CrossRef CAS.
- F. Salles, H. Jobic, A. Ghoufi, P. L. Llewellyn, C. Serre, S. Bourrelly, G. Férey and G. Maurin, Angew. Chem., Int. Ed., 2009, 48, 8335–8339 CrossRef CAS.
- P. Dauber-Osguthorpe, V. A. Roberts, D. J. Dauber-Osguthorpe, J. Wolf, M. Genest and A. T. Hagler, Proteins: Struct., Funct., Genet., 1988, 4, 31–47 CAS.
- F. L. Hirshfeld, Theor. Chim. Acta, 1977, 44, 129 CrossRef CAS.
- R. S. Mulliken, J. Chem. Phys., 1955, 23, 1833–1840 CAS.
- C. M. Breneman and K. B. Wiberg, J. Comput. Chem., 1990, 11, 361–373 CrossRef CAS.
- H. Besler, K. M. Merz and P. A. Kollman, J. Comput. Chem., 1990, 11, 431–439 CrossRef CAS.
- N. A. Ramsahye, G. Maurin, S. Bourrelly, P. L. Llewellyn, T. Loiseau and G. Ferey, Phys. Chem. Chem. Phys., 2007, 9, 1059–1063 RSC.
- N. L. Allinger, Y. H. Yuh and J.-H. Lii, J. Am. Chem. Soc., 1989, 111, 8551–8566 CrossRef CAS.
- M. Tafipolsky and R. Schmid, J. Phys. Chem. B, 2009, 113, 1341–1352 CrossRef CAS.
- N. A. Ramsahye, G. Maurin, S. Bourrelly, P. L. Llewellyn, T. Devic, C. Serre, T. Loiseau and G. Ferey, Adsorption, 2007, 13, 461–467 CrossRef CAS.
- N. A. Ramsahye, G. Maurin, S. Bourrelly, P. L. Llewellyn, T. Loiseau, C. Serre and G. Férey, Chem. Commun., 2007, 3261–3263 RSC.
- J. G. Harris and K. H. Yung, J. Phys. Chem., 1995, 99, 12021–12024 CrossRef CAS.
- G. Maurin, P. L. Llewellyn and R. G. Bell, J. Phys. Chem. B, 2005, 109, 16084–16091 CrossRef CAS.
- J. J. Potoff and J. I. Siepmann, AlChE J., 2001, 47, 1676–1682 Search PubMed.
- M. G. Martin and J. I. Siepmann, J. Phys. Chem. B, 1998, 102, 2569–2577 CrossRef CAS.
- C. S. Murthy, K. Sing, M. L. Klein and I. R. McDonald, Mol. Phys., 1980, 41, 1387–1399 CAS.
- S. Y. Jiang, K. E. Gubbins and J. A. Zollweg, Mol. Phys., 1993, 80, 103–116 CrossRef CAS.
- X. Xu, X. Zhao, L. Sun and X. Liu, J. Nat. Gas Chem., 2009, 18, 167–172 Search PubMed.
- J. A. Delgado, M. A. Uguina, J. M. Gómez and L. Ortega, Sep. Purif. Technol., 2006, 48, 223–228 CrossRef CAS.
- K. A. Fisher, K. D. Huddersman and M. J. Taylor, Chem.–Eur. J., 2003, 9, 5873–5878 CrossRef CAS.
- A. Zukal, I. Dominguez, J. Mayerová and J. Čejka, Langmuir, 2009, 25, 10314–10321 CrossRef CAS.
- C. Martin, N. Tosi-Pellenq, J. Patarin and J. P. Coulomb, Langmuir, 1998, 14, 1774–1778 CrossRef CAS.
- X. X. Zhao, X. L. Xu, L. B. Sun, L. L. Zhang and X. Q. Liu, Energy Fuels, 2009, 23, 1534–1538 CrossRef CAS.
- M. E. Rivera-Ramos, G. J. Ruiz-Mercado and A. J. Hernandez-Maldonado, Ind. Eng. Chem. Res., 2008, 47, 5602–5610 CrossRef CAS.
- I. Deroche, L. Gaberova, G. Maurin, P. L. Llewellyn, M. Castro and P. A. Wright, Adsorption, 2008, 14, 207–213 CrossRef CAS.
- H. Hayashi, A. P. Cote, H. Furukawa, M. O'Keeffe and O. M. Yaghi, Nat. Mater., 2007, 6, 501–506 CrossRef CAS.
- B. Wang, A. P. Cote, H. Furukawa, M. O'Keeffe and O. M. Yaghi, Nature, 2008, 453, 207–U6 CrossRef CAS.
- S. Bourrelly, P. L. Llewellyn, C. Serre, F. Millange, T. Loiseau and G. Férey, J. Am. Chem. Soc., 2005, 127, 13519–13521 CrossRef CAS.
- F. Debatin, A. Thomas, A. Kelling, N. Hedin, Z. Bacsik, I. Senkovska, S. Kaskel, M. Junginger, H. Müller, U. Schilde, C. Jäger, A. Friedrich and H.-J. H, Angew. Chem., Int. Ed., 2010, 49, 1258–1262 CrossRef CAS.
- A. Kapoor and R. T. Yang, Chem. Eng. Sci., 1989, 44, 1723–1733 CrossRef CAS.
- T. D. Burchell, R. R. Judkins, M. R. Rogers and A. M. Williams, Carbon, 1997, 35, 1279–1294 CrossRef CAS.
- T. C. Drage, J. M. Blackman, C. Pevida and C. E. Snape, Energy Fuels, 2009, 23, 2790–2796 CrossRef CAS.
- A. Ghosh, K. S. Subrahmanyam, K. S. Krishna, S. Datta, A. Govindaraj, S. K. Pati and C. N. R. Rao, J. Phys. Chem. C, 2008, 112, 15704–15707 CrossRef CAS.
- G. Chandrasekar, W. J. Son and W. S. Ahn, J. Porous Mater., 2009, 16, 545–551 CrossRef CAS.
- V. Zelenak, D. Halamova, L. Gaberova, E. Bloch and P. L. Llewellyn, Microporous Mesoporous Mater., 2008, 116, 358–364 CrossRef CAS.
- X. C. Xu, C. S. Song, J. M. Andresen, B. G. Miller and A. W. Scaroni, Energy Fuels, 2002, 16, 1463–1469 CrossRef CAS.
- J. C. Hicks, J. H. Drese, D. J. Fauth, M. L. Gray, G. Qi and C. W. Jones, J. Am. Chem. Soc., 2008, 130, 2902–2903 CrossRef CAS.
- H. Y. Huang, R. T. Yang, D. Chinn and C. L. Munson, Ind. Eng. Chem. Res., 2003, 42, 2427–2433 CrossRef CAS.
- S. N. Kim, W. J. Son, J. S. Choi and W. S. Ahn, Microporous Mesoporous Mater., 2008, 115, 497–503 CrossRef CAS.
- P. J. E. Harlick and A. Sayari, Ind. Eng. Chem. Res., 2007, 46, 446–458 CrossRef CAS.
- P. J. E. Harlick and A. Sayari, Ind. Eng. Chem. Res., 2006, 45, 3248–3255 CrossRef CAS.
- R. S. Franchi, P. J. E. Harlick and A. Sayari, Ind. Eng. Chem. Res., 2005, 44, 8007–8013 CrossRef CAS.
- R. Franchi, P. J. E. Harlick and A. Sayari, Stud. Surf. Sci. Catal., 2005, 156, 879–886 CAS.
- C. Chen, S. T. Yang, W. S. Ahn and R. Ryoo, Chem. Commun., 2009, 3627–3629 RSC.
- A. D. Ebner, S. P. Reynolds and J. A. Ritter, Ind. Eng. Chem. Res., 2006, 45, 6387–6392 CrossRef CAS.
- M. L. Gray, J. S. Hoffman, D. C. Hreha, D. J. Fauth, S. W. Hedges, K. J. Champagne and H. W. Pennline, Energy Fuels, 2009, 23, 4840–4844 CrossRef CAS.
- Y. Belmabkhout and A. Sayari, Chem. Eng. Sci., 2009, 64, 3729–3735 CrossRef CAS.
- Y. Belmabkhout, R. Serna-Guerrero and A. Sayari, Chem. Eng. Sci., 2009, 64, 3721–3728 CrossRef CAS.
- L. Zhao, Z. Bacsik, N. Hedin, W. Wei, Y. H. Sun, M. Antonietti and M. M. Titirici, ChemSusChem, 2010, 3, 840–845 CrossRef CAS.
- C. Baerlocher and L. B. McCusker, Database of Zeolite Structures, http://www.iza-structure.org/databases/ Search PubMed.
- R. M. Milton, US patent: 2 882 244, 1959.
- R. M. Barrer and R. M. Gibbons, Trans. Faraday Soc., 1965, 61, 948 RSC.
- S. Cavenati, C. A. Grande and A. E. Rodrigues, J. Chem. Eng. Data, 2004, 49, 1095–1101 CrossRef CAS.
- F. Brandani and D. M. Ruthven, Ind. Eng. Chem. Res., 2004, 43, 8339–8344 CrossRef CAS.
- G. K. Papadopoulos and D. N. Theodorou, Mol. Simul., 2009, 35, 79–89 CrossRef CAS.
- Y. Nakazaki, Y. Tanaka, N. Goto and T. Inui, Catal. Today, 1995, 23, 391–396 CrossRef CAS.
- W. Jia and S. Murad, J. Chem. Phys., 2004, 120, 4877–4885 CrossRef CAS.
- W. Jia and S. Murad, J. Chem. Phys., 2005, 122, 234708 CrossRef CAS.
- P. Galhotra, J. G. Navea, S. C. Larsen and V. H. Grassian, Energy Environ. Sci., 2009, 2, 401–409 RSC.
- P. J. E. Harlick and F. H. Tezel, Sep. Sci. Technol., 2005, 40, 2569–2591 CrossRef CAS.
- W. Shao, L. Z. Zhang, L. X. Li and R. L. Lee, Adsorption, 2009, 15, 497–505 CrossRef CAS.
- G. Maurin, Y. Belmabkhout, G. Pirngruber, L. Gaberova and P. L. Llewellyn, Adsorption, 2007, 13, 453–460 CrossRef CAS.
- G. Maurin, R. Bell, B. Kuchta, T. Poyet and P. L. Llewellyn, Adsorption, 2005, 11, 331–336 CrossRef.
- A. Ghoufi, L. Gaberova, J. Rouquerol, D. Vincent, P. L. Llewellyn and G. Maurin, Microporous Mesoporous Mater., 2009, 119, 117–128 CrossRef CAS.
- D. F. Plant, G. Maurin, I. Deroche and P. L. Llewellyn, Microporous Mesoporous Mater., 2007, 99, 70–78 CrossRef CAS.
- D. F. Plant, G. Maurin, H. Jobic and P. L. Llewellyn, J. Phys. Chem. B, 2006, 110, 14372–14378 CrossRef CAS.
- D. F. Plant, H. Jobic, P. L. Llewellyn and G. Maurin, Adsorption, 2007, 13, 209–214 CrossRef CAS.
- A. Pulido, M. R. Delgado, O. Bludský, M. Rubeš, P. Nachtigall and C. O. Areán, Energy Environ. Sci., 2009, 2, 1187–1195 RSC.
- D. F. Plant, G. Maurin, I. Deroche, L. Gaberova and P. L. Llewellyn, Chem. Phys. Lett., 2006, 426, 387–392 CrossRef CAS.
- A. Chatterjee and T. Iwasaki, J. Phys. Chem. A, 1999, 103, 9857–9863 CrossRef CAS.
- K. Kusakabe, T. Kuroda, A. Murata and S. Morooka, Ind. Eng. Chem. Res., 1997, 36, 649–655 CrossRef CAS.
- S. S. Liu and X. N. Yang, J. Chem. Phys., 2006, 124, 244705 CrossRef.
- E. Jaramillo and M. Chandross, J. Phys. Chem. B, 2004, 108, 20155–20159 CrossRef CAS.
- E. D. Akten, R. Siriwardane and D. S. Sholl, Energy Fuels, 2003, 17, 977–983 CrossRef CAS.
- J. Izumi, A. Yasutake, N. Tomonaga, N. Oka, H. Ota, N. Akutsu, S. Umeda and M. Tajima, Stud. Surf. Sci. Catal., 1997, 105, 2315–2322.
- Q. L. Liu, A. Mace, Z. Bacsik, J. L. Sun, A. Laaksonen and N. Hedin, Chem. Commun., 2010, 46, 4502–4504 RSC.
- E. M. Flanigen, J. M. Bennett, R. W. Grose, J. P. Cohen, R. L. Patton, R. M. Kirchner and J. V. Smith, Nature, 1978, 271, 512–516 CAS.
- J. A. Dunne, R. Mariwals, M. Rao, S. Sircar, R. J. Gorte and A. L. Myers, Langmuir, 1996, 12, 5888–5895 CrossRef CAS.
- P. J. E. Harlick and F. H. Tezel, Sep. Sci. Technol., 2002, 37, 33–60 CrossRef CAS.
- P. J. E. Harlick and F. H. Tezel, Sep. Purif. Technol., 2003, 33, 199–210 CrossRef CAS.
- A. Hirotani, K. Mizukami, R. Miura, H. Takaba, T. Miya, A. Fahmi, A. Stirling, M. Kubo and A. Miyamoto, Appl. Surf. Sci., 1997, 120, 81–84 CrossRef CAS.
- T. Yamazaki, M. Katoh, S. Ozawa and Y. Ogino, Mol. Phys., 1993, 80, 313–324 CAS.
- L. C. Geiger, B. M. Ladanyi and M. E. Chapin, J. Chem. Phys., 1990, 93, 4533–4542 CrossRef CAS.
- A. Zukal, J. Pawlesa and J. Čejka, Adsorption, 2009, 15, 264–270 CrossRef CAS.
- J. M. Leyssale, G. K. Papadopoulos and D. N. Theodorou, J. Phys. Chem. B, 2006, 110, 22742–22753 CrossRef CAS.
- X. P. Yue and X. N. Yang, Langmuir, 2006, 22, 3138–3147 CrossRef CAS.
- G. K. Papadopoulos, H. Jobic and D. N. Theodorou, J. Phys. Chem. B, 2004, 108, 12748–12756 CrossRef CAS.
- S. Himeno, M. Takenaka and S. Shimura, Mol. Simul., 2008, 34, 1329–1336 CrossRef CAS.
- E. García-Pérez, J. B. Parra, C. O. Ania, A. García-Sánchez, J. M. Van Baten, R. Krishna, D. Dubbeldam and S. Calero, Adsorption, 2007, 13, 469–476 CrossRef CAS.
- S. E. Jee and D. S. Sholl, J. Am. Chem. Soc., 2009, 131, 7896–7904 CrossRef CAS.
- N. Hedin, G. J. DeMartin, W. J. Roth, K. G. Strohmaier and S. C. Reyes, Microporous Mesoporous Mater., 2008, 109, 327–334 CrossRef CAS.
- R. R. Chance, 229 ACS Nat M San Di 2005 Search PubMed.
- R. Krishna and J. M. van Baten, Chem. Eng. J., 2007, 133, 121–131 CrossRef CAS.
- D. Selassie, D. Davis, J. Dahlin, E. Feise, G. Haman, D. S. Sholl and D. Kohen, J. Phys. Chem. C, 2008, 112, 16521–16531 CrossRef CAS.
- R. Krishna and J. M. van Baten, Sep. Purif. Technol., 2008, 61, 414–423 CrossRef CAS.
- R. Krishna and J. M. van Baten, Chem. Phys. Lett., 2007, 446, 344–349 CrossRef CAS.
- R. Krishna, J. M. Van Baten, E. García-Pérez and S. Calero, Ind. Eng. Chem. Res., 2007, 46, 2974–2986 CrossRef CAS.
- R. Krishna, J. M. van Baten, E. García-Pérez and S. Calero, Chem. Phys. Lett., 2006, 429, 219–224 CrossRef CAS.
- J. Van Den Bergh, S. A. Ban, T. J. H. Vlugt and F. Kapteijn, J. Phys. Chem. C, 2009, 113, 17840–17850 CrossRef CAS.
- R. Krishna and J. M. van Baten, Chem. Eng. Sci., 2008, 63, 3120–3140 CrossRef CAS.
- Y. Ohta, H. Takaba and S. I. Nakao, Microporous Mesoporous Mater., 2007, 101, 319–323 CrossRef CAS.
- A. Goj, D. S. Sholl, E. D. Akten and D. Kohen, J. Phys. Chem. B, 2002, 106, 8367–8375 CrossRef CAS.
- J. R. Kiovsky and P. B. Koradia, US patent: 4 059 543, 1977.
- G. Aguilar-Armenta, M. E. Patino-Iglesias and R. Leyva-Ramos, Adsorpt. Sci. Technol., 2003, 21, 81–91 Search PubMed.
- R. V. Siriwardane, M. S. Shen and E. P. Fisher, Energy Fuels, 2003, 17, 571–576 CrossRef CAS.
- S. T. Wilson, B. M. Lok, C. A. Messina, T. R. Cannan and E. M. Flanigen, J. Am. Chem. Soc., 1982, 104, 1146–1147 CrossRef CAS.
- B. M. Lok, C. A. Messina, R. L. Patton, R. T. Gajek, T. R. Cannan and E. M. Flanigen, J. Am. Chem. Soc., 1984, 106, 6092–6093 CrossRef CAS.
- X. X. Guan, F. X. Zhang, G. J. Wu and N. J. Guan, Mater. Lett., 2006, 60, 3141–3144 CrossRef CAS.
- O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705–714 CrossRef CAS.
- G. Férey, Chem. Soc. Rev., 2008, 37, 191–214 RSC.
- J.-R. Li, R. J. Kuppler and H.-C. Zhou, Chem. Soc. Rev., 2009, 38, 1477–1504 RSC.
- S. Keskin, J. Liu, R. B. Rankin, J. K. Johnson and D. S. Sholl, Ind. Eng. Chem. Res., 2009, 48, 2355–2371 CrossRef CAS.
- M. Tafipolsky, S. Amirjalayer and R. Schmid, Microporous Mesoporous Mater., 2010, 129, 304–318 CrossRef CAS.
- T. Düren, Y. S. Bae and R. Q. Snurr, Chem. Soc. Rev., 2009, 38, 1237–1247 RSC.
- M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keeffe and O. M. Yaghi, Science, 2002, 295, 469–472 CrossRef.
- A. C. Sudik, A. R. Millward, N. W. Ockwig, A. P. Cote, J. Kim and O. M. Yaghi, J. Am. Chem. Soc., 2005, 127, 7110–7118 CrossRef CAS.
- R. Banerjee, A. Phan, B. Wang, C. Knobler, H. Furukawa, M. O'Keeffe and O. M. Yaghi, Science, 2008, 319, 939–943 CrossRef CAS.
- Z. X. Zhao, Z. Li and Y. S. Lin, Ind. Eng. Chem. Res., 2009, 48, 10015–10020 CrossRef CAS.
- Q. Y. Yang, Q. Xu, B. Liu, C. L. Zhong and S. Berend, Chin. J. Chem. Eng., 2009, 17, 781–790 Search PubMed.
- B. Liu, Q. Y. Yang, C. Y. Xue, C. L. Zhong, B. Chen and B. Smit, J. Phys. Chem. C, 2008, 112, 9854–9860 CrossRef CAS.
- L. Bastin, P. S. Bárcia, E. J. Hurtado, J. A. C. Silva, A. E. Rodrigues and B. Chen, J. Phys. Chem. C, 2008, 112, 1575–1581 CrossRef CAS.
- P. S. Barcia, L. Bastin, E. J. Hurtado, J. A. C. Silva, A. E. Rodrigues and B. L. Chen, Sep. Sci. Technol., 2008, 43, 3494–3521 CrossRef CAS.
- P. S. Bárcia, J. A. C. Silva and A. E. Rodrigues, AIChE J., 2007, 53, 1970–1981 CrossRef CAS.
- P. S. Bárcia, J. A. C. Silva and A. E. Rodrigues, Ind. Eng. Chem. Res., 2006, 45, 4316–4328 CrossRef CAS.
- D. H. Liu, C. C. Zheng, Q. Y. Yang and C. L. Zhong, J. Phys. Chem. C, 2009, 113, 5004–5009 CrossRef CAS.
- R. B. Rankin, J. C. Liu, A. D. Kulkarni and J. K. Johnson, J. Phys. Chem. C, 2009, 113, 16906–16914 CrossRef CAS.
- R. Babarao and J. W. Jiang, J. Am. Chem. Soc., 2009, 131, 11417–11425 CrossRef CAS.
- R. Babarao and J. W. Jiang, Energy Environ. Sci., 2009, 2, 1088–1093 RSC.
- N. A. Ramsahye, G. Maurin, S. Bourrelly, P. L. Llewellyn, C. Serre, T. Loiseau, T. Devic and G. Férey, J. Phys. Chem. C, 2008, 112, 514–520 CrossRef CAS.
- C. Serre, S. Bourrelly, A. Vimont, N. A. Ramsahye, G. Maurin, P. L. Llewellyn, M. Daturi, Y. Filinchuk, O. Leynaud, P. Barnes and G. Férey, Adv. Mater., 2007, 19, 2246–2251 CrossRef.
- D. S. Coombes, F. Corà, C. Mellot-Draznieks and R. G. Bell, J. Phys. Chem. C, 2009, 113, 544–552 CrossRef CAS.
- L. Hamon, P. L. Llewellyn, T. Devic, A. Ghoufi, G. Clet, V. Guillerm, G. D. Pirngruber, G. Maurin, C. Serre, G. Driver, W. Van Beek, E. Jolimaître, A. Vimont, M. Daturi and G. Férey, J. Am. Chem. Soc., 2009, 131, 17490–17499 CrossRef CAS.
- A. Ö. Yazaydin, A. I. Benin, S. A. Faheem, P. Jakubczak, J. J. Low, Richard R. Willis and R. Q. Snurr, Chem. Mater., 2009, 21, 1425–1430 CrossRef.
- Q. Y. Yang, C. Y. Xue, C. L. Zhong and J. F. Chen, AIChE J., 2007, 53, 2832–2840 CrossRef CAS.
- Z. J. Liang, M. Marshall and A. L. Chaffee, Energy Fuels, 2009, 23, 2785–2789 CrossRef CAS.
- Y. Cheng, A. Rondo, H. Noguchi, H. Kajiro, K. Urita, T. Ohba, K. Kaneko and H. Kanoh, Langmuir, 2009, 25, 4510–4513 CrossRef CAS.
- S. R. Miller, P. A. Wright, T. Devic, C. Serre, G. Ferey, P. L. Llewellyn, R. Denoyel, L. Gaberova and Y. Filinchuk, Langmuir, 2009, 25, 3618–3626 CrossRef CAS.
- R. Babarao, Z. Q. Hu, J. W. Jiang, S. Chempath and S. I. Sandler, Langmuir, 2007, 23, 659–666 CrossRef CAS.
- R. Babarao and J. W. Jiang, Langmuir, 2008, 24, 5474–5484 CrossRef CAS.
- R. Babarao, J. W. Jiang and S. I. Sandler, Langmuir, 2009, 25, 5239–5247 CrossRef CAS.
- R. Babarao, J. W. Jiang and S. I. Sandler, Langmuir, 2009, 25, 6590 CrossRef CAS.
- D. Farrusseng, C. Daniel, C. Gaudillère, U. Ravon, Y. Schuurman, C. Mirodatos, D. Dubbeldam, H. Frost and R. Q. Snurr, Langmuir, 2009, 25, 7383–7388 CrossRef CAS.
- A. Martín-Calvo, E. García-Pérez, J. Manuel Castillo and S. Calero, Phys. Chem. Chem. Phys., 2008, 10, 7085–7091 RSC.
- Q. Y. Yang, C. L. Zhong and J. F. Chen, J. Phys. Chem. C, 2008, 112, 1562–1569 CrossRef CAS.
- S. Keskin and D. S. Sholl, Langmuir, 2009, 25, 11786–11795 CrossRef CAS.
- A. Demessence, D. M. D'Alessandro, M. L. Foo and J. R. Long, J. Am. Chem. Soc., 2009, 131, 8784 CrossRef CAS.
- Y. S. Bae, O. K. Farha, J. T. Hupp and R. Q. Snurr, J. Mater. Chem., 2009, 19, 2131–2134 RSC.
- Z. Q. Wang and S. M. Cohen, J. Am. Chem. Soc., 2009, 131, 16675–16677 CrossRef CAS.
- Y. S. Bae, K. L. Mulfort, H. Frost, P. Ryan, S. Punnathanam, L. J. Broadbelt, J. T. Hupp and R. Q. Snurr, Langmuir, 2008, 24, 8592–8598 CrossRef CAS.
- A. Torrisi, C. Mellot-Draznieks and R. G. Bell, J. Chem. Phys., 2009, 130, 194703 CrossRef.
- A. Torrisi, C. Mellot-Draznieks and R. G. Bell, J. Chem. Phys., 2010, 132, 044705 CrossRef.
- H. S. Choi and M. P. Suh, Angew. Chem., Int. Ed., 2009, 48, 6865–6869 CrossRef CAS.
- S. Satyapal, T. Filburn, J. Trela and J. Strange, Energy Fuels, 2001, 15, 250–255 CrossRef CAS.
- P. M. Budd, B. S. Ghanem, S. Makhseed, N. B. McKeown, K. J. Msayib and C. E. Tattershall, Chem. Commun., 2004, 230–231 RSC.
- N. Ritter, M. Antonietti, A. Thomas, I. Senkovska, S. Kaskel and J. Weber, Macromolecules, 2009, 42, 8017–8020 CrossRef CAS.
- R. Babarao and J. W. Jiang, Energy Environ. Sci., 2008, 1, 139–143 RSC.
- Q. Y. Yang and C. L. Zhong, Langmuir, 2009, 25, 2302–2308 CrossRef CAS.
- R. Babarao and J. W. Jiang, Langmuir, 2008, 24, 6270–6278 CrossRef CAS.
- L. B. Adams, C. R. Hall, R. J. Holmes and R. A. Newton, Carbon, 1988, 26, 451–459 CrossRef CAS.
- P. L. Walker, R. J. Foresti and C. C. Wright, Ind. Eng. Chem., 1953, 45, 1703–1710 CrossRef CAS.
- P. L. Walker Jr. and M. Shelef, Carbon, 1967, 5, 7–11 CrossRef.
- F. Brandani, A. Rouse, S. Brandani and D. M. Ruthven, Adsorption, 2004, 10, 99–109 CrossRef CAS.
- S. Urbonaite, J. M. Juarez-Galan, J. Leis, F. Rodriguez-Reinoso and G. Svensson, Microporous Mesoporous Mater., 2008, 113, 14–21 CrossRef CAS.
- R. V. Siriwardane, M. S. Shen, E. P. Fisher and J. A. Poston, Energy Fuels, 2001, 15, 279–284 CrossRef CAS.
- A. Montoya, F. Mondragón and T. N. Truong, Carbon, 2003, 41, 29–39 CrossRef CAS.
- D. Levesque and F. D. Lamari, Mol. Phys., 2009, 107, 591–597 CrossRef CAS.
- C. M. Tenney and C. M. Lastoskie, Environ. Prog., 2006, 25, 343–354 CrossRef CAS.
- S. H. Joo, S. Jun and R. Ryoo, Microporous Mesoporous Mater., 2001, 44–45, 153–158 CrossRef CAS.
- Y. Wan and D. Y. Zhao, Chem. Rev., 2007, 107, 2821–2860 CrossRef CAS.
- X. Peng, D. P. Cao and J. S. Zhao, Sep. Purif. Technol., 2009, 68, 50–60 CrossRef CAS.
- S. Liu, X. Yang and Z. Yang, Chin. Sci. Bull., 2008, 53, 1358–1364 CrossRef CAS.
- D. P. Cao and J. Z. Wu, Carbon, 2005, 43, 1364–1370 CrossRef CAS.
- M. Heuchel, G. M. Davies, E. Buss and N. A. Seaton, Langmuir, 1999, 15, 8695–8705 CrossRef CAS.
- D. M. Ruthven and S. C. Reyes, Microporous Mesoporous Mater., 2007, 104, 59–66 CrossRef CAS.
- S. N. Vyas, S. R. Patwardhan, S. Vijayalakshmi and K. Ganesh, J. Colloid Interface Sci., 1994, 168, 275–280 CrossRef CAS.
- M. A. Ahmad, W. M. A. W. Daud and M. K. Aroua, J. Oil Palm Res., 2008, 20, 453–460 Search PubMed.
- M. A. Ahmad, W. M. A. W. Daud and M. K. Aroua, Colloids Surf., A, 2008, 312, 131–135 CrossRef CAS.
- W. M. A. W. Daud, M. A. Ahmad and M. K. Aroua, Sep. Purif. Technol., 2007, 57, 289–293 CrossRef CAS.
- P. J. M. Carrott, I. P. P. Cansado and M. M. L. Ribeiro Carrott, Appl. Surf. Sci., 2006, 252, 5948–5952 CrossRef CAS.
- S. W. Rutherford and D. D. Do, Langmuir, 2000, 16, 7245–7254 CrossRef CAS.
- S. W. Rutherford and D. D. Do, Carbon, 2000, 38, 1339–1350 CrossRef CAS.
- S. W. Rutherford and J. E. Coons, Carbon, 2003, 41, 405–411 CrossRef CAS.
- J. M. V. Nabais, P. J. M. Carrott, M. M. L. R. Carrott, A. M. Padre-Eterno, J. A. Menendez, A. Dominguez and A. L. Ortiz, Carbon, 2006, 44, 1158–1165 CrossRef CAS.
- A. Jayaraman, A. S. Chiao, J. Padin, R. T. Yang and C. L. Munson, Sep. Sci. Technol., 2002, 37, 2505–2528 CrossRef CAS.
- M. C. Campo, F. D. Magalhães and A. Mendes, J. Membr. Sci., 2010, 346, 15–25 CrossRef CAS.
- D. S. Lafyatis, J. Tung and H. C. Foley, Ind. Eng. Chem. Res., 1991, 30, 865–873 CrossRef.
- R. K. Mariwala and H. C. Foley, Ind. Eng. Chem. Res., 1994, 33, 607–615 CrossRef CAS.
- T. X. Nguyen and S. K. Bhatia, Asia-Pac. J. Chem. Eng., 2009, 4, 557–562 Search PubMed.
- A. A. Fomkin, Prot. Met. Phys. Chem. Surf., 2009, 45, 121–136 Search PubMed.
- E. Pantatosaki, D. Psomadopoulos, T. Steriotis, A. K. Stubos, A. Papaioannou and G. K. Papadopoulos, Colloids Surf., A, 2004, 241, 127–135 CrossRef CAS.
- T. A. Steriotis, G. K. Papadopoulos, A. K. Stubos and N. Kanellopoulos, Stud. Surf. Sci. Catal., 2002, 144, 545–552 CAS.
- S. Samios, G. K. Papadopoulos, T. Steriotis and A. K. Stubos, Mol. Simul., 2001, 27, 441–456 CrossRef CAS.
- S. Samios, A. K. Stubos, G. K. Papadopoulos, N. K. Kanellopoulos and F. Rigas, J. Colloid Interface Sci., 2000, 224, 272–290 CrossRef CAS.
- A. Vishnyakov, P. I. Ravikovitch and A. V. Neimark, Langmuir, 1999, 15, 8736–8742 CrossRef CAS.
- S. Samios, A. K. Stubos, N. K. Kanellopoulos, R. F. Cracknell, G. K. Papadopoulos and D. Nicholson, Langmuir, 1997, 13, 2795–2802 CrossRef CAS.
- M. Cinke, J. Li, C. W. Bauschlicher Jr., A. Ricca and M. Meyyappan, Chem. Phys. Lett., 2003, 376, 761–766 CrossRef CAS.
- J. Zhao, A. Buldum, J. Han and J. P. Lu, Nanotechnology, 2002, 13, 195–200 CrossRef CAS.
- A. Ansón, J. Jagiello, J. B. Parra, M. L. Sanjuán, A. M. Benito, W. K. Maser and M. T. Martínez, J. Phys. Chem. B, 2004, 108, 15820–15826 CrossRef CAS.
- W. L. Yim, O. Byl, J. T. Yates Jr. and J. K. Johnson, J. Chem. Phys., 2004, 120, 5377–5386 CrossRef CAS.
- C. Matranga, L. Chen, M. Smith, E. Bittner, J. K. Johnson and B. Bockrath, J. Phys. Chem. B, 2003, 107, 12930–12941 CrossRef CAS.
- F. S. Su, C. S. Lu, W. F. Cnen, H. L. Bai and J. F. Hwang, Sci. Total Environ., 2009, 407, 3017–3023 CrossRef CAS.
- P. I. Ravikovitch, A. Vishnyakov, R. Russo and A. V. Neimark, Langmuir, 2000, 16, 2311–2320 CrossRef CAS.
- L. L Huang, L. Z. Zhang, Q. Shao, L. H. Lu, X. H. Lu, S. Y. Jiang and W. F. Shen, J. Phys. Chem. C, 2007, 111, 11912–11920 CrossRef CAS.
- M. Konstantakou, T. A. Steriotis, G. K. Papadopoulos, M. Kainourgiakis, E. S. Kikkinides and A. K. Stubos, Appl. Surf. Sci., 2007, 253, 5715–5720 CrossRef CAS.
- A. I. Skoulidas, D. S. Sholl and J. K. Johnson, J. Chem. Phys., 2006, 124, 054708 CrossRef.
- S. B. Sinnott, Z. A. Mao and K. H. Lee, CMES Comput. Model. Eng. Sci., 2002, 3, 575–587 Search PubMed.
- L. F. Xu, M. Sahimi and T. T. Tsotsis, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 2000, 62, 6942–6948 CrossRef CAS.
- L. F. Xu, M. G. Sedigh, T. T. Tsotsis and M. Sahimi, J. Chem. Phys., 2000, 112, 910–922 CrossRef CAS.
- E. A. Müller, J. Phys. Chem. B, 2008, 112, 8999–9005 CrossRef CAS.
- J. C. Su and A. C. Lua, Sep. Purif. Technol., 2009, 69, 161–167 CrossRef CAS.
- Y. X. Jia, M. Wang, L. Y. Wu and C. J. Gao, Sep. Sci. Technol., 2007, 42, 3681–3695 CrossRef CAS.
- Q. Y. Yang and C. L. Zhong, Can. J. Chem. Eng., 2004, 82, 580–589 CAS.
- P. A. Gauden and M. Wiśniewski, Appl. Surf. Sci., 2007, 253, 5726–5731 CrossRef CAS.
- A. P. Terzyk, S. Furmaniak, P. A. Gauden and P. Kowalczyk, Adsorpt. Sci. Technol., 2009, 27, 281–296 Search PubMed.
- K. Urita, S. Seki, S. Utsumi, D. Noguchi, H. Kanoh, H. Tanaka, Y. Hattori, Y. Ochiai, N. Aoki, M. Yudasaka, S. Iijima and K. Kaneko, Nano Lett., 2006, 6, 1325–1328 CrossRef CAS.
- O. Dewaele and G. F. Froment, Appl. Catal., A, 1999, 185, 203–210 CrossRef CAS.
- Z. Yong, V. Mata and A. E. Rodrigues, J. Chem. Eng. Data, 2000, 45, 1093–1095 CrossRef CAS.
- K. Pokrovski, K. T. Jung and A. T. Bell, Langmuir, 2001, 17, 4297–4303 CrossRef CAS.
- C. Knoefel, V. Hornebecq and P. L. Llewellyn, Langmuir, 2008, 24, 7963–7969 CrossRef.
- C. Schumacher, J. Gonzalez, M. Perez-Mendoza, P. Wright and N. Seaton, Ind. Eng. Chem. Res., 2006, 45, 5586–5597 CrossRef CAS.
- Y. He and N. A. Seaton, Langmuir, 2006, 22, 1150–1155 CrossRef CAS.
- T. Yoshioka, M. Asaeda and T. Tsuru, J. Membr. Sci., 2007, 293, 81–93 CrossRef CAS.
- X. N. Yang, Z. J. Xu and C. J. Zhang, J. Colloid Interface Sci., 2006, 297, 38–44 CrossRef CAS.
- H. Takaba, E. Matsuda and S. I. Nakao, J. Phys. Chem. B, 2004, 108, 14142–14147 CrossRef CAS.
- T. Yoshioka, T. Tsuru and M. Asaeda, Sep. Purif. Technol., 2001, 25, 441–449 CrossRef CAS.
- O. Leal, C. Bolivar, C. Ovalles, J. J. Garcia and Y. Espidel, Inorg. Chim. Acta, 1995, 240, 183–189 CrossRef CAS.
- G. P. Knowles, S. W. Delaney and A. L. Chaffee, Stud. Surf. Sci. Catal., 2005, 156, 887 CAS.
- G. P. Knowles, J. V. Graham, S. W. Delaney and A. L. Chaffee, Fuel Process. Technol., 2005, 86, 1435–1448 CrossRef CAS.
- S. Kim, J. Ida, V. V. Guliants and J. Y. S. Lin, J. Phys. Chem. B, 2005, 109, 6287–6293 CrossRef CAS.
- R. A. Khatri, S. S. C. Chuang, Y. Soong and M. Gray, Energy Fuels, 2006, 20, 1514–1520 CrossRef CAS.
- R. Serna-Guerrero, E. Da'na and A. Sayari, Ind. Eng. Chem. Res., 2008, 47, 9406–9412 CrossRef CAS.
- A. L. Chaffee, G. P. Knowles, Z. J. Liang, J. Zhany, P. Xiao and P. A. Webley, Int. J. Greenhouse Gas Control, 2007, 1, 11–18 CrossRef CAS.
- E. Angeletti, C. Canepa, G. Martinetti and P. Venturello, J. Chem. Soc., Perkin Trans. 1, 1989, 105–107 RSC.
- S. Che, A. E. Garcia-Bennett, T. Yokoi, K. Sakamoto, H. Kunieda, O. Terasaki and T. Tatsumi, Nat. Mater., 2003, 2, 801–805 CrossRef CAS.
- G. Y. Zhao, B. Aziz and N. Hedin, Appl. Energy, 2010, 87, 2907–2913 CrossRef CAS.
- S. Udayakumar, S. W. Park, D. W. Park and B. S. Choi, Catal. Commun., 2008, 9, 1563–1570 CrossRef CAS.
- J. M. Rosenholm, A. Penninkangas and M. Linden, Chem. Commun., 2006, 3909–3911 RSC.
- J. H. Drese, S. Choi, R. P. Lively, W. J. Koros, D. J. Fauth, M. L. Gray and C. W. Jones, Adv. Funct. Mater., 2009, 19, 3821–3832 CrossRef CAS.
- X. C. Xu, C. S. Song, B. G. Miller and A. W. Scaroni, Ind. Eng. Chem. Res., 2005, 44, 8113–8119 CrossRef CAS.
- X. C. Xu, C. S. Song, B. G. Miller and A. W. Scaroni, Fuel Process. Technol., 2005, 86, 1457–1472 CrossRef CAS.
- X. C. Xu, C. S. Song, J. M. Andrésen, B. G. Miller and A. W. Scaroni, Microporous Mesoporous Mater., 2003, 62, 29–45 CrossRef CAS.
- X. C. Xu, C. S. Song, J. M. Andresen, B. G. Miller and A. W. Scaroni, Int. J. Environ. Technol. Manage., 2004, 4, 32–52 Search PubMed.
- T. Montanari and G. Busca, Vib. Spectrosc., 2008, 46, 45–51 CrossRef CAS.
- R. W. Stevens, R. V. Siriwardane and J. Logan, Energy Fuels, 2008, 22, 3070–3079 CrossRef CAS.
- N. Hiyoshi, K. Yogo and T. Yashima, Microporous Mesoporous Mater., 2005, 84, 357–365 CrossRef CAS.
- R. A. Khatri, S. S. C. Chuang, Y. Soong and M. Gray, Ind. Eng. Chem. Res., 2005, 44, 3702–3708 CrossRef CAS.
- A. C. C. Chang, S. S. C. Chuang, M. Gray and Y. Soong, Energy Fuels, 2003, 17, 468–473 CrossRef CAS.
- C. W. Hoerr, H. J. Harwood and G. V. Ramarao, J. Org. Chem., 1944, 9, 201 CrossRef CAS.
- K. P. Battjes, A. M. Barolo and P. Dreyfuss, J. Adhes. Sci. Technol., 1991, 5, 785–799 CrossRef CAS.
- M. Aresta and E. Quaranta, Tetrahedron, 1992, 48, 1515–1530 CrossRef CAS.
- A. Dibenedetto, M. Aresta, C. Fragale and M. Narracci, Green Chem., 2002, 4, 439–443 RSC.
- Y. Belmabkhout and A. Sayari, Adsorption, 2009, 15, 318–328 CrossRef CAS.
- N. Hiyoshi, K. Yogo and T. Yashima, J. Jpn. Pet. Inst., 2005, 48, 29–36 Search PubMed.
- Z. Bacsik, R. Atluri, A. E. Garcia-Bennett and N. Hedin, Langmuir, 2010, 26, 10013–10024 CrossRef CAS.
- M. L. Gray, Y. Soong, K. J. Champagne, J. Baltrus, R. W. Stevens, P. Toochinda and S. S. C. Chuang, Sep. Purif. Technol., 2004, 35, 31–36 CrossRef CAS.
- C. Y. Lu, H. L. Bai, B. L. Wu, F. S. Su and J. F. Hwang, Energy Fuels, 2008, 22, 3050–3056 CrossRef CAS.
- E. P. Dillon, C. A. Crouse and A. R. Barron, ACS Nano, 2008, 2, 156–164 CrossRef CAS.
- A. L. Chaffee, Fuel Process. Technol., 2005, 86, 1473–1486 CrossRef CAS.
- L. J. Chen, A. Laaksonen, submitted.
- Z. Yong, V. Mata and A. E. Rodriguez, Ind. Eng. Chem. Res., 2001, 40, 204–209 CrossRef CAS.
- S. P. Reynolds, A. D. Ebner and J. A. Ritter, Environ. Prog., 2006, 25, 334–342 CrossRef CAS.
- S. P. Reynolds, A. D. Ebner and J. A. Ritter, Ind. Eng. Chem. Res., 2006, 45, 4278–4294 CrossRef CAS.
- S. P. Reynolds, A. Mehrotra, A. D. Ebner and J. A. Ritter, Adsorption, 2008, 14, 399–413 CrossRef CAS.
- A. D. Ebner, S. P. Reynolds and J. A. Ritter, Ind. Eng. Chem. Res., 2007, 46, 1737–1744 CrossRef CAS.
- C. T. Yavuz, B. D. Shinall, A. V. Iretskii, M. G. White, T. Golden, M. Atilhan, P. C. Ford and G. D. Stucky, Chem. Mater., 2009, 21, 3473–3475 CrossRef CAS.
- K. B. Lee and S. Sircar, AIChE J., 2008, 54, 2293–2302 CrossRef CAS.
- S. F. Wu, T. H. Beum, J. I. Yang and J. N. Kim, Ind. Eng. Chem. Res., 2007, 46, 7896–7899 CrossRef CAS.
- A. A. A. Solieman, J. W. Dijkstra, W. G. Haije, P. D. Cobden and R. W. van den Brink, Int. J. Greenhouse Gas Control, 2009, 3, 393–400 CrossRef CAS.
- A. Duffy, G. M. Walker and S. J. Allen, Chem. Eng. J., 2006, 117, 239–244 CrossRef CAS.
- N. Rajabbeigi, B. Elyassi, T. T. Tsotsis and M. Sahimi, J. Membr. Sci., 2009, 335, 5–12 CrossRef CAS.
- N. Rajabbeigi, T. T. Tsotsis and M. Sahimi, J. Membr. Sci., 2009, 345, 323–330 CrossRef CAS.
- F. Bulnes, A. J. Ramirez-Pastor and V. D. Pereyra, J. Mol. Catal. A: Chem., 2001, 167, 129–139 CrossRef CAS.
- C. Santschi and M. J. Rossi, J. Phys. Chem. A, 2006, 110, 6789–6802 CrossRef CAS.
- C. M. Mömming, S. Frömel, G. Kehr, R. Fröhlich, S. Grimme and G. Erker, J. Am. Chem. Soc., 2009, 131, 12280–12289 CrossRef.
- C. M. Mömming, E. Otten, G. Kehr, R. Fröhlich, S. Grimme, D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2009, 48, 6643–6646 CrossRef.
Footnote |
| † X, Y in (X, Y) describes how a graphene layer is wrapped into a single wall CNT. The integers X and Y describe the size of the CNT and the orientation of the atomic layer of carbon. Armchair: X = Y. |
| This journal is © The Royal Society of Chemistry 2010 |