Smart nanocontainers and nanoreactors†
Kyoung Taek
Kim
,
Silvie A.
Meeuwissen
,
Roeland J. M.
Nolte
and
Jan C. M.
van Hest
*
Radboud University Nijmegen, Institute for Molecules and Materials, Heyendaalseweg 135, Nijmegen, 6525, AJ, The Netherlands. E-mail: j.vanhest@science.ru.nl
First published on 12th May 2010
Abstract
We highlight recent advances in the synthesis of nanocarriers and nanoreactors from synthetic and biological building blocks with emphasis on the stimulus-responsive regulation of their function.
 Kyoung Taek Kim | Kyoung Taek Kim studied polymer chemistry at Inha University, Korea, where he obtained his BSc and MS with Prof. Chulhee Kim. He earned his Ph.D. in synthetic polymer chemistry and metal-containing polymers under supervision of Prof. Ian Manners and Prof. Mitchell A. Winnik at the University of Toronto in 2006. After his Ph.D., he was a senior scientist at Samsung Electronics, Korea. In 2008, he joined Prof. Jan van Hest's group as a post-doctoral fellow, where he has been focusing on stimuli-responsive block copolymers and polymersomes. His research interests are synthetic polymer chemistry, bio-organic chemistry, and supramolecular chemistry. |
 Silvie A. Meeuwissen | Silvie A. Meeuwissen obtained her BSc degree in chemistry at the Radboud University Nijmegen in 2006. Two years later she earned her MSc degree cum laude after traineeships in synthetic organic chemistry with Prof. Floris P. J. T. Rutjes at the Radboud University Nijmegen and chemical biology with Prof. Benjamin G. Davis at Oxford University. In 2008, she started to pursue a Ph.D in the group Prof. Jan C. M. van Hest in bio-organic chemistry. Her research involves polymersomes and their exploration as nanoreactors. |
 Roeland J. M. Nolte | Roeland J. M. Nolte is an Emeritus Professor of Organic Chemistry at the Radboud University Nijmegen, The Netherlands, and a member of the Royal Netherlands Academy of Science. His research interests span a broad range of topics at the interfaces of supramolecular chemistry, macromolecular chemistry, and biomimetic chemistry, in which he focuses on the design of catalysts and (macro) molecular materials. His contributions to science have been recognized with numerous award lectureships and several national and international prices including the Izatt-Christensen Award for Excellence in Macrocyclic Chemistry, the first Royal Netherlands Academy of Science Chair in Chemistry, and a knighthood in 2003. He has served on the editorial boards of many scientific journals, including the journal Science (Washington) and the RSC journal Chemical Communications (as Chairman). |
 Jan C. M. van Hest | Jan. C. M. van Hest conducted his doctoral research at the Eindhoven University of Technology under supervision of Prof. E. W. Meijer, for which the Ph.D. title was granted in 1996. As a postdoctoral researcher he investigated the possibilities of protein engineering for the preparation of materials under supervision of Prof. D. A. Tirrell, at the University of Massachusetts in Amherst. In 1997 he joined the chemical company DSM. In 2000 he was appointed as a full professor at the Radboud University Nijmegen to set up a new group in bio-organic chemistry. His current research efforts are aimed at developing bio-inspired materials and processes in order to combine the functionality of biological systems with the flexibility and robustness of synthetic structures. Jan van Hest is a member of The Young Academy of the Royal Netherlands Academy of Arts and Sciences. He has published around 100 papers. |
Introduction
A long-pursued goal in nanoscience is to capture the essence of structures and functions of complex biological systems, as epitomised by cells, by creating artificial nanostructures in a rational manner. In the case of cells, compartmentalisation and communication between compartments are essential to regulate biological function such as replication, metabolism, and signal transduction.1 Compartmentalisation and confinement of cellular components enable cells to control the sequential manner of biochemical reactions to maximise their precision and efficiency.2 The cellular lipid bilayer membrane encloses the complex machinery required to achieve this, and is responsible for transport of ions, water, and other molecular necessities for metabolism and replication, recognition and transduction of chemical signals. Membrane permeability is governed for a large part by the presence of channel proteins, which are regulated by external stimuli. This allows controlled mass transport and signal transduction required for the metabolic functioning of cells.3,4Artificial nanocages are the mimics of the biocompartments exhibited by Nature.5 Synthetic routes to these nanostructures have been vastly expanded by bottom-up approaches using simple building blocks to construct complex nanoarchitectures via self-assembly. From the materials point of view, the building blocks span a wide range of compounds including well-defined small molecules, polymers, inorganic materials and, recently, to biological macromolecules such as proteins and nucleic acids. Inorganic nanostructures such as mesoporous silica particles have also been utilised as cargo-carrying vehicles and nanoreactors. Recent convergence of molecular biology and materials science has enabled the direct use of biological building blocks and their assemblies as nanocages. Although numerous examples have demonstrated our ability to create such structures based on organic, biological, and inorganic materials in a wide variety of size regimes, and have shown a successful adoption of Nature's basic design principles, artificial nanostructures are still lacking the interactive properties to respond and communicate with their surroundings as is exhibited by the protein machinery in the cell membranes. This explains the current intense interest in responsive and intelligent materials that show a change in physical and chemical properties in response to external stimuli. When properly designed and assembled into nanostructures having an inner compartment that can be used for storage of guests, these smart materials may mimic the regulation behaviour exhibited by membrane proteins in cells.
In this review, we highlight recent advances in the design and construction of nanocarriers and nanoreactors with an emphasis on stimuli-responsive regulation of function. We limit our scope to three main classes of materials that are widely used for creating these architectures. In the first part, we cover block copolymers and their self-assembled structures, including polymer vesicles and micelles that exhibit stimuli-responsive behaviour. Subsequently organic-inorganic hybrid architectures functionalised with ‘gatekeepers’ or ‘nanovalves’ to regulate their release properties will be discussed. Finally, we highlight the recent developments in research in which cage-like protein assemblies and virus capsids are utilised as well-defined containers and reactors.
Polymer vesicles and micelles
Stimuli-responsive polymer vesicles
Polymer vesicles (polymersomes) have attracted increased attention in recent years due to their similarity to liposomes and cells.6 Compared to lipid vesicles that are composed of low molecular weight phospholipids or surfactants, polymersomes – based on high molecular weight amphiphilic block copolymers – consist of a bilayer membrane exhibiting superior physical and chemical stability.7 This robustness is beneficial for many applications such as cargo-storage vehicles for therapeutics and cosmetics.8 Also, the chemical structure of the amphiphilic block copolymers can easily be modified, which provides polymersomes with properties required for specific applications. For example, PEGylation of the polymersome surface leads to stealth-like behaviour to avoid immune responses when these capsules are used for in vivo applications. Furthermore, capsules with cell targeting properties can be created by immobilising specific peptides or proteins on the polymersome periphery, which enables the recognition of specific receptors.9 However, the main drawback of a physically robust polymersome membrane is the drastic decrease of its permeability for molecules both inside and outside the polymersome. For applications such as drug delivery, the controlled release of cargo molecules from the inner aqueous compartment is a crucial factor. Stimuli-responsive behaviour of polymersomes leading to transmembrane diffusion of small molecules is therefore of prime importance for the development of nanoreactors and nanocages that can be applied as controlled and targeted release vehicles.10Amphiphilic block copolymers with a degradable hydrophobic part are attractive building blocks to create polymersomes with a time-dependent release behaviour.11 Degradation of the hydrophobic block leads to changes in the architecture of the block copolymer, which causes a rearrangement of the vesicular membrane leading to cylindrical or spherical micelles.12 This degradation induces perforation of the vesicular membrane and guest molecules entrapped within the aqueous compartment and the membrane get released. Ahmed et al. performed an in-depth study on the hydrolytically cleavable amphiphiles PEG-b-(polylactic acid, PLA) and PEG-b-(polycaprolactone, PCL).12,13 Polymersomes based on a mixture of PEG-b-PLA and PEG-b-PBD were used for systematic delivery of two anticancer drugs; the hydrophilic compound doxorubicin (DOX) was positioned in the lumen, whereas the hydrophobic drug paclitaxel (TAX) was encapsulated in the bilayer. In vitro experiments at neutral pH and 37 °C showed that 80% of DOX and 60% of TAX were released from the polymersomes via perforation of the membrane after only one day. In vivo studies on human breast tumours established in nude mice showed that an increase of the deliverable maximum tolerated dose was obtained for (DOX and TAX)-loaded polymersomes compared to the free drug cocktail. As a result, the immobilised drugs caused more effective and sustainable tumour shrinkage.
Polymeric vesicular structures can also be disrupted through other chemical changes of the block copolymer, such as chemical oxidation and pH-sensitive conformational changes of the hydrophobic block.14 Stimuli-responsive polymers that are sensitive to pH and light can be introduced into the membrane to create polymersomes possessing a self-regulating property.15,16 Li and co-workers reported a strategy to remotely trigger instantaneous polymersome bursting by using a UV-sensitive liquid-crystalline (LC) copolymer with azobenzene-containing mesogenic moieties (PAzo) in the hydrophobic block (Fig. 1A).17 Upon exposure to UV-radiation, the azobenzene moieties underwent trans-to-cis isomerisation, which subsequently induced a linear-to-coil transformation in the secondary polymer structure. First, asymmetric polymersomes were designed containing an internal leaflet based upon the inert PEG-b-PBD block copolymer and an external leaflet of PEG-b-PAzo. Upon UV-radiation, the vesicles' morphology was completely destructed in less than a second (Fig. 1B,C). When polymersomes were composed of a PEG-b-PAzo inner leaflet and a PEG-b-PBD outer leaflet, the change of the curvature of the UV-responsive polymer layer upon irradiation generated an outward curling rim.
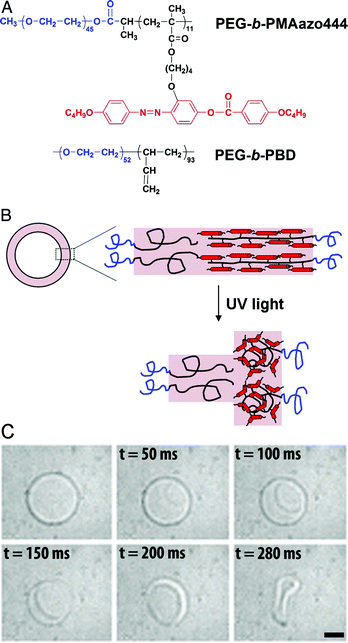 | ||
| Fig. 1 Bilayer composition and polymersome bursting. (A) Molecular structures of the PEG-b-PAzo and PEG-b-PBD block copolymers. (B) A cartoon of the asymmetric bilayer composition with PEG-b-PAzo as the outer leaflet that coils upon UV-radiation. (C) UV-radiation induces bursting of the polymersome with PEG-b-PBD in the inner leaflet and PEG-b-PAzo as the outer leaflet. (Reprinted with permission from ref. 16). | ||
Block copolymers responding to changes in pH of the medium are also of interest.15 A notable recent example of multi-compartment polymersomes that are pH-responsive was reported by Chiu et al.18 They prepared polymersomes via the double emulsion technique in a water–oil–water environment with poly(acrylic acid) polymers partially modified with hydrophobic distearin groups. Large polymersomes encapsulating small polymersomes were created by this method. During membrane formation, the acrylic acid-rich domain of the polymer phase separated within the hydrophobic membrane and formed pH-sensitive channels.
The design of reversible pH-responsive polymersomes, the so-called “breathing vesicles”, was reported by the group of Eisenberg.19 Their nano-sized polymersomes consisted of the triblock copolymer PEG-b-PS-b-(poly(2-diethylaminoethyl methacrylate), PDEAMA), which was synthesised using ATRP. After self-assembly of the PEG-b-PS-b-PDEAMA amphiphiles at pH 10.4 into polymersomes, a decrease in pH induced an impressive change in vesicle size. This was a result of the fact that the initially hydrophobic PDEAMA domain became protonated and therefore turned into a hydrophilic structure, attracting water. The concurrent phase separation between PS and protonated PDEAMA yielded a rigid PS layer in between the PDEAMA and PEG domains, keeping the self-assembled structure together (Fig. 2). At even lower pH, the progressive swelling of the pH-responsive PDEAMA block was found to crack the two PS-layers after which an even bigger increase in vesicle size was observed since the swelling of the PDEAMA was now less restrained. Remarkably, the vesicles adapted their original size within a short time when the pH was brought to 10.4 again.
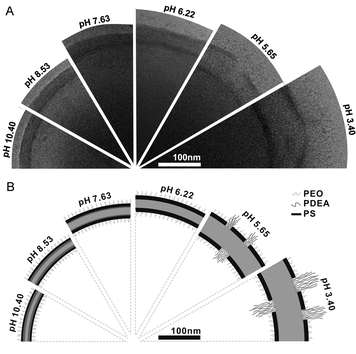 | ||
| Fig. 2 Reversible change of the PEG-b-PS-b-PDEAMA membrane upon pH change. (A) Cryo-TEM images of the vesicle wall structure at several pH values. (B) Schematic illustration of the presumed membrane structure at corresponding pH values. (Reprinted with permission from ref. 19). | ||
Electrostatic interaction between oppositely charged polyelectrolytes has widely been used to construct polymeric vesicles, or microcapsules, by templated layer-by-layer self-assembly.20 Vancso and co-workers recently reported on microcapsules built from the layer-by-layer assembly of redox-sensitive poly(ferrocenylsilane) (PFS) polyelectrolytes. The membrane of these microcapsules swells upon oxidation of the Fe(II) centres of the PFS due to the increasing repulsion between the oxidised Fe(III)+ in the membrane.21
Polymersomes with a semi-permeable vesicular membrane can be used as a nano-scaled reactor when catalytically active guests are encapsulated within their inner compartment.5,23 The semi-permeable polymer membrane should here only allow transmembrane diffusion of small reagents and products while keeping the large catalysts, such as enzymes, inside. These membrane designs are greatly beneficial towards biological and conventional chemical transformations as they protect the catalytic activity from harmful environmental effects and confine the location of chemical transformation within the nanospace.
Kataoka and coworkers developed polymersomes based on block copolypeptide polyelectrolytes (PICsome).22 The membrane of the PICsome was composed of two block copolypeptides comprising oppositely charged polypeptide blocks. The polyelectrolyte membrane of the PICsome allowed diffusion of small molecules as was shown by O2-binding of myoglobin encapsulated in the inner compartment of the PICsome. In another line of research, polymersomes were built from amphiphilic coil-helix block copolymers, polystyrene-b-poly(3-(isocyano-l-alanyl-amino-ethyl)-thiophene) (PS-b-PIAT).24 The PS-b-PIAT polymersomes were found to possess a semi-permeable membrane, presumably due to the robust, helical conformation of the hydrophilic PIAT block and the relatively low molecular weight PS block. Site-specific encapsulation of enzymes within the inner aqueous compartment and the hydrophobic membrane was successfully achieved, and created a bioreactor for enzymatic polymerisation and cascade enzymatic reactions (Fig. 3).25
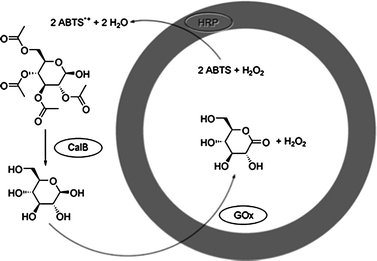 | ||
| Fig. 3 Schematic representation of the multistep reaction taking place in the three-enzyme PS-b-PIAT polymersome system with GOx in the lumen, HRP in the membrane and CalB in the surrounding medium. (Reprinted with permission from ref. 25a). | ||
Meier and co-workers developed semi-permeable polymersomes by implementing channel proteins within the membrane of polymer vesicles.28 The presence of channel proteins enables transmembrane diffusion of small substrates for the enzymes encapsulated in the inner compartment of the polymersomes. A recent example of Meier and co-workers showed that enzyme-loaded polymersomes can function as an artificial organelle within cells.29 The polymeric vesicles were functionalised on the outside with oligonucleotide polyguanylic acid (polyG), a substrate for macrophage scavenger receptors, to achieve target-specific cellular uptake by macrophages.30 These polymersomes exhibited a specific uptake by the cell and remained stable for 48 h within the cellular matrix. The polymersomes were subsequently loaded with the protease trypsin to give the “artificial organelles” a bio-functionality. The selected trypsin substrate bis-(CBZ-Ile-Pro-Arg)-rhodamine110 (BZiPAR) was able to autonomously cross the membranes of both cells and the membrane of the polymersome owing to its hydrophobicity. After sequential incubation of the macrophages with trypsin-loaded polyG-polymersomes and BZiPAR, strong fluorescence was observed in the cytoplasm. This result suggested that the substrate entered and was processed inside the polymeric vesicles.
As is also shown in the previous example, surface modification of polymersomes can readily be realised by conjugation of functional groups on the end of the hydrophilic block of a block copolymer. Bioconjugation of a cell-targeting protein or a mono-clonal antibody on the amphiphilic block copolymer has enabled the development of vesicular carriers which recognise specific target proteins and cells.13 This ‘polymer modification’ approach, however, may have an adverse effect on the self-assembly of block copolymers; the molecular architecture of amphiphiles is a crucial factor in directing the morphology of self-assembled structures. A change in block copolymer architecture especially occurs when the conjugated biomolecules are large, such as enzymes. To solve this problem, amphiphiles with a functional group on the hydrophilic block as an anchoring site have been synthesised and assembled together with the matrix polymer in the membrane of polymersomes.26 The anchoring sites on the surface of the polymersomes were subsequently modified with large biomolecules via copper(I) catalysed click chemistry. Using this approach, active enzymes were immobilised such as Candida antarctica Lipase B (CalB) and horseradish peroxidase (HRP) on the surface of the polymersome. In a follow-up experiment, three different enzymes, CalB, glucose oxidase (GOx) and HRP were respectively encapsulated in the membrane, the lumen and immobilised on the PS-b-PIAT polymersome surface. It was demonstrated that these three enzymes cooperated in a cascade reaction.27
Stimuli-responsive regulation of the permeability of vesicular polymer membranes can greatly enhance the development of controlled drug-releasing vehicles and nanoreactors with regulated reactivity. Recently a method was reported to introduce in a controllable fashion permeability in polymersomes based on PEG-b-PS block copolymers.31 The key concept lies in the use of stimuli-responsible amphiphilic polymers containing boronic acid moieties in the hydrophobic block that, upon increase of pH and concentration of monosaccharides such as D-glucose, become soluble in an aqueous environment.32 As shown in Fig. 4, PEG-b-poly(styrene boronic acid (PEG-b-PSBA) block copolymers were embedded within the inert PEG-b-PS matrix. Upon increase of pH and monosaccharide concentration, the PEG-b-PSBA domains became soluble in the aqueous polymersome medium, thereby creating pores in the vesicular membrane. The permeability of the polymersome membrane could be controlled by adjusting the weight fraction of PEG-b-PSBA compared to the matrix forming PEG-b-PS. Moreover, the enzymatic activity of encapsulated enzyme (CalB) showed to increase in a parallel fashion with larger amounts of initially incorporated PEG-b-PSBA.31
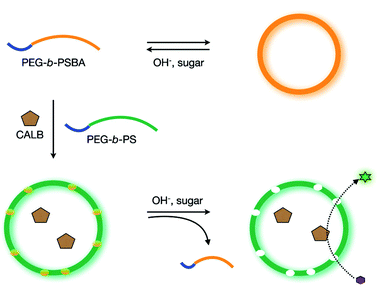 | ||
| Fig. 4 Polymersome with a controllable permeability based on a selective dissolution of sugar-responsive boronic acid block copolymers. (Reprinted with permission from ref. 31). | ||
Stimuli-responsive polymer micelles
Micelles assembled from amphiphilic block copolymers exhibit, in correspondence with polymer vesicles, greater thermodynamic and kinetic stability than their phospholipid or surfactant based analogues.33 When applied as a drug delivery system (DDS), this stability is of prime importance since polymer micelles retain their loaded drug for a longer period under the much diluted conditions experienced in vivo compared to low molecular weight micelles.34 In addition, amphiphilic block copolymers can easily be modified using a wide variety of chemical tools to construct intelligent nanodevices that are stimuli-responsive or have certain targeting abilities. In combination with encapsulation of (bio)molecules, micelles can be deployed as, for example, therapeutic gene delivery devices or nanoreactors.Block copolymers based on poly(n-isopropylacrylamide) (PNIPAAm)35 are commonly employed thermo-responsive building blocks for micelles.36 Above the lower critical solution temperature (LCST), PNIPAAm undergoes a reversible phase transition from hydrophilic to hydrophobic in water due to the entropy-driven dehydration of the polymer chain. A recent example of PNIPAAm block copolymer based micelles for controlled drug release is reported by Cao and coworkers.37 The end group of PNIPAAm was functionalised with the hydrophobic oligomer oligo(methyl methacrylate) (OMMA) to encapsulate the hydrophobic allergy suppressor prednisolone in a high initial efficiency. Upon heating the aqueous medium of the drug-loaded PNIPAAm-b-OMMA micelles above the LCST, the PNIPAAm became water-insoluble and destabilised the self-assembly leading to prednisolone release.
Micelles built from stimuli-responsive block copolymers that are sensitive to the pH change in the medium are also of interest.38 Polymers having a pKa value between 3 and 10 can be adopted as a pH-responsive segment.39 Bae and coworkers reported on polymeric micelles of poly(L-lactic acid) (PLA)-b-PEG-b-poly(L-histidine) as potential tumour pH-specific anticancer drug carrier.40 The co-presence of the pH-nonresponsive lipophilic PLA block in the core prevented the micelles from disintegration. Extensive pH-dependent swelling was observed as the size of the micelle increased to 580 nm at pH 6.6 compared to 80 nm at only a slightly higher pH of 7.4. An example in which micelles respond to a larger difference in pH is reported by Xue et al.41 The hydrophobic drug prednisone acetate was incorporated in micelles constructed from poly(acrylic acid) (PAAc)-b-poly(DL-lactide) (PDLLA) block copolymers. In vitro pH experiments showed only a small drug release at a pH of 1.4, while at pH 7.4 most of the prednisone acetate had diffused into the surrounding solution after 10 h.
Polymeric micelles can act as nanoreactors when enzymes and/or transition metal catalysts are incorporated in the micellar structure.42 Chemical transformations within confined space can be beneficial in terms of enhanced efficiency and higher reaction rate compared to reactions in a bulk environment.43
Enzyme immobilisation in reversed micelles permits enzymatic reactions to occur in organic media, as the hydrophilic core of the micelle provides a favourable aqueous environment for the enzymes while the hydrophobic part solubilises the amphiphatic colloid in an organic solvent.44 Chen et al. immobilised the lipase from Candida rugosa in reversed micelles based on PNIPAAm-co-AA polymers.45 Compared to experiments with the free enzyme in the same organic solution, superior activity towards esterification of lauric acid and 1-propanol was found for the micellar system. Interestingly, the choice of thermo-responsive PNIPAAm entailed the ability to stop the reaction by increasing the temperature above the LCST as the enzyme-holding aggregates precipitated from the reaction mixture.
Transition metal catalysts can be encapsulated inside the core of micelles built from amphiphilic block copolymers with ligand-containing hydrophobic blocks.46 Persigehl and co-workers covalently incorporated triphenylphosphine ligands in the hydrophobic segment of block copolymers, which upon self-assembly into micelles yielded a ligand-rich core.47 Elias and co-workers reported a system in which polypeptides were used as the hydrophobic block. To each of the amino acid residues a palladium diphosphine complex was attached.48 Using this system, organic molecules were hydrogenated with high yields under mild conditions via diffusion of reagents through a swollen PEG corona.
Polymer micelles can also stabilise catalytically-active transition metal nanoparticles by encapsulating the nanoparticles within the micellar core.49 The access of these embedded nanoparticles for external reagents can be regulated by stimuli-responsive behaviour of the micellar corona. Wang et al. reported on the design of catalytically-active and thermo-responsive micelle-supported gold nanoparticles.50 Reductive gold particles were loaded into PNIPAAm-b-poly(4-vinyl pyridine) (PNIPAAm-b-P4VP)-based micelles. The activity of the metallomicelles could be reversibly switched on and off by modulation of the reaction temperature (Fig. 5). The PNIPAAm chains were swollen below the LCST so reactants and products were able to easily diffuse in and out of the micelle to reach the catalytic core. At temperatures exceeding the LCST, the diffusion of reagents to the Au particles in the micelle core was hampered because of a collapse of the PNIPAAm corona.
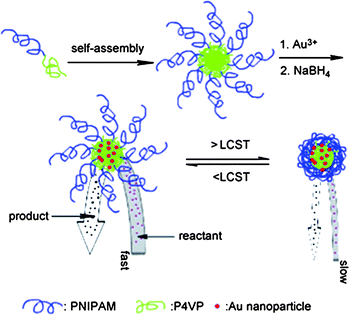 | ||
| Fig. 5 Thermoresponsive polymer micelles encapsulating Au nanoparticles in the core. Thermoresponsive PNIPAAm corona controls the access of small reagents into the nanoparticles residing in the core of the micelle. (Reprinted with permission from ref. 49). | ||
Organic-inorganic hybrid nanocages
Mesoporous silica nanoparticles
Mesoporous silica nanoparticles (MSNs) are highly interesting materials due to the presence of numerous nanochannels, which are a result of the construction of these silica structures around micellar templates.51 The 2-D hexagonal ordering of the channels, which reflects the lyotropic arrangement of the micelles, provides MSNs with an exceptionally high surface area (>900 m2 g−1) and large pore volume (>0.9 cm3 g−1), which can be used as a reservoir for small molecules.52,53 The diameter of an individual channel (typically 2–10 nm) can be tuned by using surfactants with varying sizes.54 Hydroxyl groups on the surface of MSNs can easily be modified by simple chemical reactions with various silanes having additional functional groups.55 The site-specific modification of the inner walls of the nanochannels can also be achieved by using organic silanes which have a selective solubility in the hydrophobic domain of the templating micelles.56 These modified mesoporous silica particles have been used as a solid support for anchoring heterogeneous catalysts within the nanochannels.57 For example, Aida and coworkers reported that mesoporous silica particles having titanocene catalysts covalently held within the nanopores performed polymerisation of ethylene.58 Polyethylene (PE) was extruded out of the 5 nm wide channels as polymerisation proceeded because the diameter of the pore (27 Å) was much smaller than the dimension of an ordinary PE crystal (∼100 Å). Nanofibers of highly crystalline PE with an ultrahigh molecular weight were obtained in this way.MSNs of varying sizes (50–300 nm) have been shown to be biocompatible, and were taken up by cells without invoking acute immune responses.59 MSNs have been used as containers for hydrophobic drug molecules, which could be released in a sustained fashion. Drug molecules which were contained in the nanopores solely by electrostatic or hydrophobic interactions, however, were released through the open pores, which typically generates a ‘premature release’ problem. This premature release is a phenomenon also observed for drug delivery vehicles based on simple micelles and liposomes with relatively soft diffusion barriers.
Recently, the concept of ‘gatekeeping’ or ‘nanovalves’ has therefore emerged. Based on facile site-specific chemistry around MSNs, stimuli-sensitive ‘smart’ functionality was introduced on the orifices of the open pores or on the inner walls of the nanochannels.60 These stimuli-responsive functionalities physically block free diffusion of encapsulated small molecules out of the nanopores of MSNs (gatekeeping). Such gatekeepers are mounted by labile chemical bonds or non-covalent interactions, which are subject to dissociation by applied stimuli, such as pH, ionic strength, temperature, electromagnetic waves, enzyme-catalysed reactions, and non-covalent interactions.60–65 Stimuli-responsive motifs that have long been developed for organic molecules and polymers can readily be adopted as gatekeeper moieties. This nanovalve-like function realises a controlled release of encapsulated guest molecules from MSNs at a specific time and location. Fujiwara and coworkers first demonstrated this concept by introducing coumarin moieties on the orifices of nanopores of MCM-41, which photo-dimerise upon irradiation by UV light (Fig. 6).65 The photodimerised coumarin works as the gatekeeper to block unwanted release of guest molecules from the nanopores because the dimer of coumarin (∼3.8 nm) is large enough to cover the open pore of MCM-41 (2.7 nm). This gatekeeper, upon irradiation by shorter wavelength UV (250 nm), dissociates into monomeric coumarin, which opens the gate of the nanopores. This photo-dimerisation and dissociation is reversible, and, thus, the accessibility of nanopores of MCM-41 is regulated by irradiation.
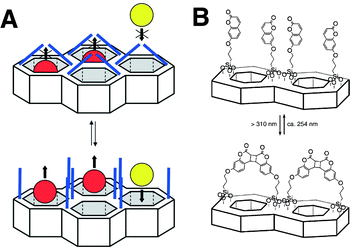 | ||
| Fig. 6 Gatekeeping of nanochannels by photo-dimerization of coumarins attached to the surface of mesoporous silica materials. (Reprinted with permission from ref. 51). | ||
Lin and coworkers introduced disulfide linkers with a terminal amine on the surface of the inner channel of MSNs, which were subsequently reacted with CdS nanoparticles with carboxylic acid moieties on their surface.60 These covalently-bound CdS nanoparticles blocked the open channel of MSNs and prevented unwanted leakage of encapsulated guest molecules. CdS nanoparticles were released by reducing the disulfide bonds in the linker with a chemical reducing agent (Fig. 7). This allowed a controlled release of guests from the inner channel triggered by a specific external signal. Magnetic nanoparticles such as iron oxide66 and PAMAM dendrimers67 have also been used as gatekeepers, regulating the release property of MSNs. The cellular uptake of MSNs was achieved by modifying the surface with triethylene glycol.68 These MSNs could deliver both interior-loaded drugs and plasmid DNA anchored on the surface to plant cells, which was confirmed by the expression of the green fluorescent protein (GFP) gene.
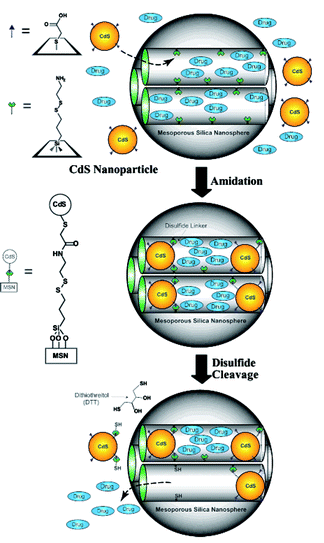 | ||
| Fig. 7 CdS gatekeepers held covalently in nanochannels of mesoporous silica nanoparticles. CdS gatekeepers are released by oxidation of disulfide linkers by dithiothreitol (DTT). (Reprinted with permission from ref. 60). | ||
Lin and coworkers recently developed boronic acid-functionalised MSNs which encapsulated cyclic adenosine monophosphate (cAMP) within nanochannels by capping the boronic acid functionalities with glutamic acid-modified insulin (G-Ins).69 The reversible covalent bonds between boronic acid and the hydroxyl groups of G-Ins readily dissociated when cyclic vicinal diols such as monosaccharides were introduced, because of the higher binding affinity of monosaccharides toward boronic acid. When G-Ins was removed from the MSNs, the entrapped guest molecule, cAMP was released.
Photocleavable linkages between MSNs and gatekeeping moieties were also used to regulate the release property of MSNs. Kim et al. employed a UV-cleavable nitrophenyl ester spacer between MSNs and the bulky β-cyclodextrin (β-CD) to block the open channels of MSNs.64 Upon irradiation with UV light, the nitrophenyl ester linkage was cleaved and, thus, the encapsulated molecules were released through the open channels. β-CD gatekeeping groups on MSNs were further used as a host to accommodate hydrophobic guest molecules. β-CD decorated MSNs were mixed with six-arm PEG star polymers which were end-functionalised with hydrophobic dodecyl chains. The specific incorporation of the dodecyl chain in the hydrophobic cavity of β-CD induced a reversible gelation process in the aqueous phase.
A non-covalent nanovalve was developed by Kim and coworkers who applied the pH-dependent threading of β-CD on PEG as a gatekeeper.61 In an alternative approach Stoddart and Zink introduced cucurbit[6]uril-diaminoalkyl moieties as pH-responsive nanovalves for MSNs.70 The same group also introduced a β-CD-azobenzene pseudorotaxane as a light-actuated nanovalve.71 The β-CD remained threaded when the azobenzene functional post was in a trans-conformation. Upon irradiation and subsequent isomerisation to the cis conformation, the β-CD lock was released.
Enzyme-mediated cleavage of the linker group between gatekeeper and MSNs was also demonstrated.72 Azide-tagged triethylene glycol (TEG) posts were introduced on the surface of MSNs, which were subsequently threaded by β-CD. The azide tag of the complex was subsequently capped with an alkyne-functional adamantyl group by click chemistry. The resulting rotaxane was selectively cleaved and disassembled by porcine liver esterase (PLE), which hydrolysed the ester linkage holding the bulky adamantyl group. Upon hydrolysis and removal of β-CD, the encapsulated guest molecule, congo red, was released
Recently Stoddart, Zink and coworkers reported on the use of mesoporous silica particles as smart containers that can regulate their release properties by using mechanised nanovalves.73,74 Utilising site-specific chemistry, they introduced mechanically bistable functional groups (pseudorotaxane and rotaxane) as nanovalves near the orifices of the particle. As shown in Fig. 8, the 2-D mesoporous silica structures were pre-filled with a fluorescent Ir(ppy)3 (∼1 nm diameter) dye, and a 1,5-dioxynaphthalene derivative (DNPD) was immobilised on the silica surface. Upon addition of the cyclobis-(paraquat-p-phenylene) (CBPQT4+) ring a pseudorotaxane [DNPD ⊂ CBPQT]4+ was formed, which worked as a closed valve. When the CBPQT4+ ring was reduced with a chemical reducing agent, the interaction between CBPQT and DNPD weakened, and subsequently the CBPQT ring flipped away from the DNPD gatepost. This mechanical move opened the nanochannels of mesoporous silica, and entrapped guest molecules were released. Once demonstrated on a 2-D surface, the authors expanded this concept to 3-D silica nanoparticles (Fig. 9). Another concept to mechanically trigger release was demonstrated by tethering photoresponsive azobenzene moieties on the inner walls of the nanochannels of silica particles.75cis–trans isomerisation of azobenzene within these nanochannels drove the encapsulated molecules mechanically out of the channel, similar to impellers within the barrel. Recently, pH-responsive nanovalves and nano-impellers were combined in mesoporous silica nanoparticles, which only showed the release of entrapped guests when both light-actuated azobenzene groups within the nanochannels and pH-responsive cucurbit[6]uril (CB[6])-diaminoalkyl pseudorotaxane valves were activated.76 This ‘dual-controlled’ MSNs exhibited a behaviour as an ‘AND’ logic device, which is a basic device widely used in electronic circuitry.
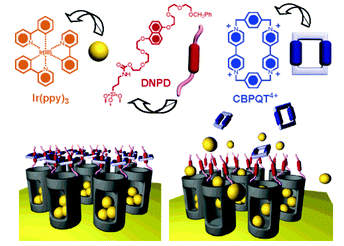 | ||
| Fig. 8 Psuedorotaxane nanovalves responsive upon chemical reduction of CBPQT ring that causes dethreading of the ring from the post and releasing of entrapped guest molecules from nanochannels of mesoporous silica. (Reprinted with permission from ref. 73a). | ||
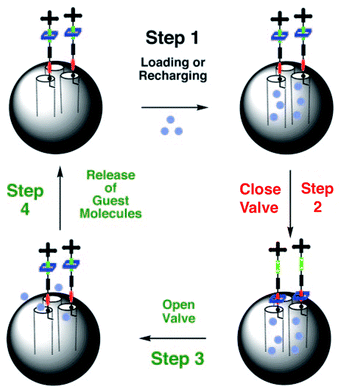 | ||
| Fig. 9 Reversible rotaxane nanovalves anchored to the surface mesoporous silica nanoparticles. (Reprinted with permission from ref. 73b). | ||
Inorganic nanocages
Caged nanostructures constructed from inorganic and organometallic building blocks are also emerging as a way to achieve nanocontainers with unique functions arising from electro-optic and magnetic properties of inorganic materials. Föster and coworkers recently devised a self-assembly strategy of amphiphilic semiconductor nanoparticles based on the well-established packing factor theory developed for amphiphiles.77 They treated CdSe/CdS nanoparticles (NPs) which were covered on the surface with hydrophobic trioctylphosphine/trioctylphosphine oxide (TOP/TOPO) ligands with poly(ethylene glycol) with a terminal amino ligand. The terminal amino groups of PEG replaced the weak TOP/TOPO binding ligands, and induced an amphiphilic character to the nanoparticles. By adjusting the ratio between the number of introduced PEG chains and the nanoparticles, they could control the volume ratio between hydrophilic PEG chains and the residual hydrophobic TOP/TOPO ligands. The resulting amphiphilic semiconductor nanoparticles self-assembled into nanostructures with different morphologies such as spheres, cylinders, and vesicles, as often observed from the self-assembly of amphiphilic block copolymers with varying ratio between the volumes of hydrophobic and hydrophilic blocks (Fig. 10). Upon aggregation, PEG and TOP/TOPO ligands presumably rearrange throughout the surface of nanoparticles, which increases the density of hydrophilic ligands on the water-contacting particle area and of hydrophobic ligands between NP–NP contacting areas. The amount of PEG ligands replacing TOP/TOPO ligands and the rearrangement of PEG ligands at the interface between water and the nanoparticle are crucial factors to decide the morphology of self-assembled structures of these amphiphilic nanoparticles.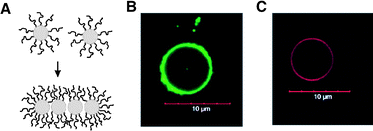 | ||
| Fig. 10 (A) Self-assembling amphiphilic nanoparticles caused by reorganization of surface-bound hydrophilic polymer chains. (B and C) Vesicles from the self-assembly of amphiphilic CdSe/CdS nanoparticles. The diameter of nanoparticles is 2.6 nm for B and 3.1 nm for C. (Reprinted with permission from ref. 77). | ||
Wang and coworkers synthesised nanocages of a Prussian blue (PB)-based cyanometallate polymer, by polymerising a precursor, pentacyano(4-(dimethylamino)pyridine)ferrate (EPE-Fe), on the surface of oil droplets prepared by a mini-emulsion technique.78 This mini-emulsion periphery polymerisation (MEEP) was performed using a telechelic triblock copolymer (PEG-b-PPG-b-PEG) with polymerisable EPE-Fe precursors at the PEG chain ends. The use of a spherical oil droplet template resulted in the formation of a thin crust of polymerised PB (∼2 nm thickness). The diameter of PB nanocages could be controlled by changing the conditions for the miniemulsion polymerisation, such as the ratio of water and organic solvents.
Cage-like protein assemblies
Nanostructures that are constructed from well-defined biological building blocks, such as proteins and DNAs, are receiving increased attention in recent years.79 The well-defined 3-D structure of these biological building blocks allows the self-assembly processes to be designed and controlled in precision. The resulting discrete nano objects not only possess structural robustness but also can exhibit biological activity. This topic has been well-covered in previous reviews;79,80 herein, we specifically focus on protein-based cage-like nanostructures which show promise in applications such as drug delivery vehicles and nanoreactors.Protein assemblies often exist as nanocages in their native forms in vivo. A well-studied example is ferritin, a 450 kDa protein assembly of 24 subunits.81 Ferritin, ubiquitous in cells and in extracellular matrices, is a cage-like protein having an inner space of 8 nm in diameter (Fig. 11). The inner space of ferritin allows storage of up to 4500 Fe atoms as ferric oxyhydroxide clusters, which serves as a reservoir providing Fe atoms for metabolic use.82 The protein cage has 8 hydrophilic channels (∼4 Å) located at the intersections of three subunits, which enables transfer of Fe atoms and molecules through the protein coat. The Fe clusters within the ferritin cage can be removed leaving only a protein cage (apo-ferritin). Due to the presence of the pores in the protein coat, apo-ferritin has been used as a scaffold for the synthesis of inorganic nanoparticles within the inner space.
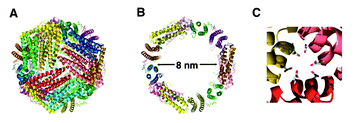 | ||
| Fig. 11 Sturctures of L-ferritin (PDB 1DAT). (A) 24 subunit assembled cage. (B) The inner cavity (8 nm diameter). (C) 3-fold axis hydrophilic channel. (Reprinted with permission from ref. 91). | ||
This possibility was first demonstrated by Mann and coworkers who used the ferritin cage as a scaffold for the growth of non-natural metal oxides.83 The ferrihydrite (5Fe2O3·9H2O) core of ferritin was transformed into FeS nanoparticles by reacting native ferritin with H2S or Na2S in an aqueous buffer. They also showed that the reconstitution of subunits of apo-ferritin in the presence of Mn2+ ions led to the synthesis of manganese oxide within the cage.84 Based on these approaches, various inorganic nanoparticles such as quantum dots, metal oxides, transition metals, and metal alloys have been synthesised within apo-ferritin cages.85–88 Native ferritin has its limitations. Due to the diameter of the channels (∼4 Å) of the ferritin cage, large precursors other than transition metal salts can not be easily encapsulated by pre-constructed ferritin and apo-ferritin. Dominguez-Vera and Colacio therefore reconstituted subunits of ferritin in the presence of hexacyanoferrate (III) ([FeCN6]3−) to encapsulate bulky [FeCN6]3− monomers for the synthesis of Prussian blue nanoparticles within the ferritin cages.89 After self-assembly, ferritin cages filled with cyanoferrate precursors were treated with Fe(II) to turn the precursors into Prussian blue nanoparticles. The smaller Fe(II) reagents could easily access the inner space of the ferritin cages and induce the Prussian blue formation process within the cage.
The open pores of the apo-ferritin cage allow small molecules to access nanoparticles residing in the inner space. Watanabe et al. used these bio-inorganic hybrids as nanoreactors by introducing transition metal nanoparticles within the ferritin cages.87a Pd(0) nanoparticles were synthesised in the cavity by reduction of encased Pd2+ with NaBH4 (Fig. 12). Next, this hybrid metal catalyst was used for the hydrogenation of a series of olefins. The Pd(0)-apo-ferritin nanocomposites remained stable during and after hydrogenation without aggregation. The turnover frequency of these hybrid reactors was comparable to Pd(0) nanoparticles encapsulated within dendrimers. Since the pores present in the ferritin cages are anionically charged, the effect of substrate charge on catalytic efficiency was investigated. For this purpose a series of acrylic acid substrates modified with different functional groups on the carboxylic acid site were tested. A high turnover frequency only was observed when the acrylic acid substrate was modified with a cationic functionality (acrylamide) or small neutral alkyl chains (isopropyl and tert-butyl acrylamides). When the substrate possessed an anionic functionality (acrylic acid) or more bulky substituents (acrylic acid glycine methylester and alanine methylester), only low turnover frequencies were observed. These results indicate that anionic substrates may not penetrate the anionic channels of ferritin cages due to electrostatic repulsion, and the size-selective inclusion through 4 Å channels prevents bulky substrates to reach the inside of the ferritin cage.
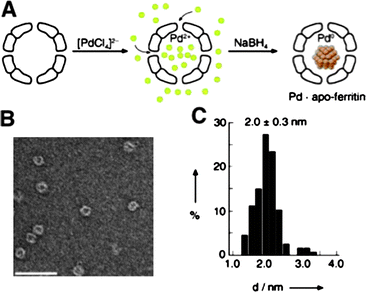 | ||
| Fig. 12 Pd nanoparticles grown in the cavity of apoferritin. (A) A scheme for encapsulation of Pd2+ and subsequent reduction with NaBH4. (B) A negatively stained TEM picture of Pd-ferritin after reduction (scale bar 50 nm). (C) A histogram of diameters of Pd nanoparticles. (Reprinted with permission from ref. 87a). | ||
Catalytic transition metal complexes can also be incorporated within ferritin cages by site-specific coordination of transition metal complexes to the unit proteins. Watanabe et al. coordinated a dinuclear Pd complex ([PdII(allyl)Cl2]2) to the apo-ferritin cage.90 The crystal structures of the Pd-ferritin complex revealed that the binding sites for the Pd complexes were located at the 3-fold axis channel and the internal accumulation center of each coat protein. Therefore, the apo-ferritin cage can coordinate to 48 complexes and 96 Pd atoms. The resulting Pd-ferritin complex was able to show catalytic activity toward Suzuki cross-coupling of 4-iodoaniline and phenylboronic acid to 4-phenylaniline. Based on a comparison between the catalytic activities of complexes built from wild-type and engineered ferritin cages, of which the internal accumulation center was modified to be incapable of binding, it was concluded that both Pd complexes bound inside the cage and near the channel involved in the coupling reaction. The idea of coordinating transition metal complexes within the ferritin cage was also applied to Rh(II) complexes, which were used as a nanoreactor for the polymerisation of phenylacetylene.91 The confinement of this reaction within the cage was found to affect the polymerisation behaviour, which produced a polymer with a narrower molecular weight distribution than that of polyphenylacetylene polymerised with the same Rh complex without the ferritin cages in solution. About two polymer chains of molecular weight of 13 kg mol−1 could be contained within the ferritin cage, which indicates that polymerisation is limited by the available space within the nanoreactor.
Barrel-shaped protein assemblies have also been used as a host to accommodate nanoparticles. For example, GroEL is a protein assembly (800 kDa) of 14 identical subunits (57 kDa) which stack into rings, yielding a barrel-shape structure with a cavity of 4.5 nm in diameter.92 GroEL is a molecular chaperonin which binds denatured proteins and helps their refolding within its cavity. After refolding, the folded proteins are released from GroEL by a conformational change of the chaperonin to the open form, which takes place upon binding with ATP.93 Aida and coworkers used chaperonin proteins with barrel-like structures as a host for encapsulating nanoparticles (Fig. 13).94 When barrel-shaped chaperonin GroEL and T.th cpn proteins were mixed with CdS nanoparticles (2–4 nm), the encapsulation of CdS particles within the barrel of chaperonin proteins occured in ∼75% yield, as observed by TEM. Pristine CdS nanoparticles were prone to lose fluorescence upon contact with a quenching agent (methylviologen, MV2+). However, CdS nanoparticles encapsulated within chaperonin proteins did not suffer from fluorescence quenching due to the protective protein cage surrounding the particles. The encapsulated particle could be ejected from the chaperonin cage by using the ATP-induced conformational change of the chaperonin proteins to the open form.
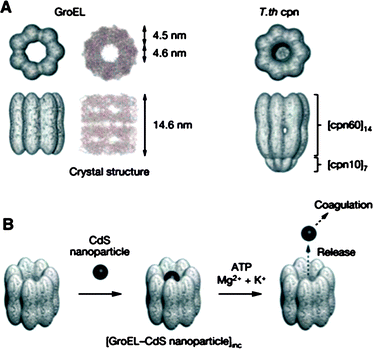 | ||
| Fig. 13 (A) Structures of chaperonin proteins GroEL and T.th cpn. (B) A schematic illustration of encapsulation of CdS nanoparticles within a chaperonin protein and ATP mediated release of nanoparticles. (Reprinted with permission from ref. 94). | ||
The same authors recently reported that genetically modified GroEL with 6 cysteine residues on the rim of the barrel could be linearly assembled into micrometre long fibers by the interaction of cysteines with the photochromic molecule spiropyran, in the presence of divalent cations such as Mg2+.95 Spiropyran spontaneously undergoes isomerisation to merocyanine in aqueous buffers. The isomerisation of spiropyran to merocyanine seemed an important parameter to induce this self-assembly, as evidenced by the fact that the self-assembly of GroELsp/mc was rendered to be less effective when GroELsp/mc was irradiated by visible light (>400 nm), which induces the reverse isomerisation of merocyanine to spiropyran. Notably, GroELsp/mc which had denatured α-lactalbumin as a guest within its cavity could also self-assemble into nanofibers, showing promise as a guest releasing system with possibly better pharmacokinetics and efficiency in vivo (Fig. 14).
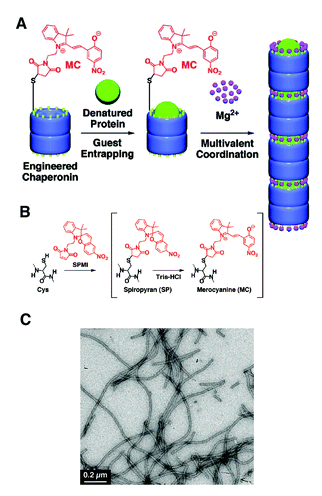 | ||
| Fig. 14 (A) Merocyanine-modified GroEL and Mg2+ mediated self-assembly of modified GroEL with guest proteins into 1-D turular aggregates. (B) Preparation of spiropyran/merocyanine modified GroEL from genetically modified GroEL. (C) A TEM micrograph of Mg2+ triggered self-assembled structures of modified GroEL. (Reprinted with permission from ref. 95). | ||
The heat shock protein, Hsp60, which is 17 nm in diameter, has also been used as a container, hosting Au and CdSe nanoparticles.96 By genetic engineering, the pore diameter of Hsp60 could be expanded from 3 nm to 9 nm. This genetically modified Hsp60 was used as a template for the synthesis of Ni–Pd alloy nanoparticles. Hsp60 with nanoparticles readily assembled into 2-D hexagonal crystalline lattices.
Another small heat shock protein (MjHsp) forms a spherical assembly (diameter of 12 nm) composed of 24 protein subunits. The assembly of MjHsp has large pores (∼3 nm), which allow the diffusion of small molecules to the interior of the protein assembly. Young, Douglas and coworkers showed that the protein subunit of MjHsp could be modified to have specific functional groups on the interior and exterior by replacing amino acid residues at the required locations by genetic engineering.97–99 The interior of MjHsp assembly was modified with a cysteine residue. Due to the large pores of MjHsp assembly, these functional groups could readily react with reactive guest molecules such as fluorophores and drug molecules such as doxorubicin. Transition metals, iron oxide, and alloy nanoparticles were trapped and synthesised within the cavity of MjHsp assembly. The exterior of MjHsp also could be modified by implementing a cancer cell recognising peptide, RGD-C4 at the dangling C-terminus of the subunit protein via genetic engineering. Selective uptake of modified MjHsp assembly by cancer cells (C32 Melanoma cell) was observed.
Virus capsids
Virus capsids (empty protein cages of viruses) have recently turned into one of the most extensively studied biostructures in materials science.100 Due to their well-defined architectures formed by assembly of simple subunit proteins and their readily accessible genetic information they offer potentially unlimited possibilities for structural modification by genetic engineering and post-modification of coat proteins. Virus capsids have extensively been studied as scaffolds for biomineralisation and for the templated synthesis of inorganic and metallic nanostructures.101 The internal space of viral capsids, normally reserved for their genome, can be used as a nanoscaled reservoir for external guests, which opens new possibilities to use them as nanocages for storage and chemical reactions.The cowpea chlorotic mottle virus (CCMV) is an icosahedral virus composed of 180 identical coat proteins.102 The viral capsid has a 28 nm outer diameter and an inner diameter of 18 nm. The coat proteins can self-assemble into a cage structure without the viral RNA chain, making the CCMV capsid an attractive object for use as a nanocontainer. The viral capsid undergoes a pH-dependent reversible swelling behavior (Fig. 15). The capsid increases its dimensions about 10% compared to the non-swollen form when the pH of the medium is ca. 6.5, which provides 60 open pores (∼2 nm) in the viral capsid. Therefore, a CCMV capsid in its swollen state allows small external guest molecules to diffuse into the internal space.
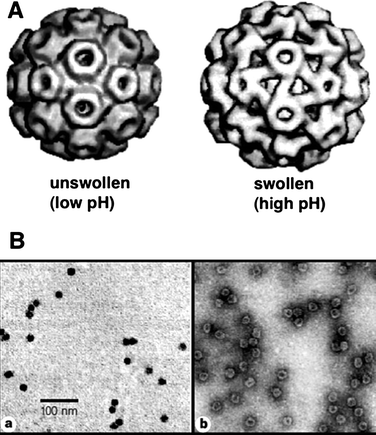 | ||
| Fig. 15 (A) Unswollen and swollen forms of cowpea chlorotic mosaic virus. The swollen form demonstrates 60 copies of pores in the viral coat proteins. (B) TEM images of CCMV capsids with crystallized minerals in the cavity. The comparison of images obtained without staining (a) and with negative staining (b) shows that the viral capisd surrounds nanoparticles and stays intact. (Reprinted with permission from ref. 103). | ||
Douglas and Young pioneered the approach of using viral capsids as nanoscaled reaction vessels by demonstrating crystallisation of minerals within the CCMV capsids.103 The capsid without an RNA chain was incubated in its swollen form with inorganic precursors such as aqueous molecular tungstate (WO42−). The inner wall of the CCMV capsid is highly positively charged because the N-termini of the protein subunits residing inside the capsid have 9 basic amino acid residues. This highly charged interior allowed the anionic molecular precursor to be accumulated within the capsid. After incubation, the capsid was reverted to the pristine form by lowering the pH of the medium, and, subsequently, the inorganic precursors crystallised into highly crystalline minerals having ∼15 nm diameter. Under the same conditions, the tungstate precursors did not crystallise without CCMV capsids, indicating that the local confinement and the cationic nature of the inner wall of the capsid are important factors for the mineralisation to be successful. In a similar approach native CCMV capsids were used as a nanoreactor to synthesise magnetic Prussian blue nanoparticles.104 The cationically charged interior of CCMV attracted and accumulated the precursors, [FeCN6]3− and [Fe(C2O4)3]3− within the cavity, which subsequently were polymerised by a photo-initiated stepwise reaction. Well-defined Prussian blue nanoparticles (∼18 nm) were formed within the CCMV cage. By genetic engineering, the amino acid residues of the N-termini of the coat proteins of CCMV were replaced by glutamates (Glu), which provided anionic charge to the inner wall of the CCMV capsid. This modification did not appreciably alter the self-assembly of the coat protein into the icosahedral capsid. This modified CCMV capsid was used as a nanoreactor for the synthesis of Fe(II) oxide nanoparticles.105
The interior of the CCMV capsid provides a unique opportunity for studying the effect of confinement on chemical reactions. Due to the pH-responsive swelling of the capsid and the resulting change in the pore diameter of the capsid, CCMV is an ideal platform to study chemical reactions of site-isolated catalysts with controllable diffusion rates of reactants and products through the barrier. Nolte and coworkers first demonstrated a single-enzyme nanoreactor based on a CCMV capsid.106 A single enzyme was encapsulated by assembling the subunit proteins of CCMV capsid in the presence of horseradish peroxidase (HRP). The pores of the CCMV capsid allowed diffusion of the fluorogenic substrate, dihydrorhodamine 6G through the protein coat. The enzymatic activity of encapsulated HRP was monitored by confocal fluorescence microscopy. A diffusion controlled enzymatic reaction of HRP was observed, which differed from the reaction of non-encapsulated HRP enzyme. The measured diffusion rate of the product out of the CCMV capsid clearly showed a discontinuity at pH = 5.7, which corresponds to the sudden increase of the pore diameter of the CCMV capsid at the corresponding pH (Fig. 16).
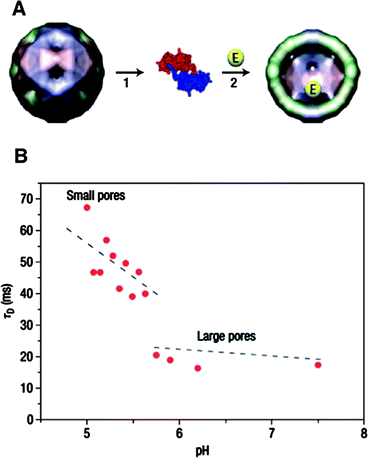 | ||
| Fig. 16 (A) Encapsulation of enzymes in the CCMV capsid by assembling coat proteins in the presence of enzyme molecules. (B) Diffusion time (τD) of fluorescent products produced by enzymes residing in the virus capsid at varying pH. (Reprinted with permission from ref. 106). | ||
Another icosahedral virus capsid, MS2 was studied by Francis and coworkers.107 The MS2 bacteriophage capsid is an assembly of 180 identical protein units of 27 nm diameter. The RNA genome can be removed from the capsid by hydrolysis of the RNA chain at high pH conditions. The capsid possesses 32 pores (∼1.8 nm), which allow diffusion of small molecules into and out of the interior of the capsid. Francis and coworkers developed an efficient procedure to functionalise tyrosine residues on the inner wall of the capsid based on diazo-transfer and hetero Diels–Alder reactions (Fig. 17).106 The site-specific chemical modification of the interior surface of the MS2 capsid enabled molecular cargos such as Gd3+-binding ligands to be confined within the capsid. The MS2 capsid with internally confined Gd3+-ligand complexes outperformed as an MRI contrasting agent the capsid having Gd3+-ligand complex on the exterior.108 Site-selective functionalisation of both the interior and the exterior of the MS2 capsid was used to prepare biomolecular drug delivery systems which specifically bind to the target proteins and cells. A capsid, covalently attached cargo molecules on the inside, was modified on the exterior with aptamers (protein-binding DNA fragments) to direct uptake of the capsid drug carrier by specific cells (Fig. 18).109
 | ||
| Fig. 17 (A) Exterior and interior of the MS2 capsid. (B) A dimer of unit proteins showing the locations of tyrosine residues. (C) Reaction scheme used for the modification of tyrosine by diazo-transfer and a hetero Diels–Alder reaction. For details, see ref. 107 (Reprinted with permission from ref. 107). | ||
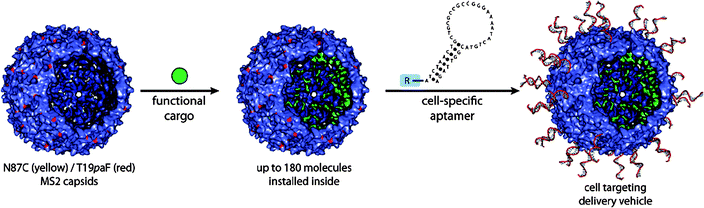 | ||
| Fig. 18 Covalent loading of functional cargos within the interior of MS2 capsid and exterior decoration of the capsid with cell specific aptamer. (Reprinted with permission from ref. 109). | ||
Most virus capsids require the genetic polymer as a prerequisite for self-assembly and structural integrity. Cowpea mosaic virus (CPMV) has been used as a nanosized platform to bind various surface functional groups or compounds such as Au nanoparticles, polymers, and carbohydrates at reactive sites introduced by mutagenesis of the protein subunits.110 The coat protein of red clover necrotic mosaic virus (RNCMV) recognises a viral RNA sequence, the origin of assembly (OAS), which initiates the self-assembly of the coat proteins into the cage structure. Franzen et al. encapsulated Au nanoparticles within the RCNMV capsid by hybridising a viral RNA with Au nanoparticles that have DNA strands, with a complementary sequence of the viral RNA, on the surface.111 This RNA-DNA Au nanoparticle complex initiated the self-assembly of coat proteins of RCNMV, yielding the encapsulation of one Au nanoparticle per capsid.
Concluding remarks
In this review many different examples of smart nanocages have been described. These architectures can be prepared from organic or inorganic building blocks which are preferentially self-assembled into the final functional structure.Smart behaviour is, in most of the approaches, coupled to stimulus responsiveness: the accessibility of the nanocage can be modulated by varying external conditions such as pH or temperature, or by using irradiation. In some cases, such as with the viral capsids, the exploration of the possibilities of these self-assembled protein structures has just begun and many new opportunities lie ahead and still have to be discovered. In other cases, in particular with mesoporous silica nanoparticles and polymeric vesicles switching behaviour has reached a high level of sophistication and applications such as controlled drug releasing vehicles and bioreactors have come within reach.
Although the field that we have highlighted in this review has shown remarkable progress, particularly with respect to the construction of synthetic nanocages possessing regulated permeation, still a number of challenges have to be met in order to realise the full potential of these nanostructures. Reversibility of gating as exhibited by membrane proteins has been realised by designing stimuli-responsive moieties based on conformational changes rather than applying chemical degradation or solubilization of the molecules. Nevertheless, synthetic polymers and gatekeeping groups showing reversible conformational changes upon stimuli have to be explored further and in more detail. Also, stabilization of nanostructures such as micelles and polymersomes may become more beneficial when the stimuli-responsive conformational changes of the constituting polymers is better utilised. For constructing nanoreactors, encapsulation of catalysts such as transition metal nanoparticles and enzymes has been limited to statistical entrapment during cage formation. Immobilisation of catalysts on block copolymers and protein building blocks may help to develop nanoreactors that show better catalytic activities.112 In conclusion, smart nanocages built from polymers, inorganic materials, and proteins become available now displaying the properties and function of natural cells. This highly exciting field can be expected to flourish greatly in the coming years as our ability to create smart materials advances.
References
- L. H. Hartwell, J. J. Hopfield, S. Leibler and A. W. Murray, Nature, 1999, 402, C47 CrossRef CAS.
- B. Alberts, Cell, 1998, 92, 291 CrossRef CAS.
- S. Mann, Angew. Chem., Int. Ed., 2008, 47, 5306 CrossRef CAS.
- K. Simons and E. Ikonen, Nature, 1997, 387, 569 CrossRef CAS.
- D. M. Vriezema, M. C. Aragones, J. A. A. W. Elemans, J. J. L. M. Cornelissen, A. E. Rowan and R. J. M. Nolte, Chem. Rev., 2005, 105, 1445 CrossRef CAS.
- D. E. Disher and A. Eisenberg, Science, 2002, 297, 967 CrossRef CAS.
- (a) B. M. Discher, Y.-Y. Won, D. S. Ege, J. C.-M. Lee, F. S. Bates, D. E. Discher and D. A. Hammer, Science, 1999, 284, 1143 CrossRef; (b) D. E. Discher and F. Ahmed, Annu. Rev. Biomed. Eng., 2006, 8, 323 CrossRef CAS.
- Medical Applications of Liposomes, Ed. D. D. Lasic and D. Papahadjopoulos, Elsevier, Amsterdam 1998 Search PubMed.
- (a) P. Dalhaimer, A. J. Engler, R. Parthasarathy and D. E. Discher, Biomacromolecules, 2004, 5, 1714 CrossRef CAS; (b) D. A. Hammer, G. P. Robbins, J. B. Haun, J. J. Lin, W. Qi, L. A. Smith, P. P. Ghoroghchian, M. J. Therien and F. S. Bates, Faraday Discuss., 2008, 139, 129 RSC; (c) P. J. Photos, L. Bacakova, B. Discher, F. S. Bates and D. E. Discher, J. Controlled Release, 2003, 90, 323 CrossRef; (d) T. M. Allen and R. R. Cullis, Science, 2004, 303, 1818 CrossRef CAS.
- (a) F. Meng, Z. Zhong and J. Feijen, Biomacromolecules, 2009, 10, 197 CrossRef CAS; (b) M.-H. Lee and P. Keller, Soft Matter, 2009, 5, 927 RSC.
- (a) F. Meng, C. Hiemstra, G. H. M. Engbers and J. Feijen, Macromolecules, 2003, 36, 3004 CrossRef CAS; (b) F. Ahmed and D. E. Discher, J. Controlled Release, 2004, 96, 37 CrossRef CAS; (c) F. Meng, G. H. M. Engbers and J. Feijen, J. Controlled Release, 2005, 101, 187 CrossRef CAS.
- (a) F. Ahmed, R. I. Pakunlu, A. Brannan, F. Bates, T. Minko and D. E. Discher, J. Controlled Release, 2006, 116, 150 CrossRef CAS; (b) F. Ahmed, A. Hategan, D. E. Discher and B. M. Discher, Langmuir, 2003, 19, 6505 CrossRef CAS.
- F. Ahmed, R. I. Pakunlu, G. Srinivas, A. Brannan, F. Bates, M. L. Klein, T. Minko and D. E. Discher, Mol. Pharmaceutics, 2006, 3, 340 CrossRef CAS.
- (a) A. Napoli, M. Valentini, N. Tirelli, M. Müller and J.A. Hubbell, Nat. Mater., 2004, 3, 183 CrossRef CAS; (b) E. G. Bellomo, M. D. Wyrsta, L. Pakstis, D. J. Pochan and T. J. Deming, Nat. Mater., 2004, 3, 244 CrossRef CAS.
- (a) J. Du, Y. Tang, A. L. Lewis and S. P. Armes, J. Am. Chem. Soc., 2005, 127, 17982 CrossRef CAS; (b) H. Lomas, I. Canton, S. MacNeil, J. Du, S. P. Armes, A. J. Ryan, A. L. Lewis and G. Battaglia, Adv. Mater., 2007, 19, 4238 CrossRef CAS; (c) U. Borchert, U. Lipprandt, M. Bilang, A. Kimpfler, A. Rank, R. Peschka-Süss, R. Schubert, P. Lindner and S. Förster, Langmuir, 2006, 22, 5843 CrossRef CAS.
- E. Mabrouk, D. Cuvelier, F. Brochard-Wyart, P. Nassoy and M.-H. Li, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 7294 CrossRef CAS.
- (a) X. Liu and M. Jiang, Angew. Chem., Int. Ed., 2006, 45, 3846 CrossRef CAS; (b) J. Yang, D. Levy, W. Deng, P. Keller and M.-H. Li, Chem. Commun., 2005, 4345 RSC.
- H.-C. Chiu, Y.-W. Lin, Y.-F. Huang, C.-K. Chuang and C.-S. Chern, Angew. Chem., Int. Ed., 2008, 47, 1875 CrossRef CAS.
- S. Yu, T. Azzam, I. Rouiller and A. Eisenberg, J. Am. Chem. Soc., 2009, 131, 10557 CrossRef CAS.
- (a) F. Caruso, R. A. Caruso and H. Möhwald, Science, 1998, 282, 1111 CrossRef CAS; (b) G. Ibarz, L. D. ähne, E. Donath and H. Möhwald, Adv. Mater., 2001, 13, 1324 CrossRef CAS; (c) E. Donath, G. B. Sukhorukov, F. Caruso, S. A. Davis and H. Möhwald, Angew. Chem., Int. Ed., 1998, 37, 2201 CrossRef.
- Y. Ma, W.-F. Dong, M. A. Hempenius, H. Möhwald and G. J. Vancso, Nat. Mater., 2006, 5, 724 CrossRef CAS.
- (a) A. Koide, A. Kishimura, K. Osada, W.-D. Jang, Y. Yamasaki and K. Kataoka, J. Am. Chem. Soc., 2006, 128, 5988 CrossRef CAS; (b) A. Kishimura, A. Koide, K. Osada, Y. Yamasaki and K. Kataoka, Angew. Chem., Int. Ed., 2007, 46, 6085 CrossRef CAS.
- (a) D. Lensen, D. M. Vriezema and J. C. M. van Hest, Macromol. Biosci., 2008, 8, 991 CrossRef CAS; (b) J. A. Opsteen, J. J. L. M. Cornelissen and J. C. M. van Hest, Pure Appl. Chem., 2004, 76, 1309 CrossRef CAS.
- (a) D. M. Vriezema, J. Hoogboom, K. Velonia, K. Takazawa, P. C. M. Christianen, J. C. Maan, A. E. Rowan and R. J. M. Nolte, Angew. Chem., Int. Ed., 2003, 42, 772 CrossRef CAS; (b) D. M. Vriezema, A. Kros, R. de Gelder, J. J. L. M. Cornelissen, A. E. Rowan and R. J. M. Nolte, Macromolecules, 2004, 37, 4736 CrossRef CAS; (c) M. Nallani, R. Woestenenk, H.-P. M. de Hoog, S. F. M. van Dongen, J. Boezeman, J. J. L. M. Cornelissen, R. J. M. Nolte and J. C. M. van Hest, Small, 2009, 5, 1138 CAS.
- (a) D. M. Vriezema, P. M. L. Garcia, N. S. Oltra, N. S. Hatzakis, S. M. Kuiper, R. J. M. Nolte, A. E. Rowan and J. C. M. van Hest, Angew. Chem., Int. Ed., 2007, 46, 7378 CrossRef CAS; (b) M. Nallani, H.-P. M. de Hoog, J. J. L. M. Cornelissen, A. R. A. Palmans, J. C. M. van Hest and R. J. M. Nolte, Biomacromolecules, 2007, 8, 3723 CrossRef CAS.
- (a) S. F. M. van Dongen, M. Nallani, S. Schoffelen, J. J. L. M. Cornelissen, R. J. M. Nolte and J. C. M. vah Hest, Macromol. Rapid Commun., 2008, 29, 321 CrossRef CAS; (b) J. A. Opsteen, R. P. Brinkhuis, R. L. M. Teeuwen, D. W. P. M. Löwik and J. C. M. van Hest, Chem. Commun., 2007, 3136 RSC.
- S. F. M. van Dongen, M. Nallani, J. J. L. M. Cornelissen, R. J. M. Nolte and J. C. M. van Hest, Chem. Eur. J., 2009, 15, 1107 CAS.
- (a) C. Nardin, S. Thoeni, J. Widmer, M. Winterhalter and W. Meier, Chem. Commun., 2000, 1433 RSC; (b) A. Mecke, C. Dittrich and W. Meier, Soft Matter, 2006, 2, 751 RSC.
- N. Ben-Haim, P. Broz, S. Marsch, W. Meier and P. Hunziker, Nano Lett., 2008, 8, 1368 CrossRef CAS.
- P. Broz, S. M. Benito, C. Saw, P. Burger, H. Heider, M. Pfisterer, S. Marsch, W. Meier and P. Hunziker, J. Controlled Release, 2005, 102, 475 CrossRef CAS.
- K. T. Kim, J. J. L. M. Cornelissen, R. J. M. Nolte and J. C. M. van Hest, Adv. Mater., 2009, 21, 2787 CrossRef CAS.
- (a) K. T. Kim, J. J. L. M. Cornelissen, R. J. M. Nolte and J. C. M. van Hest, J. Am. Chem. Soc., 2009, 131, 13908 CrossRef CAS; (b) D. Roy, J. N. Cambre and B. S. Sumerlin, Chem. Commun., 2009, 2106 RSC; (c) J. N. Cambre, D. Roy, S. R. Gondi and B. S. Sumerlin, J. Am. Chem. Soc., 2007, 129, 10348 CrossRef CAS.
- G. Kwon, S. Suwa, M. Yokoyama, T. Okano, Y. Sakurai and K. Kataoka, Langmuir, 1993, 9, 945 CrossRef CAS.
- (a) V. P. Torchilin, J. Controlled Release, 2001, 73, 137 CrossRef CAS; (b) D. Le Garrec, M. Ranger and J.-C. Leroux, Am. J. Drug Delivery, 2004, 2, 15 Search PubMed.
- Y. Hirokawa and T. Tanaka, J. Chem. Phys., 1984, 81, 6379 CrossRef.
- J. E. Chung, M. Yokoyama and T. Okano, J. Controlled Release, 2000, 65, 93–103 CrossRef CAS.
- W. Li, W. Tu and D. Cao, J. Appl. Polym. Sci., 2009, 111, 701 CAS.
- S. Dai, P. Ravi and K. C. Tam, Soft Matter, 2008, 4, 435–449 RSC.
- D. Schmaljohann, Adv. Drug Delivery Rev., 2006, 58, 1655 CrossRef CAS.
- E. S. Lee, K. T. Oh, D. Kim, Y. S. Youn and Y. H. Bae, J. Controlled Release, 2007, 123, 19 CrossRef CAS.
- Y.-N. Xue, Z.-Z. Huang, J.-T. Zhang, M. Liu, M. Zhang, S.-W. Huang and R.-X. Zhuo, Polymer, 2009, 50, 3706 CrossRef CAS.
- (a) K. Tonova and Z. Lazarova, Biotechnol. Adv., 2008, 26, 516 CrossRef CAS; (b) E. M. Sulman, V. G. Matveeva, M. G. Sulman, G. N. Demidenko, P. M. Valetsky, B. Stein, T. Mates and L. M. Bronstein, J. Catal., 2009, 262, 150 CrossRef CAS.
- D. Astruc, F. Lu and J. R. Aranzaes, Angew. Chem., Int. Ed., 2005, 44, 7852 CrossRef CAS.
- M. A. Biasutti, E. B. Abuin, J. J. Silbera, N. M. Correa and E. A. Lissi, Adv. Colloid Interface Sci., 2008, 136, 1 CrossRef CAS.
- H. Chen, L.-S. Wang, C.-B. Ching, H.-W. Yu and Y.-Y. Yang, Adv. Funct. Mater., 2008, 18, 95 CrossRef CAS.
- J.-F. Gohy, Coord. Chem. Rev., 2009, 253, 2214 CrossRef CAS.
- P. Persigehl, R. Jordan and O. Nuyken, Macromolecules, 2000, 33, 6977 CrossRef CAS.
- S. Elias and A. Vigalok, Adv. Synth. Catal., 2009, 351, 1499 CrossRef CAS.
- V. Uskoković and M. Drofenik, Adv. Colloid Interface Sci., 2007, 133, 23 CrossRef CAS.
- Y. Wang, G. Wei, W. Zhang, X. Jiang, P. Zheng, L. Shi and A. Dong, J. Mol. Catal. A: Chem., 2007, 266, 233 CrossRef CAS.
- A. B. Descalzo, R. Martinez-Manez, F. Sancenon, K. Hoffmann and K. Rurack, Angew. Chem., Int. Ed., 2006, 45, 5924 CrossRef CAS.
- C. T. Kresge, M. E. Leonowicz, W. J. Roth, J. C. Vartuli and J. S. Beck, Nature, 1992, 359, 710 CrossRef CAS.
- J. S. Beck, J. C. Vartuli, W. J. Roth, M. E. Leonowicz, C. T. Kresge, K. D. Schmitt, C. T.-W. Chu, D. H. Olson, E. W. Sheppard, S. B. McCullen, J. B. Higgins and J. L. Schlenker, J. Am. Chem. Soc., 1992, 114, 10834 CrossRef CAS.
- Q. Huo, D. I. Margolese, U. Ciesla, P. Feng, T. E. Gler, P. Sieger, R. Leon, P. M. Petroff, F. Schüth and G. D. Stucky, Nature, 1994, 368, 317 CrossRef CAS.
- (a) S. L. Burkett, S. D. Sims and S. Mann, Chem. Commun., 1996, 1367 RSC; (b) M. H. Lim, C. F. Blanford and A. Stein, J. Am. Chem. Soc., 1997, 119, 4090 CrossRef CAS; (c) X. Feng, G. E. Fryxell, L.-Q. Wang, A. Y. Kim, J. Liu and K. M. Kemmer, Science, 1997, 276, 923 CrossRef CAS.
- T. Asefa, M. J. MacLachlan, N. Coombs and G. A. Ozin, Nature, 1999, 402, 867 CAS; R. Hernandez, A.-C. Franville, P. Minoofar, B. Dunn and J. I. Zink, J. Am. Chem. Soc., 2001, 123, 1248 CrossRef CAS.
- T. Maschmeyer, F. Rey, G. Sankar and J. M. Thomas, Nature, 1995, 378, 159 CrossRef CAS.
- K. Kageyama, J. Tamazawa and T. Aida, Science, 1999, 285, 2113 CrossRef CAS.
- I. I. Slowing, J. L. Vivero-Escoto, C.-W. Wu and V. S.-Y. Lin, Adv. Drug Delivery Rev., 2008, 60, 1278 CrossRef CAS.
- C.-Y. Lai, B. G. Trewyn, D. M. Jeftinija, K. Jeftinija, S. Xu, S. Jeftinija and V. S.-Y. Lin, J. Am. Chem. Soc., 2003, 125, 4451 CrossRef CAS.
- C. Park, K. Oh, S. C. Lee and C. Kim, Angew. Chem., Int. Ed., 2007, 46, 1455 CrossRef CAS.
- R. Casasus, M. D. Marcos, R. Martinez-Manez, R. V. Ros-Lis, J. Soto, L. A. Villaescusa, P. Amoros, D. Beltran, C. Guillem and J. Latorre, J. Am. Chem. Soc., 2004, 126, 8612 CrossRef CAS.
- Q. Fu, G. V. Rama Rao, L. K. Ista, Y. Wu, B. P. Andrzejewski, L. A. Sklar, T. L. Ward and G. P. Lopez, Adv. Mater., 2003, 15, 1262 CrossRef CAS.
- C. Park, K. Lee and C. Kim, Angew. Chem., Int. Ed., 2009, 48, 1275 CrossRef CAS.
- N. K. Mal, M. Fujiwara and Y. Tanaka, Nature, 2003, 421, 350 CrossRef CAS.
- S. Giri, B. G. Trewyn, M. P. Stellmaker and V. S.-Y. Lin, Angew. Chem., Int. Ed., 2005, 44, 5038 CrossRef CAS.
- D. R. Radu, C.-Y. Lai, K. Jeftinija, E. W. Rowe, S. Jeftinjia and V. S.-Y. Lin, J. Am. Chem. Soc., 2004, 126, 13216 CrossRef CAS.
- F. Torney, B. G. Trewyn, V. S.-Y. Lin and K. Wang, Nat. Nanotechnol., 2007, 2, 295 CrossRef CAS.
- Y. Zhao, B. G. Trewyn, I. I. Slowing and V. S.-Y. Lin, J. Am. Chem. Soc., 2009, 131, 8398 CrossRef CAS.
- S. Angelos, Y.-W. Yang, K. Patel, J. F. Stoddart and J. I. Zink, Angew. Chem., Int. Ed., 2008, 47, 2222 CrossRef CAS.
- D. P. Ferris, Y.-L. Zhao, N. M. Khashab, H. A. Khatib, J. F. Stoddart and J. I. Zink, J. Am. Chem. Soc., 2009, 131, 1686 CrossRef CAS.
- K. Patel, S. Angelos, W. R. Dichtel, A. Coskun, Y.-W. Yang, J. I. Zink and J. F. Stoddart, J. Am. Chem. Soc., 2008, 130, 2382 CrossRef CAS.
- (a) R. Hernandez, H.-R. Tseng, J. W. Wong, J. F. Stoddart and J. I. Zink, J. Am. Chem. Soc., 2004, 126, 3370 CrossRef CAS; (b) T. D. Nguyen, H.-R. Tseng, P. C. Celestre, A. H. Flood, Y. Liu, J. F. Stoddart and J. I. Zink, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10029 CrossRef CAS; (c) T. D. Nguyen, Y. Liu, S. Saha, K. C.-F. Leung, J. F. Stoddart and J. I. Zink, J. Am. Chem. Soc., 2007, 129, 626 CrossRef CAS.
- (a) K. K. Coti, M. E. Belowich, M. Liong, M. W. Ambrogio, Y. A. Lau, H. A. Khatib, J. I. Zink, M. Khashab and J. F. Stoddart, Nanoscale, 2009, 1, 16 RSC; (b) S. Angelos, E. Johansson, J. F. Stoddart and J. I. Zink, Adv. Funct. Mater., 2007, 17, 2261 CrossRef CAS.
- (a) S. Angelos, E. Choi, F. Vögtle, L. De Cola and J. I. Zink, J. Phys. Chem. C, 2007, 111, 6589 CrossRef CAS; (b) J. Lu, F. Tamanoi and J. I. Zink, Small, 2008, 4, 421 CrossRef CAS.
- S. Angelos, Y.-W. Yang, N. M. Khashab, J. F. Stoddart and J. I. Zink, J. Am. Chem. Soc., 2009, 131, 11344 CrossRef CAS.
- M. S. Nikolic, C. Olsson, A. Salcher, A. Kornowski, A. Rank, R. Schubert, A. Frömsdorf, H. Weller and S. Förster, Angew. Chem., Int. Ed., 2009, 48, 2752 CrossRef CAS.
- G. Liang, J. Xu and X. Wang, J. Am. Chem. Soc., 2009, 131, 5378 CrossRef CAS.
- (a) J. C. M. van Hest and D. A. Tirrell, Chem. Commun., 2001, 1897 RSC; (b) R. Langer and D. A. Tirrell, Nature, 2004, 428, 487 CrossRef CAS.
- C. M. Niemeyer, Angew. Chem., Int. Ed., 2001, 40, 4128 CrossRef CAS.
- G. C. Ford, P. M. Harrison, D. W. Rice, J. M. A. Smith, A. Treffry, J. L. White and J. Yariv, Philos. Trans. R. Soc. London, Ser. B, 1984, 304, 551 CrossRef CAS.
- B. Gallois, B. L. d'Estainot, M. A. Michaux, A. Dautant, T. Granier, G. Precigoux, J. A. Soruco, F. Roland, O. Chavas-Alba, A. Herbas and R. R. Chrichton, JBIC, J. Biol. Inorg. Chem., 1997, 2, 360 CrossRef CAS.
- F. C. Meldrum, V. J. Wade, D. L. Nimmo, B. R. Heywood and S. Mann, Nature, 1991, 349, 684 CrossRef CAS.
- (a) F. C. Meldrum, B. R. Heywood and S. Mann, Science, 1992, 257, 522 CrossRef CAS; (b) P. Mackle, J. M. Charnock and C. D. Garner, J. Am. Chem. Soc., 1993, 115, 8471 CrossRef CAS.
- (a) F. C. Meldrum, T. Douglas, S. Levi, P. Arosio and S. Mann, J. Inorg. Biochem., 1995, 58, 59 CrossRef CAS; (b) T. Douglas and V. T. Stark, Inorg. Chem., 2000, 39, 1828 CrossRef CAS; (c) M. Okuda, K. Iwahori, I. Yamashita and H. Yoshimura, Biotechnol. Bioeng., 2003, 84, 187 CrossRef CAS; (d) M. Okuda, Y. Kobayashi, K. Suzuki, K. Sonoda, T. Kondoh, A. Wagawa, A. Kondo and H. Yoshimura, Nano Lett., 2005, 5, 991 CrossRef CAS.
- (a) T. Douglas, D. P. E. Dickson, S. Betteridge, J. Charnock, C. D. Garner and S. Mann, Science, 1995, 269, 54 CrossRef CAS; (b) K. K. W. Wong and S. Mann, Adv. Mater., 1996, 8, 928 CrossRef CAS; (c) I. Yamashita, J. Hayashi and M. Hara, Chem. Lett., 2004, 33, 1158 CrossRef CAS; (d) K. Iwahori, K. Yoshizawa, M. Muraoka and I. Yamashita, Inorg. Chem., 2005, 44, 6393 CrossRef CAS.
- (a) T. Ueno, M. Suzuki, T. Goto, T. Matsumoto, K. Nagayama and Y. Watanabe, Angew. Chem., Int. Ed., 2004, 43, 2527 CrossRef CAS; (b) R. M. Kramer, C. Li, D. C. Carter, M. O. Stone and R. R. Naik, J. Am. Chem. Soc., 2004, 126, 13282 CrossRef CAS; (c) B. Warne, O. I. Kasyutich, E. L. Mayers, J. A. L. Wiggins and K. K. W. Wong, IEEE Trans. Magn., 2000, 36, 3009 CrossRef CAS; (d) M. Suzuki, M. Abe, T. Ueno, S. Abe, T. Goto, Y. Toda, T. Akita, Y. Yamada and Y. Watanabe, Chem. Commun., 2009, 4871 RSC.
- J. Niemeyer, S. Abe, T. Hikage, T. Ueno and G. Erker, Chem. Commun., 2008, 6519 RSC.
- J. M. Dominquez-Vera and E. Colacio, Inorg. Chem., 2003, 42, 6983 CrossRef.
- S. Abe, J. Niemeyer, M. Abe, Y. Takezawa, T. Ueno, T. Hikage, G. Erker and Y. Watanabe, J. Am. Chem. Soc., 2008, 130, 10512 CrossRef CAS.
- S. Abe, K. Hirata, T. Ueno, K. Morino, N. Shimizu, M. Yamamoto, M. Takata, E. Yashima and Y. Watanabe, J. Am. Chem. Soc., 2009, 131, 6958 CrossRef CAS.
- K. Braig, Z. Otwinowski, R. Hegde, D. C. Bolsvert, A. Joachimiak, A. L. Horwich and P. B. Sigler, Nature, 1994, 371, 578 CrossRef CAS.
- N. A. Ranson, G. W. Farr, A. M. Roseman, B. Gowen, W. A. Fenton, A. L. Horwich and H. R. Saibil, Cell, 2001, 107, 869 CrossRef CAS.
- D. Ishii, K. Kinbara, Y. Ishida, N. Ishii, M. Okochi, M. Yohda and T. Aida, Nature, 2003, 423, 628 CrossRef CAS.
- S. Biswas, K. Kinbara, N. Oya, N. Ishii, H. Taguchi and T. Aida, J. Am. Chem. Soc., 2009, 131, 7556 CrossRef CAS.
- (a) R. A. McMillan, C. D. Paavola, J. Howard, S. L. Chan, N. J. Zaluzec and J. D. Trent, Nat. Mater., 2002, 1, 247 CrossRef CAS; (b) R. A. McMillan, J. Howard, N. J. Zaluzec, H. K. Kagawa, R. Mogul, Y.-F. Li, C. D. Paavola and J. D. Trent, J. Am. Chem. Soc., 2005, 127, 2800 CrossRef CAS.
- M. T. Klem, D. Willits, D. J. Solis, A. M. Belcher, M. Young and T. Douglas, Adv. Funct. Mater., 2005, 15, 1489 CrossRef CAS.
- M. L. Flenniken, D. A. Willits, S. Brumfield, M. J. Young and T. Douglas, Nano Lett., 2003, 3, 1573 CrossRef CAS.
- M. L. Flenniken, D. A. Willits, A. L. Harmsen, L. O. Liepold, A. G. Harmsen, M. J. Young and T. Douglas, Chem. Biol., 2006, 13, 161 CrossRef CAS.
- (a) T. Douglas and M. Young, Science, 2006, 312, 873 CrossRef CAS; (b) M. Fischlechner and E. Donath, Angew. Chem., Int. Ed., 2007, 46, 3184 CrossRef CAS; (c) M. Uchida, M. T. Klem, M. Allen, P. Suci, M. Flenniken, E. Gillitzer, Z. Varpness, L. O. Liepold, M. Young and T. Douglas, Adv. Mater., 2007, 19, 1025 CrossRef CAS; (d) A. de la Escosura, R. J. M. Nolte and J. J. L. M. Cornelissen, J. Mater. Chem., 2009, 19, 2274 RSC.
- (a) S. Mann, Nature, 1993, 365, 499 CrossRef CAS; (b) S. Mann, Nat. Mater., 2009, 8, 781 CrossRef CAS.
- J. A. Speir, S. Munshi, G. Wang, T. S. Baker and J. E. Johnson, Structure, 1995, 3, 63 CrossRef CAS.
- T. Douglas and M. Young, Nature, 1998, 393, 152 CrossRef CAS.
- A. de la Escosura, M. Verwegen, F. D. Sikkema, M. Conellas-Aragones, A. Kirilyuk, T. Rasing, R. J. M. Nolte and J. J. L. M. Cornelissen, Chem. Commun., 2008, 1542 RSC.
- T. Douglas, E. Strable, D. Willits, A. Aitouchen, M. Libera and M. Young, Adv. Mater., 2002, 14, 415 CrossRef CAS.
- M. Comellas-Aragones, H. Engelkamp, V. I. Claessen, N. A. J. M. Sommerdijk, A. E. Rowan, P. C. M. Christianen, J. C. Maan, D. J. M. Verduin, J. J. L. M. Cornelissen and R. J. M. Nolte, Nat. Nanotechnol., 2007, 2, 635 CrossRef CAS.
- (a) J. M. Hooker, E. W. Kovacs and M. B. Francis, J. Am. Chem. Soc., 2004, 126, 3718 CrossRef CAS; (b) E. W. Kovacs, J. M. Hooker, D. W. Romanini, P. G. Holder, K. E. Berry and M. B. Francis, Bioconjugate Chem., 2007, 18, 1140 CrossRef CAS.
- J. M. Hooker, A. Datta, M. Botta, K. N. Raymond and M. B. Francis, Nano Lett., 2007, 7, 2207 CrossRef CAS.
- G. J. Tong, S. C. Hsiao, Z. M. Carrico and M. B. Francis, J. Am. Chem. Soc., 2009, 131, 11174 CrossRef CAS.
- (a) Q. Wang, T. Lin, L. Tang, J. E. Johnson and M. G. Finn, Angew. Chem., Int. Ed., 2002, 41, 459 CrossRef CAS; (b) E. Strable and M. G. Finn, Curr. Top. Microbiol. Immunol., 2009, 327, 1 Search PubMed; (c) Q. Wang, T. Lin, J. E. Johnson and M. G. Finn, Chem. Biol., 2002, 9, 813 CrossRef CAS; (d) K. S. Raja, Q. Wang, M. J. Gonzalez, M. Manchester, J. E. Johnson and M. G. Finn, Biomacromolecules, 2003, 4, 472 CrossRef CAS; (e) S. S. Gupta, K. S. Raja, E. Kaltgrad, E. Strable and M. G. Finn, Chem. Commun., 2005, 4315 RSC; (f) D. E. Prasuhn, Jr., R. M. Yeh, A. Obenaus, M. Manchester and M. G. Finn, Chem. Commun., 2007, 1269 RSC.
- (a) L. Loo, R. H. Guenther, V. R. Basnayake, S. A. Lommel, S. Franzen and J. Am, J. Am. Chem. Soc., 2006, 128, 4502 CrossRef CAS; (b) L. Loo, R. H. Guenther, S. A. Lommel and S. Franzen, J. Am. Chem. Soc., 2007, 129, 11111 CrossRef CAS.
- N. Carette, H. Engelkamp, E. Akpa, S. J. Pierre, N. R. Canmeron, P. C. M. Christianen, J. C. Maan, J. C. Thies, R. Weberskirch, A. E. Rowan, R. J. M. Nolte, T. Michon and J. C. M. van Hest, Nat. Nanotechnol., 2007, 2, 226 CrossRef CAS.
Footnote |
| † This work was supported by NWO-ABTS, IBOS, and Encapson. |
| This journal is © The Royal Society of Chemistry 2010 |