Postsynthetic functionalization of mesoporous silica
Dominik
Brühwiler
*
Institute of Inorganic Chemistry, University of Zurich, Winterthurerstrasse 190, CH-8057, Zurich, Switzerland. E-mail: bruehwi@aci.uzh.ch
First published on 13th April 2010
Abstract
Functionalized mesoporous silica offers promising possibilities for numerous applications, including drug delivery, catalysis, and adsorption. This minireview focuses on recent developments related to the postsynthetic positioning of functional groups on mesoporous silica. After briefly introducing the reagents that are commonly used for this purpose, methods to control and to analyze the distribution of the grafted functional groups are discussed, with particular emphasis on concepts that allow the placement of the groups at specific distances from each other, as well as on approaches towards the selective functionalization of the external particle surface.
 Dominik Brühwiler | Dominik Brühwiler received his PhD from the University of Bern in 2001, followed by a postdoctoral stay at the Lawrence Berkeley National Laboratory. Since 2004, he has led a research group at the University of Zürich, specializing in the synthesis and functionalization of zeolites and mesoporous silica, with a particular focus on the supramolecular organization and controlled spatial distribution of luminescent guests. |
Introduction
Following a recommendation by IUPAC, porous materials are divided into three classes, namely microporous (with a pore diameter below 2 nm), mesoporous (2–50 nm), and macroporous (larger than 50 nm) materials.1 Zeolites, the most prominent members of the microporous class, have a long history in terms of their synthesis,2 beginning with the pioneering work of Barrer in the late 1940s.3 Mesoporous silicas with ordered pores (hereafter referred to simply as mesoporous silica) are young by comparison, with first reports dating back to the early 1990s.4 Since then, progress has been remarkable, leading to a rich palette of structure-directing agents (SDAs) and a variety of synthetic pathways.5 The functionalization of mesoporous silica has attracted particular interest, mainly due to potential applications of these materials in catalysis,6 drug delivery,7 and adsorption.8,9There are two general approaches towards the functionalization of mesoporous silica (Fig. 1). In co-condensation, also termed direct or one-pot synthesis, a condensable precursor bearing the desired functional group is added to the mixture containing the components for the formation of the mesoporous silica. In most cases, silanes of the type R–Si(OR′)3 are used as precursors. The distribution of the functional groups in the final products is typically homogeneous, but the addition of organoalkoxysilanes can have a pronounced effect on the pore structure and morphology of the mesoporous material.10 High functionalization degrees often lead to decreasing mesoscopic order. The SDA must ultimately be removed by extraction, as calcination would destroy the grafted organic moieties. Extending the co-condensation approach to bridged organosilica precursors of the type (R′O)3Si–R–Si(OR′)3 yields periodic mesoporous organosilicas (PMOs).11Postsynthetic methods (often referred to as grafting) introduce the functional groups after the formation of the mesoporous silica, either before or after removal of the SDA. This method exploits the abundant silanol groups present on the mesoporous silica surface.12 Postsynthetic functionalization offers possibilities for functional group placement. Specific parts of the mesoporous silica surface (external surface, pore surface, pore entrances) can be modified and high functionalization degrees are feasible without compromising the mesoscopic order. This minireview focuses on recent progress in postsynthetic functionalization of mesoporous silica, with particular emphasis on the control and the analysis of the distribution of surface-anchored organic functionalities.
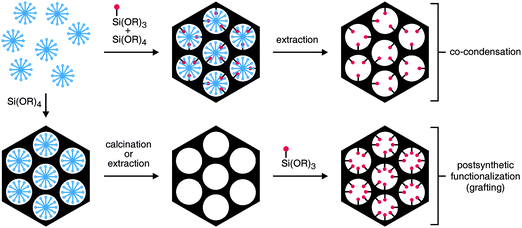 | ||
| Fig. 1 Functionalization of mesoporous silica by co-condensation and by postsynthetic treatment. A trialkoxysilane molecule bearing a functional moiety (red) is shown as an example of a precursor. The structure-directing agent (SDA) is represented by the blue micelles. | ||
Anchoring of functional groups
Precursors for the postsynthetic functionalization of mesoporous silica are most often based on chlorosilanes or alkoxysilanes. Disilazane compounds of the type HN(SiR′R′′2)2 have been reported as an interesting alternative.13 A further possibility, resulting in the formation of a Sisurface–C bond instead of the more common attachment via siloxane bridges, is the use of Grignard reagents after esterification of the surface silanol groups.14 Grignard reagents can also be employed in combination with a dehydroxylation of the silica surface. This postsynthetic modification route relies on the formation of strained siloxane bridges upon vacuum degassing at elevated temperatures, thereby generating electron deficient surface silicon atoms.15 In addition to Grignard reagents, organolithium compounds can be used to functionalize mesoporous silica surfaces. Reactions are performed at relatively low temperatures compared to the classical alkoxysilane-based grafting techniques.16 The concept of functionalization via metalorganic reagents has also been applied to colloidal suspensions containing mesoporous silica nanoparticles.17The postsynthetic functionalization of mesoporous silica is usually performed in a solvent. In the case of 3-aminopropyltrimethoxysilane (APTMS), functionalization by deposition from the vapor phase has been reported, leading to materials that compare favorably with those prepared by deposition from toluene.18 As a further alternative to the common solution-based reactions, materials containing Sisurface–NH2 sites have been obtained by treatment of mesoporous silica with gaseous NH3 at elevated temperatures.19
The effect of water on the functionalization of mesoporous silica with 3-aminopropyltriethoxysilane (APTES) was investigated by nitrogen sorption and confocal laser scanning microscopy (CLSM). It was observed that clustering of the silanes is promoted by the presence of water, leading to non-uniform distributions of the grafted amino groups with higher concentrations at the pore entrances.20 If a uniform distribution of the amino groups is desired, the reaction of 3-aminopropyltrialkoxysilanes with mesoporous silica is therefore usually performed in an anhydrous solvent. It should, however, be mentioned that for the formation of self-assembled monolayers on mesoporous silica by reaction with 3-mercaptopropyltrimethoxysilane, the presence of water (approximately 2 monolayers) in the reaction mixture was found to be of critical importance for generating the respective hydroxysilane, which acts as the key species in the self-assembly process. Furthermore, a hydrated mesoporous silica surface provides the large number of silanol groups that is needed for the formation of self-assembled monolayers.9,21
A postsynthetic approach not explicitly shown in Fig. 1 introduces functional groups by exchanging the SDA in the as-synthesized mesoporous material with suitable organosilanes. Functionalization according to this approach has, for example, been achieved by exchanging the cetyltrimethylammonium ions in as-synthesized MCM-41 with positively charged organoalkoxysilanes.22 It has further been shown that any suitable organoalkoxy- or organochlorosilane is able to displace the SDA from as-synthesized MCM-41 and covalently bind to the pore surface in the process. By following this approach and conducting the reactions without additional solvent, high functionalization degrees were obtained.23 It should, however, be noted that materials synthesized by methods based on such exchange processes usually contain residual amounts of non-exchanged SDA.
Many postsynthetic modification routes make use of additional reaction steps to convert grafted functional groups. Examples include the oxidation of –SH to –SO3H by treatment with H2O2,24 the cleavage of grafted disulfide species of the type [Si](CH2)3SS(CH2)3[Si], leading to pairs of –(CH2)3SH groups on the mesoporous silica surface,25 or the hydrolysis of –CN to –COOH.26 Countless possibilities exist for attaching further moieties to grafted functional groups. In the extreme case, a series of such steps can lead to highly complex structures, as demonstrated by the synthesis of melamine-based dendrimers in the pores of amino-functionalized mesoporous silica.27
Conventional spectroscopic techniques can be used to characterize grafted functional groups. To confirm the integrity of the anchored organic moieties, FTIR, Raman, and 13C MAS NMR spectroscopy is most commonly used,13,17,25,28 whereas 29Si MAS NMR data can provide evidence for the presence of Si–C bonds14–16 and give information on the degree of organic functionalization.28 The amount of grafted organic moieties is usually determined by elemental or thermogravimetric analysis.
Distribution of functional groups
It is often proposed that postsynthetic modification leads to preferential functionalization of the most readily accessible sites, i.e., sites on the external particle surface and sites on the pore surface close to the pore entrances. The degree of non-uniformity of the final functional group distribution strongly depends on the size of the grafting reagent relative to the pore diameter. Lim and Stein compared vinyl-functionalized MCM-41 samples prepared either by postsynthetic grafting or by co-condensation.28 A kinetic study of the bromination of the samples revealed that the bromination rate of the postsynthetically functionalized sample was significantly faster than for the co-condensed material, despite a similar pore diameter. One possible explanation for this effect is based on the distribution of the vinyl groups. Contrary to the uniform distribution in the co-condensed sample, vinyl groups in the postsynthetically modified sample are present mainly on the external surface and on the pore surface close to the openings. This hypothesis was supported by X-ray photoelectron spectroscopy (XPS). As the escape depth of electrons is only a few nanometres, XPS emphasizes contributions from the external surface. Measurements on the brominated samples indeed indicated a higher bromine content for the postsynthetically modified sample, whereas bromine contents obtained from bulk chemical analysis were almost equal for co-condensed and postsynthetically functionalized samples. A similar comparison between the two synthetic methods was made by Yokoi et al., focusing on the functionalization with mono-, di-, and tri-amino-organoalkoxysilanes.29 Adsorption of Co2+ and Fe3+ was significantly different for the co-condensed and postsynthetically modified samples. The results were explained by a non-uniform distribution of the amino groups in the postsynthetically modified sample, with higher concentrations on the external surface and on the pore surface near the openings. It should be noted that in these comparative studies, postsynthetic functionalization was performed by refluxing a dispersion of the mesoporous silica in toluene containing the respective trichloro- or trialkoxysilane. Several authors have found that in terms of obtaining a uniform distribution of surface-anchored amino groups, toluene is not an ideal solvent. Sharma et al. grafted 3-aminopropyl and N-(2-aminoethyl)-3-aminopropyl groups to MCM-41 using different solvents and analyzed the degree of aggregation of the surface-anchored functional groups colorimetrically with Cu2+ complexes.30 Particularly in the case of N-(2-aminoethyl)-3-aminopropyl moieties, reaction in toluene produced clusters, whereas reaction in alcohols resulted in a higher degree of site isolation. We have made a similar observation for the reaction of bis(trimethoxysilylpropyl)amine with MCM-41.31 While grafting in toluene caused an accumulation of anchored groups on the external surface and at the pore entrances, reaction in THF produced a significantly higher degree of site isolation and led to an increased tendency towards a more uniform functionalization of the pore surface. Such solvent effects are less pronounced in the case of weakly interacting silanes, such as APTMS,30,31 but are nevertheless likely to play an important role in determining the properties of the functionalized products. A very distinct solvent effect was observed upon reaction of 3-aminopropyltris(methoxyethoxyethoxy)silane (APTMEES) with hexagonal MCM-41 type particles.32 Fluorescent labeling of the grafted amino groups and subsequent analysis by CLSM showed that labeled amino groups were mainly present on the external particle surface and at the pore entrances when the grafting reaction was carried out in toluene or hexane, whereas a uniform distribution was obtained for ethanol or acetone. Using THF as a solvent led to an intermediate behavior, featuring a decrease of the concentration of labeled amino groups towards the center of the pores.For catalytic applications of functionalized mesoporous silica, it is important to establish molecular scale structure-property relationships. This process is greatly facilitated if the grafted catalytically active groups experience similar environments and are isolated from each other. The molecular spacer approach, which is schematically shown in Fig. 2, is very versatile in this regard. Obtaining site-isolated surface-grafted amino groups is particularly challenging, as they tend to interact with adjacent surface silanol groups or arrange in clusters with amine-amine hydrogen bonding. McKittrick and Jones have used a tritylimine patterning agent to control the minimum distance between the surface-anchored amino groups. After grafting of the tritylimine, the remaining silanol groups were capped with hexamethyldisilazane, and the tritylimine was hydrolyzed to yield the primary amino groups.33 The use of a benzyl instead of a trityl protecting group led to higher grafting densities, approaching those of highly loaded amino-functionalized silicas synthesized by conventional techniques, while still retaining a high degree of site isolation.34 In an approach more direct than the concept outlined in Fig. 2, site-isolated amino groups have also been obtained by reacting a mixture of APTMS and methyltrimethoxysilane with mesoporous silica.35
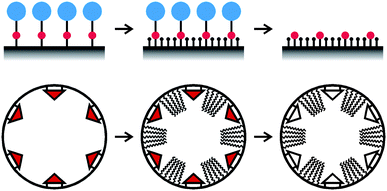 | ||
| Fig. 2 Top: schematic illustration of the molecular spacer approach. A functional group (red) bearing a bulky protecting group (blue) is grafted to the pore surface. The remaining silanol groups are then capped to obtain the desired surface properties and the protecting groups are removed. The size of the protecting group determines the minimum distance between the functional groups. Bottom: molecular imprinting approach for the creation of defined cavities in a pore surface coated monolayer. | ||
Imprinting36 and site pairing37 methods have been applied to mesoporous silica to control the relative positioning of functional groups. A mesoporous silica coated by a molecular monolayer containing cavities of defined size and shape can be prepared by such an imprinting approach, as shown schematically in Fig. 2. In this particular case, a triangular template molecule was grafted to the pore surface, followed by the reaction of the unoccupied surface with a long-chain alkyltrimethoxysilane. The template molecules were then removed to leave the desired cavities in the monolayer coating.38 Interestingly, it was found that the accessibility of these cavities can be changed. In toluene, the long hydrocarbon chains assume an extended conformation, thus making the cavities accessible. In ethanol, on the other hand, the chains form a more compact conformation, resulting in a blocking of the cavities.38
Independent of the grafting method, the assessment of the degree of site isolation is a key issue. Amino-functionalized samples can be labeled with fluorescein isothiocyanate under mild conditions. The luminescence intensity of the labeled samples is a direct measure of the degree of site isolation, as short distances between the amine sites lead to self-quenching.31 This method is particularly useful for samples with loadings on the order of 0.1 mmol g−1. For higher amino loadings, the adsorption of 1-pyrenecarboxylic acid can provide information on site isolation.34 Short distances between adsorbed pyrene moieties cause excimer emission, which can be easily identified by a broad band in the luminescence spectrum around 470 nm. A further method relies on the formation of Cu2+ complexes with grafted amino groups. The d-d absorption spectrum of these complexes allows conclusions regarding the number and arrangement of amine ligands on the Cu2+.30 Site isolation in amino-functionalized mesoporous silica has also been probed by reaction with terephthaloyl chloride.33 A short distance between surface-anchored amino groups allows the formation of a bridged species by reaction with both acyl chloride functionalities on the terephthaloyl chloride, leading to two amide bonds. However, if the grafted amino groups are site-isolated, only one of the acyl chloride functionalities is expected to react. The materials were characterized by Raman spectroscopy to identify the relative amount of amide bonds.33
Selective functionalization
Functional groups on the external particle surface define the interaction of mesoporous silica particles with the surrounding medium. Regarding applications in the field of drug delivery, the selective functionalization of the external surface is an essential synthetic step to obtain multifunctional carriers. Regulation of the cellular uptake39 or targeting40 are examples of tasks that can be solved by functional groups on the external surface. Additionally, the internal surface needs to be functionalized independently, for example to enable high drug loading capacity and controlled release.7One of the first reports of a site-directed surface derivatization of mesoporous silica was motivated by the potential of mesopore confinement effects in catalysis. To exclude binding of catalytic centers to external surface sites, Shephard et al. developed a method to passivate the external surface.41 Based on the assumption that the most kinetically available silanol groups are those on the external surface, treatment of MCM-41 with small amounts of diphenyldichlorosilane (Ph2SiCl2, 20 μL g−1) was expected to deactivate the external surface for further reaction. Subsequent grafting of APTMS would then lead to an exclusive amino-functionalization of the pore surface. The position of the surface-grafted amino groups was determined by high-resolution transmission electron microscopy following acidification (to generate surface-anchored ammonium groups) and adsorption of a ruthenium cluster compound. Indeed, a sample without Ph2SiCl2 treatment showed strong contrast on the external surface of the particles, whereas for a passivated sample, ruthenium clusters were found in the mesopores.
A strategy for the differential functionalization of acid-prepared mesoporous spheres (3 nm average pore diameter, 5 μm particle size) employing diffusion-based deprotection was proposed by Cheng and Landry.42 The material was first reacted with Fmoc-modified organosilanes, leading under certain conditions to a complete filling of the pores. As the cleavage of the Fmoc groups is much slower within the pores, the external surface groups could be deprotected selectively and modified further. CLSM can be used to visualize the spatial distribution of the functional groups after appropriate fluorescent labeling. Large particles of defined morphology are advantageous for this type of analysis. Arrays of silica nanochannels (ASNCs)43 and large spherical mesoporous silica particles42,44 are examples for materials that are ideally suited for such studies.
Grafting of 3-aminopropyltrialkoxysilanes to as-synthesized mesoporous silica, i.e., before the removal of the SDA, is a straightforward method for external surface modification. Intuitively, one would assume that the SDA blocks the pores and therefore prevents the silane molecules from grafting to the pore surface (Fig. 3). Following this approach, we found that the degree of pore surface grafting strongly depends on the organosilane. In case of the frequently used APTES, significant derivatization of the pore surface was found for ASNCs (MCM-41 type material, 2.9 nm average pore diameter) and SBA-15 type spheres (5.5 nm average pore diameter) despite the presence of the respective SDA in the pores. APTMEES, on the other hand, grafted preferentially to external surface sites (Fig. 3). The mesopores remained accessible for further modification after functionalization of the external surface with APTMEES.32
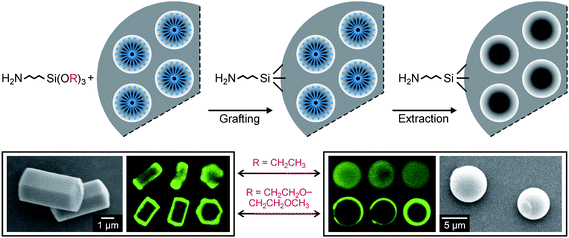 | ||
| Fig. 3 Top: schematic representation of external surface functionalization by grafting to as-synthesized mesoporous silica. Bottom: scanning electron microscopy images of ASNCs (left) and mesoporous silica spheres (right). The corresponding CLSM images taken after fluorescent labeling of the amino groups indicate that the degree of pore surface grafting strongly depends on the type of silane. Three particles are shown for each silane/silica combination. Optical slices in the center of the particles were selected.32 | ||
The concept of reacting alkoxysilanes with mesoporous silica still containing the SDA has been utilized to attach molecular gates at the pore entrances, enabling controlled uptake and release.45 The positioning of molecules at the channel entrances of mesoporous silica is a challenging task. In the case of zeolite L, a microporous aluminosilicate, a wide variety of molecules has been developed that can be selectively adsorbed or covalently attached to the channel entrances.46 As this approach typically relies on a moiety that is just slightly too bulky to enter the zeolite L channels, direct application to the much larger pore openings of mesoporous materials is hardly possible. However, the recent development of molecular nanovalves based on redox-switchable bistable rotaxanes shows that highly sophisticated materials can be prepared by carefully controlling the location of functional moieties on the mesoporous silica surface.47
Studies on the selective functionalization of external surfaces are usually conducted with micrometre-sized mesoporous silica particles, allowing for example the visualization of the functional group distribution by CLSM. It should, however, be noted that procedures developed for such particles are not necessarily successful when working with nanometre-sized mesoporous silica particles. As a consequence of the shorter channels, the overall accessibility of the pore surface sites is much higher in nanometre-sized particles than in micrometre-sized particles having the same pore diameter.48 For mesoporous silica nanoparticles, a sequential co-condensation approach has been proposed as an alternative to postsynthetic routes. It has been shown that functional groups can be concentrated in parts of the mesopores, or exclusively placed on the external surface, by adding the respective functionalized triethoxysilane during a specific time of the particle growth.49
Depending on the synthesis conditions, mesoporous silica of the SBA-15 type50 exhibits a significant amount of disordered micropores.51 There is evidence for the preferential adsorption of APTMS in the intrawall micropores of SBA-15.48 In fact, micropores in SBA-15 can be functionalized independently from the primary mesopores by first cleaving and removing the accessible part of the SDA, thus exposing the mesopore surface. The latter is then functionalized and the material is heated to vacate the micropores. In a final step, the open micropores are modified to yield a bifunctional material.52
Conclusions
Given the variety of reagents for the postsynthetic modification of mesoporous silica, including alkoxysilanes, chlorosilanes, disilazanes, organolithium and Grignard reagents, the possibilities for preparing mesoporous materials with specific functionalities are enormous. For many potential applications of these materials, a controlled functional group placement is crucial. Grafting methods that position functional groups in a defined environment or at specific distances from each other are of particular interest for the development of catalysts and sensors, whereas the selective functionalization of specific regions of the mesoporous silica surface (external surface, pore surface, pore entrances) constitutes an integral part of the concept of using these materials as drug delivery devices. Recent years have seen significant progress in this regard, in part due to the application of sophisticated methods for the analysis of the spatial distribution of the grafted functional groups. Considering the wide pore size and particle size range covered by mesoporous silica, the development of generally applicable methods for the selective functionalization of mesoporous silica surfaces remains a challenge.Acknowledgements
Our work in the field of functionalized silicate-based porous materials is financially supported by the Swiss National Science Foundation (Project 200020-117591), the Swiss Commission of Technology and Innovation (KTI/CTI, Project 9231.2 PFNM-NM), and the European Commission through the Human Potential Programme (Marie-Curie RTN Nanomatch, Grant No. MRTN-CT-2006-035884).References
- J. Rouquerol, D. Avnir, C. W. Fairbridge, D. H. Everett, J. H. Haynes, N. Pernicone, J. D. F. Ramsay, K. S. W. Sing and K. K. Unger, Pure Appl. Chem., 1994, 66, 1739 CrossRef CAS
.
- C. S. Cundy and P. A. Cox, Microporous Mesoporous Mater., 2005, 82, 1 CrossRef CAS
- R. M. Barrer, J. Chem. Soc., 1948, 127 RSC
- T. Yanagisawa, T. Shimizu, K. Kuroda and C. Kato, Bull. Chem. Soc. Jpn., 1990, 63, 988 CrossRef CAS
- Y. Wan and D. Zhao, Chem. Rev., 2007, 107, 2821 CrossRef CAS
- A. Taguchi and F. Schüth, Microporous Mesoporous Mater., 2005, 77, 1 CrossRef CAS
- M. Vallet-Regí, F. Balas and D. Arcos, Angew. Chem., Int. Ed., 2007, 46, 7548 CrossRef CAS
- H. Yoshitake, New J. Chem., 2005, 29, 1107 RSC
- G. E. Fryxell, S. V. Mattigod, Y. Lin, H. Wu, S. Fiskum, K. Parker, F. Zheng, W. Yantasee, T. S. Zemanian, R. S. Addleman, J. Liu, K. Kemner, S. Kelly and X. Feng, J. Mater. Chem., 2007, 17, 2863 RSC
- S. Huh, J. W. Wiench, B. G. Trewyn, S. Song, M. Pruski and V. S.-Y. Lin, Chem. Commun., 2003, 2364 RSC
- T. Asefa, C. Yoshina-Ishii, M. J. MacLachlan and G. A. Ozin, J. Mater. Chem., 2000, 10, 1751 RSC
- J. Chen, Q. Li, R. Xu and F. Xiao, Angew. Chem., Int. Ed., 1995, 34, 2694 CrossRef CAS
- R. Anwander, I. Nagl, M. Widenmeyer, G. Engelhardt, O. Groeger, C. Palm and T. Röser, J. Phys. Chem. B, 2000, 104, 3532 CrossRef CAS
- K. Yamamoto and T. Tatsumi, Microporous Mesoporous Mater., 2001, 44–45, 459 CrossRef CAS
- J. E. Lim, C. B. Shim, J. M. Kim, B. Y. Lee and J. E. Yie, Angew. Chem., Int. Ed., 2004, 43, 3839 CrossRef CAS
- S. Angloher, J. Kecht and T. Bein, Chem. Mater., 2007, 19, 3568 CrossRef
- J. Kecht and T. Bein, Langmuir, 2008, 24, 14209 CrossRef CAS
- H. Ritter, M. Nieminen, M. Karppinen and D. Brühwiler, Microporous Mesoporous Mater., 2009, 121, 79 CrossRef CAS
- Y. Inaki, Y. Kajita, H. Yoshida, K. Ito and T. Hattori, Chem. Commun., 2001, 2358 RSC
- N. Gartmann, C. Schütze, H. Ritter and D. Brühwiler, J. Phys. Chem. Lett., 2010, 1, 379 Search PubMed
- J. Liu, X. Feng, G. E. Fryxell, L.-Q. Wang, A. Y. Kim and M. Gong, Adv. Mater., 1998, 10, 161 CrossRef CAS
- A. B. Bourlinos, T. Karakostas and D. Petridis, J. Phys. Chem. B, 2003, 107, 920 CrossRef CAS
- V. Antochshuk and M. Jaroniec, Chem. Mater., 2000, 12, 2496 CrossRef CAS
- D. Das, J.-F. Lee and S. Cheng, Chem. Commun., 2001, 2178 RSC
- V. Dufaud and M. E. Davis, J. Am. Chem. Soc., 2003, 125, 9403 CrossRef CAS
- K. Y. Ho, G. McKay and K. L. Yeung, Langmuir, 2003, 19, 3019 CrossRef CAS
- E. J. Acosta, C. S. Carr, E. E. Simanek and D. F. Shantz, Adv. Mater., 2004, 16, 985 CrossRef CAS
- M. H. Lim and A. Stein, Chem. Mater., 1999, 11, 3285 CrossRef CAS
- T. Yokoi, H. Yoshitake and T. Tatsumi, J. Mater. Chem., 2004, 14, 951 RSC
- K. K. Sharma, A. Anan, R. P. Buckley, W. Ouellette and T. Asefa, J. Am. Chem. Soc., 2008, 130, 218 CrossRef CAS
- H. Salmio and D. Brühwiler, J. Phys. Chem. C, 2007, 111, 923 CrossRef CAS
- N. Gartmann and D. Brühwiler, Angew. Chem., Int. Ed., 2009, 48, 6354 CrossRef CAS
- M. W. McKittrick and C. W. Jones, Chem. Mater., 2003, 15, 1132 CrossRef CAS
- J. C. Hicks and C. W. Jones, Langmuir, 2006, 22, 2676 CrossRef CAS
- M. Luechinger, R. Prins and G. D. Pirngruber, Microporous Mesoporous Mater., 2005, 85, 111 CrossRef CAS
- S. Dai, M. C. Burleigh, Y. Shin, C. C. Morrow, C. E. Barnes and Z. Xue, Angew. Chem., Int. Ed., 1999, 38, 1235 CrossRef CAS
- E. L. Margelefsky, R. K. Zeidan and M. E. Davis, Chem. Soc. Rev., 2008, 37, 1118 RSC
- Y. Shin, J. Liu, L.-Q. Wang, Z. Nie, W. D. Samuels, G. E. Fryxell and G. J. Exarhos, Angew. Chem., Int. Ed., 2000, 39, 2702 CrossRef CAS
- I. Slowing, B. G. Trewyn and V. S.-Y. Lin, J. Am. Chem. Soc., 2006, 128, 14792 CrossRef CAS
- C.-P. Tsai, C.-Y. Chen, Y. Hung, F.-H. Chang and C.-Y. Mou, J. Mater. Chem., 2009, 19, 5737 RSC
- D. S. Shephard, W. Zhou, T. Maschmeyer, J. M. Matters, C. L. Roper, S. Parsons, B. F. G. Johnson and M. J. Duer, Angew. Chem., Int. Ed., 1998, 37, 2719 CrossRef CAS
- K. Cheng and C. C. Landry, J. Am. Chem. Soc., 2007, 129, 9674 CrossRef CAS
- Y. Kievsky and I. Sokolov, IEEE Trans. Nanotechnol., 2005, 4, 490 CrossRef
- A. Katiyar and N. G. Pinto, Small, 2006, 2, 644 CrossRef CAS
- N. K. Mal, M. Fujiwara and Y. Tanaka, Nature, 2003, 421, 350 CrossRef CAS
- D. Brühwiler, G. Calzaferri, T. Torres, J. H. Ramm, N. Gartmann, L.-Q. Dieu, I. Lopez-Duarte and M. V. Martinez-Diaz, J. Mater. Chem., 2009, 19, 8040 RSC
- T. D. Nguyen, Y. Liu, S. Saha, K. C.-F. Leung, J. F. Stoddart and J. I. Zink, J. Am. Chem. Soc., 2007, 129, 626 CrossRef CAS
- H. Ritter and D. Brühwiler, J. Phys. Chem. C, 2009, 113, 10667 CrossRef CAS
- J. Kecht, A. Schlossbauer and T. Bein, Chem. Mater., 2008, 20, 7207 CrossRef CAS
- D. Zhao, J. Feng, Q. Huo, N. Melosh, G. H. Fredrickson, B. F. Chmelka and G. D. Stucky, Science, 1998, 279, 548 CrossRef CAS
- M. Kruk, M. Jaroniec, C. H. Ko and R. Ryoo, Chem. Mater., 2000, 12, 1961 CrossRef CAS
- C.-M. Yang, H.-A. Lin, B. Zibrowius, B. Spliethoff, F. Schüth, S.-C. Liou, M.-W. Chu and C.-H. Chen, Chem. Mater., 2007, 19, 3205 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2010 |