ESR spectroscopy as a tool to investigate the properties of self-assembled monolayers protecting gold nanoparticles
Marco
Lucarini
*a and
Lucia
Pasquato
*b
aDepartment of Organic Chemistry “A. Mangini”, University of Bologna, via San Giacomo 11, I-40126, Bologna, Italy. E-mail: marco.lucarini@unibo.it
bDepartment of Chemical Sciences and INSTM, Trieste Unit, University of Trieste, via L. Giorgieri 1, I-34127, Trieste, Italy. E-mail: lpasquato@units.it
First published on 22nd March 2010
Abstract
Electron spin resonance (ESR) has emerged as a powerful spectroscopic technique to study the properties of metal nanoparticles (NPs) protected by a self-assembled monolayer (SAM) of organic molecules. This technique has been employed to explore the capacity of homoligand monolayers to bind to a hydrophobic probe or to “sense” the hydrophobicity of mixed-ligand monolayers. Moreover, spin labels anchored to the metal surface enable the investigation of the dynamic of the ligands that form the monolayer. Here we review these applications with the aim of unravelling the many features of monolayer-protected metal NPs.
 Marco Lucarini | Marco Lucarini joined the Dept. of Organic Chemistry “A. Mangini” at the University of Bologna in 1992 as an assistant lecturer. He became Associate Professor in 2001 and Full Professor of Organic Chemistry in 2006 at the Faculty of Pharmacy. In 1996 he spent some time as Honorary Research Follow at the University College in London. In 1998 he received the Ciamician Medal awarded by the Italian Chemical Society to young researchers in the field of organic chemistry. His current research interests focus on mechanistic and ESR spectroscopic aspects of free-radical chemistry. |
 Lucia Pasquato | Lucia Pasquato obtained her PhD degree in 1983 from the University of Padova under the supervision of Professor Giorgio Modena. In 1985 she was appointed as researcher with the CNR (National Council of Research). In 2002 she moved to the University of Trieste to become Associate Professor. She was a research associate at the University of Würzburg with Professor Waldemar Adam during 1985–86. Her current research interests focus on functional- and naked-metal nanoparticles for applications in catalysis, in biochemistry and in material science. |
1. Introduction
The most intensively studied nanosized materials are gold NPs (AuNPs) in the form of colloids and nanocrystals that typically are 1–50 nm in diameter. These NPs find useful applications in many fields of science such as, for example, in electronics and energy storage; for the development of tags or staining materials, for the analysis of biological samples by electron and optical microscopic techniques, as carriers of biomolecules and as nanosized drug-delivery systems. NPs may also act as catalysts, mainly in oxidation processes, or as seeds for the generation of more sophisticated nanostructures such as nanowires of semiconducting materials.1,2These systems display unusual and unique size-dependant chemical and physical properties, and one of the striking differences between NPs and the corresponding bulk materials is the percentage of atoms that are interfacial.1,3 Non-protected, “naked” clusters possess surface atoms that are coordinatively highly unsaturated, interacting only with atoms inside the particle and having free valences outside. Their contribution to the electronic structure of the particle is different from that of fully coordinated inner atoms.
The surface of AuNPs may be passivated by organic ligands, forming a self-assembled monolayer (SAM) on a curved surface.4–7 These systems are, usually named monolayer-protected clusters (MPCs). SAMs on NPs simultaneously stabilize the reactive metal and present organic functional groups at the particle–solvent interface. Tailored organic surfaces on AuNPs are useful for applications in nanotechnology, in different fields of material science, chemistry, biology, and medicine.
The protection of the metal surface is of paramount importance to enable the facile manipulation of these systems, to tune their solubility properties, thus allowing for the analysis of individual particles and their assembly in two- and three-dimensional structures without the onset of uncontrolled aggregation. For instance, functional AuNPs have been implemented as powerful analytical tools2,8–10 for nucleic acids,11 proteins,12 and bacteria13 as well as in intracellular probes and gene regulators.14 Enzyme biosensor devices based on peptide–AuNP conjugates have been conceived in recent years.15,16 Peptide–AuNP conjugates behave also as artificial proteins presenting an unique pseudoenzymatic activity.17 Moreover, recent work shows that DNA–AuNP conjugates might serve as basic building blocks to assemble highly ordered macroscopic materials through programmable base-pairing interactions.18,19
Since the first report by Brust and Schiffrin,20 on the synthesis of protected AuNPs, we have been witness to an unprecedented revolution in particle syntheses, which has led to a spectacular variety of systems of different shapes, composition, patterns and functionalities. This has been possible thanks to the merging of synthetic innovation and development of methodologies that enable to investigate how molecules arrange around a particle core. Among the different methods of investigations electron spin resonance has played a fundamental role. Here we describe the major achievements obtained by the use of ESR to study the properties of protected AuNPs and the potential of this technique. The manuscript presents a first section reporting a summary of the synthetic approaches and characterization of passivated AuNPs to understand why ESR is useful in this field whereas the potential of this spectroscopy is underlined in section 3 and applications aimed to investigate protected AuNPs are reported in sections 4 and 5.
2. Synthesis and characterization of passivated AuNPs
The development of simple strategies to produce robust AuNPs begun with the seminal work20 of Schiffrin and Brust in 1994 and strategies have been reported in many reviews and books.1–7 The extension to systems that are stable in biological media is an active field of research as illustrated in a recent review by Rotello et al.21Protected AuNPs may be obtained essentially in two manners by i) formation of an organic layer around preformed AuNPs or colloids or ii) synthesis of AuNPs in the presence of organic ligands such as thiols, amines or phosphines. The different methodologies to prepare monolayer-protected metal NPs have been reviewed.1,5,22–27
One of the most attractive features of monolayer-protected AuNPs is the possibility of the facile introduction of a wide variety of different thiolated molecules into the ligand shell. This may be achieved using different strategies: 1) direct synthetic methods,6,28,29 2) the so-called place-exchange reaction,30 3) post-synthetic modifications31–34 or 4) displacement of weak ligands (or protective agents) by functional ligands.35,36 None of these methodologies allow control over the number of functional groups per nanoparticle and their topology within the monolayer. The control of these properties is mandatory for “clever,” programmable biological devices with applications ranging from pattern-based triggering of intracellular signaling pathways37 to the self-assembly of supramolecular catalysts.38 Few strategies have so far been reported that enable the functionalization of only one hemisphere or of one distinct point on a particle.39,40 It has been possible to introduce the concept of valency into NPs thereby allowing greater control over the assembly process of NPs. These strategies introduce anisotropy into particles and have enabled the assembly of dimeric and trimeric structures not attainable with isotropically functionalized particles. Accordingly, a key application of anisotropically patterned NPs is to use regulated functional group spacing and layout as “supermolecules” in the “bottom-up” self-assembly of new materials.41
MPCs behave as large molecules and consequently they may be studied using all the spectroscopic techniques employed for large organic molecules as polymers and biopolymers. Several studies have been reported, mainly concerning NPs protected by alkylthiolates, using FTIR,7,42,43 non-linear surface-selective vibrational sum-frequency generation (VSFG) spectroscopy,44 NMR,42,45 neutron scattering,46 and DSC.47 These studies have shown that SAMs on NPs are at least as ordered as SAMs on flat surfaces.42,48,49 However, local disorder is present, and this disorder increases with the distance from the metal surface. UV–Vis spectra show the optical properties of the nanoparticle due to the collective oscillation of conduction electrons resulting from the interaction with electromagnetic radiation. The oscillation wavelength depends on a number of factors, among which particle size and shape, as well as the nature of the surrounding medium, are the most important.50,51 Only recently, it has been reported that some small AuNPs exhibit molecular step-like transitions in their absorption spectra due to the electronic interactions between the metal core and the ligands forming the monolayer.52–58
A precise picture of MPCs in the solid state has been obtained by X-ray diffraction analysis of small AuNPs and gold clusters with a gold core composed of 102 and 25 gold atoms.52,53,59,60 These studies have revealed the unexpected nature of the sulfur–gold (S–Au) interaction, in agreement with recent studies on self-assembled monolayers on flat gold surfaces.61 In particular, two types of Au–SR (R = alkyl or aryl group) interactions are present: the first type is the “staple motif”, characterized by SR bridges between two gold atoms. In the other motif, Au atoms on the outermost external shell (adatoms) bind two SR groups forming a RS–Au–SR structure. Focusing on the monolayer characteristics, in the case of Au102(SR)44 (where SR is the para-mercaptobenzoic acid, p-MBA) particularly interesting are the three different interactions present between the ligands that stabilize the monolayer: 1) π–π stacking between two close and parallel phenyl rings, 2) H–π phenyl rings interacting at right angles (T-stacking) and 3) a sulfur–phenyl ring interaction. Almost all sulfur atoms are also engaged in lone pair bonding to a phenyl edge. Most p-MBAs are linked through chains of such interactions extending from one pole of the nanoparticle to the other. The chirality observed on the gold atoms extends to the monolayer and to interparticle interactions.
Whereas the S–Au interactions may be extended to larger AuNPs, the interactions between thiolates, and consequently the packing arrangement of the thiolates on the gold surface, is expected to change when alky thiolates are used to form the self-assembled monolayer.
Scanning tunneling microscopy (STM) has emerged as a powerful technique that has the capability of visualizing ligands of self-assembled monolayers on AuNPs.61,62 STM, being a microscopy tool, has the distinct advantage of providing single-particle data as opposed to the average ensemble data that spectroscopy techniques produce. However, STM imaging of NPs with molecular resolution is nontrivial and, being a microscopy tool, needs averaging over multiple images to result in representative data. These analyses have been carried out on NPs immobilized on gold surfaces so that they are not free to move during imaging. Dithiol molecules were used to act as linkers, binding the NPs to the underlying substrate.
The success of DFT calculations53,54 in reproducing experimental structures of AuNPs characterized by X-ray analysis has shown the potential of this approach in understanding and predicting structures and physical properties of small NPs.
3. ESR in supramolecular chemistry
ESR and related methods offer distinct opportunities to study SAM-protected AuNPs. The main advantages of ESR are the sensitivity of the method; the possibility of obtaining kinetic information in the submicrosecond time range; the ability to measure tumbling rates on the nanosecond timescale and distances between spin labels in close proximity. The strength of ESR lies also in its possible application to heterogeneous and solid samples. Due to the short relaxation times of paramagnetic species, ESR gives information which is not accessible by NMR,63 allowing for the study of the dynamic and structural features of supramolecular systems. The basic principles and applications of ESR may be found in many reviews and books.64–66While single gold atoms are paramagnetic because of the unpaired 6s electron, bulk gold is diamagnetic because the paramagnetism of the conduction electrons is counteracted by the orbital and ionic core diamagnetism.67 This implies that AuNPs protected by a self-assembled monolayers are ESR silent due to the absence of paramagnetic molecules.68
Thus, in order to get an ESR signal it is necessary to add a paramagnetic material to the sample. All the work reviewed in this paper was carried out on diamagnetic materials which are doped with paramagnetic species that must be stable radicals either in the form of added molecules (spin probes) or in the form of covalently bonded compounds (spin labels).69 The most widely used class of stable spin probes and labels is given by the nitroxide compounds containing the paramagnetic N–O unit.70Fig. 1 shows the chemical structures of some nitroxides used as spin probes and as reactants for the preparation of spin labels.
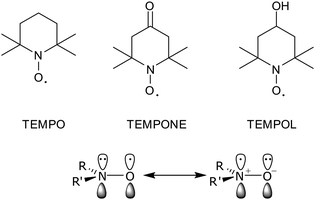 | ||
| Fig. 1 Some representative nitroxide radicals and their resonance structures. | ||
Typically, the ESR spectrum of a nitroxide is characterized by the hyperfine interaction between the unpaired electron with the14N nucleus (I = 1) and by the g-factor shifts due to spin–orbit coupling. For a dialkyl nitroxide, the isotropic nitrogen hyperfine splitting constants range from 13.5 to 18 G and are known to be very sensitive to the nature of the environment. In particular, the greater the polarity71 and/or the hydrogen bonding capability of the medium,72 the more important it is the dipolar structure having both a larger spin density on the nitrogen and a larger value of the nitrogen hyperfine coupling, Fig. 1.
When radicals are used as the guest, they show an “environmental” sensitivity, with a subsequent change of the spectroscopic parameters upon complexation. As a consequence, ESR spectroscopy, being characterized by a relatively short timescale, detects different signals from the complexed and uncomplexed radicals.73,74 In this way it is possible to extend the time range accessible to resonance methods of supramolecular chemical kinetics into the microsecond region.75 Through this approach, we can obtain kinetic information on different host–guest complexation elementary processes. These comprise also the exchange of an organic radical probe between the aqueous phases and the monolayer of water-soluble MPCs.
The mobility of a spin probe in fluid and semi-fluid systems depends mainly on the local viscosity. For a spin label the mobility depends on the tumbling of the macromolecule as a whole and on the flexibility of the backbone to which it is bound. The correlation time, τc which corresponds to the characteristic time during which a molecule turns over ca. 1 radian, is the parameter generally used for describing mobility in ESR studies. The correlation time is usually obtained by the analysis of the ESR line shape.
The use of paramagnetic probes or labels, provides a lot of information on the properties of MPCs. In particular ESR has been used for studying the exchange of an organic radical probe between the aqueous phase and a monolayer of water-soluble MPCs; for probing electrostatic phenomena or ligand organization of 3D mixed monolayers; for investigating the mechanistic aspects of ligand exchange in AuNPs. In sections 4 and 5 these results are reviewed.
4. Passivated AuNPs doped with spin probes
Several researchers employed ESR spectroscopy for the investigation of the interaction between stable nitroxide radicals, such as TEMPO, TEMPOL, and unprotected AuNPs of various sizes.76 The exchange interactions between the unpaired electrons of the adsorbed organic radicals and conduction-band electrons of the metallic particles were found to hide the ESR signal in several cases.In the presence of an organic monolayer covering the gold core of the nanoparticle, the addition of suitable spin probes gives rise to ESR signals that have been used to investigate the properties of self-assembled organic monolayers. These investigations rely on a series of nitroxide probes, obtained by in situ oxidation of amine precursors, based on the benzyl tert-butyl nitroxide moiety.77 These probes have been used in aqueous solutions to study the complexation behaviour of cyclodextrins,78,79 calixarenes,80 micelles,81 and cucurbiturils.82 The formation of a host–guest complex was manifested by large ESR spectral changes comprising a change in the value of the nitrogen hyperfine splitting, a(N), induced by the different polar environment experienced in the presence of the host, and the strong reduction of the benzylic protons coupling, a(2Hβ), due to conformational changes occurring upon complexation.
Modification of the original skeleton of benzyl tert-butyl nitroxide by introducing a hydrocarbon chain in the aromatic ring and by substituting one of the methyl groups with a hydroxymethyl group, Fig. 2, enables to investigate the dynamic of exchange of an organic probe between the aqueous phases and the monolayer of different classes of water-soluble protected AuNPs.
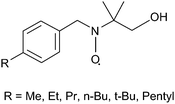 | ||
| Fig. 2 Structures of nitroxide probes employed to investigate protected NPs. | ||
The first monolayer investigated83 was composed of an amphiphilic thiolate (C8-TEG) containing a hydrocarbon chain close to the gold surface which provides a closely packed protective shield to the gold surface and a poly(ethylene glycol) chain which provides high solubility in water and other polar solvents. The ESR spectra of the para-substituted nitroxides, recorded in the presence of protected NPs are characterized by the presence of two set of signals due to the radical located in the monolayer, in equilibrium with the free nitroxide, Fig. 3. The former species was attributed to the radical hosted in the AuNP monolayer, on the basis of the following experimental evidence.
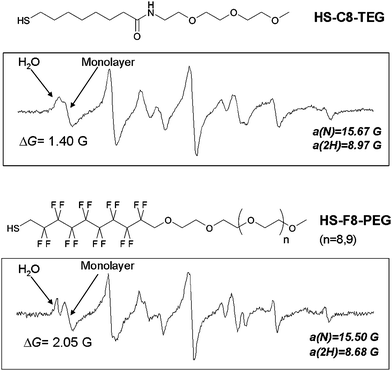 | ||
| Fig. 3 ESR spectra of p-pentylbenzyl-(1-hydroxy-2-methyl-2-propyl)nitroxide in the presence of AuNPs passivated by HS-C8-TEG (top) and HS-F8-PEG (bottom). | ||
i) The ESR nitrogen hyperfine splitting a(N) was significantly smaller, remaining approximately constant by increasing the length of the linear alkyl chain of the free radical, this indicates that the nitroxide group is located in a less polar environment shielded from the aqueous solvent. This signifies that the polarity of the environment surrounding the nitroxide function is similar for all the radicals and that the N–O group is deeply inserted inside the monolayer with the polar O–H group close to the hydrophilic region.
ii) The high-field ESR lines of the new species were characterized by a lower height, due to the slower motion of the radical probe when hosted in the monolayer, resulting in incomplete averaging of the anisotropic components of the hyperfine and g-tensors.
iii) The molar ratio between the complexed and free radical, obtained from the ESR spectra, increases linearly with increasing concentration of the dissolved MPC-C8-TEG.
In contrast to micelles where it is impossible to know precisely how the monomers aggregate due to the disorder of the system, the monolayer shows well defined regions due to the low mobility and to the specific orientation of the constituent thiols. From the value of the nitrogen splitting the locations of the probes in the monolayer was identified at the boundary between the hydrocarbon and polyether regions. These results confirmed the general view that it is possible to put functional groups deeply inside the monolayer so that they can interact with molecules in desolvated conditions similar to what is observed in enzymes where reactions take place mostly in a hydrophobic environment. The alkanethiol monolayer of the nanoparticle coupled with the radial nature of the ligands creates “hydrophobic pockets” inside the monolayer of the AuNPs where organic solutes can be partitioned. This property has been recently exploited by Rotello and co-workers who demonstrated that hydrophobic dyes/drugs can be stably entrapped in a hydrophobic pocket of AuNPs and released into the cell by membrane-mediated diffusion without uptake of the carrier nanoparticle.84
The ESR spectra also showed a strong alternating linewidth dependence on temperature, indicating that the lifetime of the nitroxide in the monolayer and in the bulk water is comparable to the ESR timescale. Thus, by analysis of ESR lineshape it is possible to obtain information on the kinetics of the exchange process (k+ and k−, Fig. 4). In particular the rate constant for the partition of the radical probe in the amphiphilic monolayer is independent of the length of the para-alkyl chain and very close to the diffusion limit while the dissociation rate constant depends on the length of the alkyl chain in the para position, that is on the lipophilicity of the probe with the exception of the bulky tert-butyl group. Actually, comparison of the values found when the substituent is a n- or tert-butyl indicates that for similar hydrophobicities an increased steric hindrance of the substituent reduces the residence time in the monolayer.
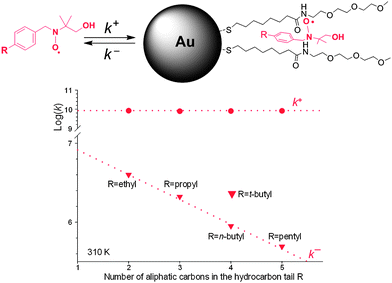 | ||
| Fig. 4 Rate constants for the nitroxide exchange between water and the monolayer. | ||
The NPs are generally prepared by reacting hydrogen tetrachloroaurate with the appropriate thiol and by adding sodium borohydride to the aqueous solution. By changing both the gold/thiol molar ratio and NaBH4 addition rate it is possible to prepare, in homogeneous solution, NPs with different core diameters ranging from 1.6 to 3.4 nanometres.85 Larger NPs having an average core diameter of about 5 nm can also be prepared following the two-step procedure described by Brust.34
By measuring the concentration ratio between the nitroxide solubilized in the monolayer and the free species by ESR as a function of the nanoparticle size it was possible to measure the affinity of the probe as a function of the nanoparticle diameter.86 Inspection of these data clearly shows that the solubilization of the organic probe in the monolayer strongly depends on the nanoparticle diameter. In particular, the affinity of the probe for the monolayer increases as the nanoparticle diameter decreases. The decrease of the core size of the nanoparticle determines a decrease of the packing of the thiol chains upon moving away from the Au surface. Accordingly, it is conceivable that the thiol chains bound to the smaller NPs experience a larger conformational freedom so that the accommodation of the hydrophobic guest requires a smaller energetic cost than that required in the presence of thiol chains bound to larger particles in which the monolayer is much more packed.
Very recently the first example of water-soluble AuNPs protected by a monolayer of fluorinated amphiphilic thiolates (F8-PEG) containing a perfluorinated portion and a short poly(ethylene glycol) unit was reported.87 The ESR spectrum of the nitroxide probe, containing the pentyl chain on the aromatic ring, when located in the monolayer is characterized by nitrogen and hydrogen splittings significantly smaller than those measured when the same radical is located in a hydrocarbon monolayer like C8-TEG, Fig. 3. This difference reflects a lower polarity of the environment experienced by the nitroxide function when dissolved in the F8-PEG monolayer. The possibility of “distinguishing” the hydrophobicity of the monolayer by ESR has been exploited to study the topology of mixed monolayers composed of thiolates that differ by an alkyl- or a perfluorinated chain.88
Although the differences in the value of hyperfine splitting constants are not sufficient to permit the resolution of the spectra due to the radicals partitioned in the two phases, the measurement of the apparent overall splitting of the spectrum allows the calculation of the ratio between the amount of the radical partitioned in the two phases. Actually, the field separation (ΔG) between the low-field lines of the spectrum due to the radical being partitioned in water (which can be considered as field markers) and in the monolayer decreases from 2.05 G, when the sample contains only alkyl monolayers, and to 1.40 G, in the presence of pure fluorinated monolayers, Fig. 5. When the ratio between the number of C8-TEG and F8-PEG chains (RSAM) is less than 2.5 the value of ΔG is nearly independent of the monolayer composition and equivalent to the value found in pure fluorinated monolayers. The equivalence of spectroscopic parameters strongly suggests exposure of the probe mainly in islands having a polarity similar to that of a homogenous phase of perfluorinated chains. This behavior corresponds to that expected for a phase-separated system different from a homogeneous arrangement of the binary mixture of the ligands.
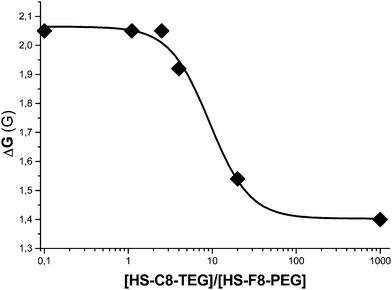 | ||
| Fig. 5 Dependence of ΔG (in Gauss) as a function of the ratio between the number of C8-TEG and F8-PEG chains (RSAM) in the monolayer. | ||
These data also indicate that the nitroxide probe is partitioned exclusively in the fluorinated islands, thus confirming the greater affinity of the probe for the perfluorinated phase relative to that for the nonfluorinated one.
In the presence of a larger amount of hydrocarbon chains (RSAM > 2.5) the field separation between the lines of the radical partitioned in water and in the monolayer starts to decrease indicating that the probe experiences a monolayer containing also hydrocarbon ligands. When RSAM = 20 the spectrum of the probe closely resembles the one recorded in the MPC-C8-TEG, indicating that the radical probes are practically surrounded by hydrocarbon chains.
These ESR experiments demonstrated that in heteroligand 3D SAMs phase separation occurs forming islands of homoligands triggered by the lipophobicity of perfluorocarbons.88
5. AuNPs passivated with spin labels
Anchoring of spin-labeled thiolates to the gold surface of NPs makes these systems suitable for investigation by ESR spectroscopy. Actually, in 1998 Murray and co-workers reported the first example of MPCs bearing multiple spin labels (ca. 13 nitroxide units for each cluster).89 In this pioneering report, 4-amino TEMPO was coupled with an MPC having ω-carboxylic acid groups, Fig. 6.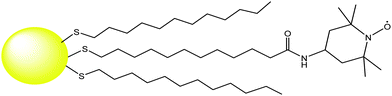 | ||
| Fig. 6 Structure of a TEMPO-functionalized MPC reported in ref. 89. | ||
In 2002 Chechik and coworkers showed that simple mixing of AuNPs protected by weakly bound ligands, such as phosphines, amines or short alkyl thiolates, with a disulfide diradical leads to spin-labeled NPs.90,91
By following the changes in the ESR spectrum shape observed during the exchange reaction of a spin-labeled disulfides with a short thiol linked to AuNPs, Chechik and coworkers were able to investigate the ligand dynamic on the gold surface and the ligand place-exchange reactions.92–94
When a spin-labeled disulfide reacts with a thiol-protected nanoparticle, the two branches of the disulfide molecule do not adsorb adjacent to each other on the surface, as is evident from the absence of spin–spin interactions between the two branches after ligand exchange, Fig. 7. A spin-labeled intermediate was identified in the reaction mixture by ESR, and was assigned to the mixed disulfide with the outgoing ligand. This mixed disulfide intermediate can of course participate in further ligand exchange steps, so that the final product of the reaction is the symmetrical disulfide of the outgoing ligand.
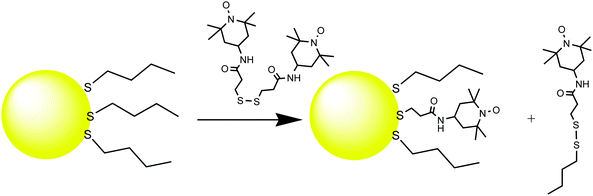 | ||
| Fig. 7 Ligand exchange of spin-labeled disulfides with thiol-protected AuNPs. | ||
A related reaction is the exchange of ligands between NPs. Also in this case ESR spectroscopy has been used to monitor the interparticle exchange reaction between AuNPs functionalized with a nitroxide spin label and a series of unlabeled NPs.95 The reaction was observed to be too slow at room temperature and could be monitored conveniently at 70 °C. NPs with different coverage of spin label have very different ESR spectra. One can observe that with decreased average coverage the line shape gradually changes from a spectral pattern dominated by a single broad line to a typical pattern of isolated nitroxide (three lines). This is due to dipole–dipole and exchange interactions between adjacent nitroxides adsorbed on the same nanoparticle. Chechik and co-workers suggested that the EPR spectra of NPs with more than five nitroxide groups per particle are dominated by a broad line. On the other hand, particles coated with one or two nitroxide ligands give predominantly a three-line spectrum.95 The kinetic studies suggest a dissociative mechanism with ligand desorption from the nanoparticle surface in the rate-determining step.
Analysis of the powder EPR spectra of spin-labeled NPs made it possible to find the distance between the nitroxide and the Au surface in spin-labeled NPs which provides direct information about the conformation of spin labels adsorbed on AuNPs.96
The lateral mobility of the thiolate ligands on the surface of AuNPs has also been probed by ESR spectroscopy. This was achieved by using bisnitroxide ligands, which contained a disulfide group (to ensure attachment to the Au surface) and a cleavable ester bridge connecting the two spin-labeled branches of the molecule, Fig. 8.97 Upon adsorption of these ligands on the surface of AuNPs, the two spin-labeled branches were held next to each other by the ester bridge as evidenced by the spin–spin interactions. Cleavage of the bridge removed the link that kept the branches together. The through-space dipole–dipole interactions between spin labels in the range of 1–2.5 nm can be directly observed by conventional continuous wave methods. Pulsed EPR (e.g., electron–electron double resonance, DEER) was also employed to extend the range to ca. 7–8 nm.98
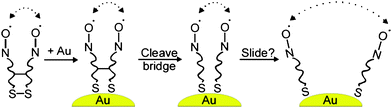 | ||
| Fig. 8 Schematic drawing of the EPR experiment directly probing the lateral diffusion of ligands on the surface of AuNPs. The dipole–dipole interactions between nitroxide spin labels are shown with dotted arrows. Reprinted with permission from ref. 97. Copyright American Chemical Society 2008. | ||
These experiments showed that the average distance between the adjacent thiolate branches on the Au nanoparticle surface only marginally increased after cleaving the bridge and thermal treatment. This implies that the lateral diffusion of thiolate ligands on the nanoparticle surface is very slow at room temperature and takes hours even at elevated temperatures (90 °C).
To the best of our knowledge this study is the only example reporting the use of pulsed ESR techniques to investigate the properties of monolayer-protected gold nanoparticles.94
ESR spectroscopy has also been employed for accessing interfacial surface electrostatics of ligand-protected AuNPs.99 These measurements are based on the properties of imidazoline and imidazolidine nitroxides containing a basic functional group in the heterocyclic ring to show pH-sensitive ESR spectra.100 In particular protonation of the amidine group affects both the rotational dynamics of the nitroxide and its magnetic parameters. The latter is attributed to the electron-withdrawing effect of the amidinium cation.
A series of ligands with variable length of the hydrocarbon bridge between the anchoring sulfur and a pH-sensitive nitroxide unit of the imidazoline series, Fig. 9, have been linked to tiopronin-protected AuNPs. The negatively charged environment in the carboxylate-functionalized AuNPs was found to shift the pKa of the amidine nitroxide probes by up to ca. 1.1 pH units, this pKa shift being affected by the position of the spin probe with respect to the nanoparticle polar interface.
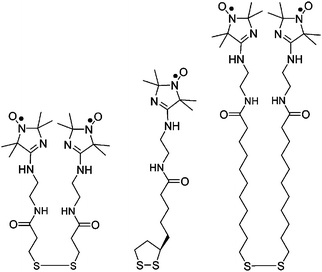 | ||
| Fig. 9 Structures of nitroxides containing the imidazoline group. | ||
6. Conclusions
The evolution of metal NPs in nanoscience is determined by the understanding of the properties of these supramolecular systems and by the capacity to perfectly control the morphology of the monolayer. These achievements may be reached by implementation of synthetic strategies but also finding the appropriate methods to look into protected metal NPs.ESR has proved to be an important tool for the investigation of SAM-protected AuNPs in solution. Although paramagnetic probes must be added to the samples, the reported examples provided considerable information on the properties of the monolayer that are difficult to obtain with more common techniques. We expect that this technique will significantly improve our ability to study more deeply monolayer-protected NPs.
Acknowledgements
We gratefully acknowledge financial support from MIUR [PRIN2006, Prot. 2006039071 (L. P.) 006033539 (M.L.)], Progetto Regione FVG 2008 and the University of Bologna.Notes and references
- Nanoparticles: From Theory to Application, ed. G. Schmid, Wiley-VCH, 2004, Weinheim Search PubMed.
- G. A. Ozin and A. C. Arsenault, Nanochemistry. A chemical approach to nanomaterials, RSC Publishing, Cambridge, 2005 Search PubMed.
- G. Schmid, Chem. Rev., 1992, 92, 1709–1727 CrossRef CAS.
- J. C. Love, L. A. Estroff, J. K. Kriebel, R. G. Nuzzo and G. M. Whitesides, Chem. Rev., 2005, 105, 1103–1169 CrossRef CAS.
- M. C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293–346 CrossRef CAS.
- A. C. Templeton, W. P. Wuelfing and R. W. Murray, Acc. Chem. Res., 2000, 33, 27–36 CrossRef CAS.
- A. Badia, R. B. Lennox and L. Reven, Acc. Chem. Res., 2000, 33, 475–481 CrossRef CAS.
- Nanobiotechnology, ed. C. M. Niemeyer and C. A. Mirkin, Wiley-VCH, Weinheim, 2004 Search PubMed; Nanobiotechnology II, ed. C. A. Mirkin and C. M. Niemeyer, Wiley-VCH, Weinheim, 2007 Search PubMed.
- Nanoparticles. Building blocks for Nanotechnology, ed. V. Rotello, Kluwer Academic/Plenum Publishers, 2004, New York Search PubMed.
- N. L. Rosi and C. A. Mirkin, Chem. Rev., 2005, 105, 1547–1562 CrossRef CAS.
- James J. Storhoff and Chad A. Mirkin, Chem. Rev., 1999, 99, 1849–1862 CrossRef CAS.
- (a) C.-C. You, A. Chompoosor and V. M. Rotello, Nano Today, 2007, 2, 34–43 CrossRef; (b) C.-C. You, A. Verma and V. M. Rotello, Soft Matter, 2006, 2, 190–204 RSC; (c) C.-C. You, O. R. Miranda, B. Basar Gider, P. S. Ghosh, IK.-B. Kim, B. Erdogan, S. A. Krovi, U. H. F. Bunz and V. M. Rotello, Nat. Nanotechnol., 2007, 2, 318–323 CrossRef CAS.
- R. L. Phillips, O. R. Miranda, C.-C. You, V. M. Rotello and U. H. F. Bunz, Angew. Chem., Int. Ed., 2008, 47, 2590–2594 CrossRef.
- G. Han, P. Ghosh, M. De and V. M. Rotello, NanoBiotechnology, 2007, 3, 40–45 CrossRef CAS.
- For a review see: J. E. Ghadiali and M. M. Stevens, Adv. Mater., 2008, 20, 4359–4363 Search PubMed.
- C. Guarise, L. Pasquato, V. De Filippis and P. Scrimin, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 3978–3982 CrossRef CAS.
- F. Manea, F. Bodar Houillon, L. Pasquato and P. Scrimin, Angew. Chem., Int. Ed., 2004, 43, 6165–6169 CrossRef CAS; L. Pasquato, P. Pengo and P. Scrimin, J. Mater. Chem., 2004, 14, 3481–3487 RSC; P. Pengo, S. Polizzi, L. Pasquato and P. Scrimin, J. Am. Chem. Soc., 2005, 127, 1616–1617 CrossRef CAS; P. Pengo, L. Baltzer, L. Pasquato and P. Scrimin, Angew. Chem., Int. Ed., 2007, 46, 400–404 CrossRef CAS.
- D. Nykypanchuk, M. M. Maye, D. van der Lelie and O. Gang, Nature, 2008, 451, 549–552 CrossRef CAS.
- S. Y. Park, A. K. R. Lytton-Jean, B. Lee, S. Weigand, G. C. Schatz and C. A. Mirkin, Nature, 2008, 451, 553–556 CrossRef CAS.
- M. Brust, M. Walker, D. Bethell, D. J. Schiffrin and R. Whyman, J. Chem. Soc., Chem. Commun., 1994, 801–802 RSC.
- M. De, P. S. Ghosh and V. M. Rotello, Adv. Mater., 2008, 20, 4225–4241 CrossRef CAS.
- P. Pengo and L. Pasquato in The Supramolecular Chemistry of Organic–Inorganic Hybrid Materials, ed. K. Rurack and R. Mártínez-Máñez, Wiley, Chichester, 2010, ch. 4 Search PubMed.
- R. Sardar, A. M. Funston, P. Mulvaney and R. W. Murray, Langmuir, 2009, 25, 13840–13851 CrossRef CAS.
- E. Boisselier and D. Astruc, Chem. Soc. Rev., 2009, 38, 1759–1782 RSC.
- J. Shan and H. Tenhu, Chem. Commun., 2007, 4580–4598 RSC.
- R. Lévy, ChemBioChem, 2006, 7, 1141–1145 CrossRef CAS.
- J. M. de la Fuente and S. Penades, Biochim. Biophys. Acta, Gen. Subj., 2006, 1760, 636–651 Search PubMed.
- K. G. Thomas and P. V. Kamat, Acc. Chem. Res., 2003, 36, 888–898 CrossRef CAS.
- F. Stellacci, C. A. Bauer, T. Meyer-Friedrichsen, W. Wenseleers, S. R. Marder and J. W. Perry, J. Am. Chem. Soc., 2003, 125, 328–329 CrossRef CAS.
- (a) M. J. Hostetler, S. J. Green, J. J. Stokes and R. W. Murray, J. Am. Chem. Soc., 1996, 118, 4212–4213 CrossRef CAS; (b) R. S. Ingram, M. J. Hostetler and R. W. Murray, J. Am. Chem. Soc., 1997, 119, 9175–9178 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- For example: (a) H. Nandivada, X. Jiang and J. Lahann, Adv. Mater., 2007, 19, 2197–2208 CrossRef CAS; (b) J. L. Brennan, N. S. Hatzakis, T. R. Tshikhudo, N. Dirvianskyte, V. Razumas, S. Patkar, J. Vind, A. Svendsen, R. J. M. Nolte, A. E. Rowan and M. Brust, Bioconjugate Chem., 2006, 17, 1373–1375 CrossRef CAS.
- (a) M. Fischler, A. Sologubenko, J. Mayer, G. Clever, G. Burley, J. Gierlich, T. Carell and U. Simon, Chem. Commun., 2008, 169–171 RSC; (b) W. J. Sommer and M. Weck, Langmuir, 2007, 23, 11991–11995 CrossRef CAS.
- R. Voggu, P. Suguna, S. Chandrasekaran and C. N. R. Rao, Chem. Phys. Lett., 2007, 443, 118–121 CrossRef CAS.
- M. Brust, D. Bethell, D. J. Schiffrin and C. J. Kiely, Adv. Mater., 1995, 7, 795–797 CAS.
- C. J. Loweth, W. B. Caldwell, X. Peng, A. P. Alivisatos and P. G. Schultz, Angew. Chem., Int. Ed., 1999, 38, 1808–1812 CrossRef CAS.
- M. M. Stevens and J. H. George, Science, 2005, 310, 1135–1138 CrossRef CAS.
- J.-M. Lehn, Science, 2002, 295, 2400–2403 CrossRef CAS.
- Q. Huo and J. G. Worden, J. Nanopart. Res., 2007, 9, 1013–1025 CrossRef CAS.
- C. Gentilini and L. Pasquato, J. Mater. Chem., 2010, 20, 1403–1412 RSC.
- S. Kinge, M. Crego-Calma and D. N. Reinhoudt, ChemPhysChem, 2008, 9, 20–42 CrossRef CAS.
- M. J. Hostetler, J. E. Wingate, C. J. Zhong, J. E. Harris, R. W. Vachet, M. R. Clark, J. D. Londono, S. J. Green, J. J. Stokes, G. D. Wignall, G. L. Glish, M. D. Porter, N. D. Evans and R. W. Murray, Langmuir, 1998, 14, 17–30 CrossRef CAS.
- R. Paulini, B. L. Frankamp and V. M. Rotello, Langmuir, 2002, 18, 2368–2373 CrossRef CAS.
- C. Weeraman, A. K. Yatawara, A. N. Bordenyuk and A. V. Benderskii, J. Am. Chem. Soc., 2006, 128, 14244–14245 CrossRef CAS; A. N. Bordenyuk, C. Weeraman, A. Yatawara, H. D. Jayathilake, I. Stiopkin, Y. Liu and A. V. Benderskii, J. Phys. Chem. C, 2007, 111, 8925–8933 CrossRef CAS.
- A. Badia, W. Gao, S. Singh, L. Demers, L. Cuccia and L. Reven, Langmuir, 1996, 12, 1262–1269 CrossRef CAS; H. Schmitt, A. Badia, L. Dickinson, L. Reven and R. B. Lennox, Adv. Mater., 1998, 10, 475–480 CrossRef CAS.
- T. Pradeep, S. Mitra, A. S. Nair and R. Mukhopadhyay, J. Phys. Chem. B, 2004, 108, 7012–7020 CrossRef CAS.
- A. Badia, S. Singh, L. Demers, L. Cuccia, G. R. Brown and R. B. Lennox, Chem.–Eur. J., 1996, 2, 359–363 CrossRef CAS.
- A. Badia, L. Cuccia, L. Demers, F. Morin and R. B. Lennox, J. Am. Chem. Soc., 1997, 119, 2682–2692 CrossRef CAS.
- M. J. Hostetler, J. J. Stokes and R. W. Murray, Langmuir, 1996, 12, 3604–3612 CrossRef CAS.
- C. M. Aikens, S. Li and G. C. Schatz, J. Phys. Chem. C, 2008, 112, 11272–11279 CrossRef CAS.
- J. P. Wilcoxon, J. E. Martin and P. Provencio, J. Chem. Phys., 2001, 115, 998–1008 CrossRef.
- M. W. Heaven, A. Dass, P. S. White, K. M. Holt and R. W. Murray, J. Am. Chem. Soc., 2008, 130, 3754–3755 CrossRef CAS.
- M. Zhu, C. M. Aikens, F. J. Hollander, G. C. Schatz and R. Jin, J. Am. Chem. Soc., 2008, 130, 5883–5885 CrossRef CAS.
- J. Akola, M. Walter, R. L. Whetten, H. Hakkinen and H. Grönbeck, J. Am. Chem. Soc., 2008, 130, 3756–3757 CrossRef CAS.
- M. Walter, J. Akola, O. Lopez-Acevedo, P. D. Jadzinsky, G. Calero, C. J. Ackerson, R. L. Whetten, H. Grönbeck and H. Hakkinen, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 9157–9162.
- Y. Negishi, N. K. Chaki, Y. Shichibu, R. L. Whetten and T. Tsukuda, J. Am. Chem. Soc., 2007, 129, 11322–11323 CrossRef CAS.
- Y. Negishi, K. Nobusada and T. Tsukuda, J. Am. Chem. Soc., 2005, 127, 5261–5270 CrossRef CAS.
- Y. Shichibu, Y. Negishi, T. Tsukuda and T. Teranishi, J. Am. Chem. Soc., 2005, 127, 13464–13465 CrossRef CAS.
- P. D. Jadzinsky, G. Calero, C. J. Ackerson, D. A. Bushnell and R. D. Kornberg, Science, 2007, 318, 430–433 CrossRef CAS.
- M. Zhu, W. T. Eckenhoff, T. Pintaeur and R. Jin, J. Phys. Chem. C, 2008, 112, 14221–14224 CrossRef CAS.
- (a) R. Mazzarello, A Cossaro, A Verdini, R. Rousseau, L. Casalis, M. F. Danisman, L. Floreano, S. Scandolo, A. Morgante and G. Scoles, Phys. Rev. Lett., 2007, 98, 016102 CrossRef CAS; (b) A Cossaro, R. Mazzarello, R. Rousseau, L. Casalis, A. Verdini, A. Kohlmeyer, L. Floreano, S. Scandolo, A. Morgante, M. L. Klein and G. Scoles, Science, 2008, 321, 943–946 CrossRef CAS.
- (a) A. M. Jackson, J. W. Myerson and F. Stellacci, Nat. Mater., 2004, 3, 330–336 CrossRef CAS; (b) A. M. Jackson, Y. Hu, P.J. Silva and F. Stellacci, J. Am. Chem. Soc., 2006, 128, 11135–11149 CrossRef CAS.
- Relaxation times of organic free radicals are in the scale of μs whereas relaxation times of nuclei observed by nuclear magnetic resonance (NMR) are in the scale of seconds.
- J. A. Weil, J. R. Bolton and J. E. Wertz, Electron Paramagnetic Resonance: Elementary Theory and Practical Applications, John Wiley & Sons Inc., New York, 1994 Search PubMed.
- N. M. Atherton, Principles of Electron Spin Resonance, Ellis Horwood and Prentice Hall, Chichester, 1993 Search PubMed.
- Electron Paramagnetic Resonance: a practitioner's toolkit, ed. M. Brustolon, E. Giamello, John Wiley & Sons Inc., Hoboken, New Jersey, 2009 Search PubMed.
- R. M. White, Quantum Theory of Magnetism, 3rd edn, Springer, New York, 2007 Search PubMed.
- Recently the first example of paramagnetic protected Au nanoparticles has been reported. The unpaired spin in this system, consisting of 25 gold atoms stabilized by 18 thiolate ligands, resides in the Kohn–Sham highest occupied molecular orbital. In this case the Au25(SR)18 nanoparticle should be considered as a ligand-protected superatom. M. Zhu, C. M. Aikens, M. P. Hendrich, R. Gupta, H. Qian, G. C. Schatz and R. Jin, J. Am. Chem. Soc., 2009, 131, 2490–2492 Search PubMed.
- Spin Labeling II. Theory and Applications, ed. L. J. Berliner, Academic Press, New York, 1979 Search PubMed.
- G. Likhtenshtein, J. Yamauchi, S. Nakatsuji, A. I. Smirnov and R. Tamura, Nitroxides: Applications in Chemistry, Biomedicine, and Materials Science, Wiley-VCH, Weinheim, 2009 Search PubMed.
- B. R. Knauer and J. J. Napier, J. Am. Chem. Soc., 1976, 98, 4395–4400 CrossRef CAS.
- P. Franchi, M. Lucarini, P. Pedrielli and G. F. Pedulli, ChemPhysChem, 2002, 3, 789–793 CrossRef CAS.
- P. Franchi, M. Lucarini and G. F. Pedulli, Curr. Org. Chem., 2004, 8, 1831–1849 CrossRef CAS.
- G. Martini and L. Ciani, Phys. Chem. Chem. Phys., 2009, 11, 211–254 RSC.
- W. M. Nau and X. Wang, ChemPhysChem, 2002, 3, 393–398 CrossRef CAS.
- Z. Zhang, A. Berg, H. Levanon, R. W. Fessenden and D. Meisel, J. Am. Chem. Soc., 2003, 125, 7959–7963 CrossRef CAS.
- M. Lucarini and B. P. Roberts, Chem. Commun., 1996, 1577–1578 RSC.
- P. Franchi, M. Lucarini, E. Mezzina and G. F. Pedulli, J. Am. Chem. Soc., 2004, 126, 4343–4354 CrossRef CAS.
- M. Lucarini, B. Luppi, G. F. Pedulli and B. P. Roberts, Chem.–Eur. J., 1999, 5, 2048–2054 CrossRef CAS.
- P. Franchi, M. Lucarini, G. F. Pedulli and D. Sciotto, Angew. Chem., Int. Ed., 2000, 39, 263–266 CrossRef CAS.
- E. Mileo, P. Franchi, R. Gotti, C. Bendazzoli, E. Mezzina and M. Lucarini, Chem. Commun., 2008, 1311–1313 RSC.
- E. Mezzina, F. Cruciani, G. F. Pedulli and M. Lucarini, Chem.–Eur. J., 2007, 13, 7223–7233 CrossRef CAS.
- M. Lucarini, P. Franchi, G. F. Pedulli, P. Pengo, P. Scrimin and L. Pasquato, J. Am. Chem. Soc., 2004, 126, 9326–9329 CrossRef CAS.
- C. K. Kim, P. Ghosh, C. Pagliuca, Z.-J. Zhu, S. Menichetti and V. M. Rotello, J. Am. Chem. Soc., 2009, 131, 1360–1361 CrossRef CAS.
- P. Pengo, S. Polizzi, M. Battagliarin, L. Pasquato and P. Scrimin, J. Mater. Chem., 2003, 13, 2471–2478 RSC.
- M. Lucarini, P. Franchi, G. F. Pedulli, C. Gentilini, P. Polizzi, P. Pengo, P. Scrimin and L. Pasquato, J. Am. Chem. Soc., 2005, 127, 16384–16385 CrossRef CAS.
- C. Gentilini, F. Evangelista, P. Rudolf, P. Franchi, M. Lucarini and L. Pasquato, J. Am. Chem. Soc., 2008, 130, 15678–15682 CrossRef CAS.
- C. Gentilini, P. Franchi, E. Mileo, S. Polizzi, M. Lucarini and L. Pasquato, Angew. Chem., Int. Ed., 2009, 48, 3060–3064 CrossRef CAS.
- A. C. Templeton, M. J. Hostetler, E. K. Warmoth, S. Chen, C. M. Hartshorn, V. M. Krishnamurthy, M. D. E. Forbes and R. W. Murray, J. Am. Chem. Soc., 1998, 120, 4845–4849 CrossRef CAS.
- P. Ionita, A. Caragheorgheopol, B. C. Gilbert and V. Chechik, J. Am. Chem. Soc., 2002, 124, 9048–9049 CrossRef CAS.
- V. Chechik, H. J. Wellsted, A. Korte, B. C. Gilbert, H. Caldararu, P. Ionita and A. Caragheorgheopol, Faraday Discuss., 2004, 125, 279–291 RSC.
- A. Caragheorgheopol and V. Chechik, Phys. Chem. Chem. Phys., 2008, 10, 5029–5041 RSC.
- P. Ionita, A. Caragheorgheopol, B. C. Gilbert and V. Chechik, Langmuir, 2004, 20, 11536–11544 CrossRef CAS.
- P. Ionita, B. C. Gilbert and V. Chechik, Angew. Chem., Int. Ed., 2005, 44, 3720–3722 CrossRef CAS.
- M. Zachary and V. Chechik, Angew. Chem., Int. Ed., 2007, 46, 3304–3307 CrossRef CAS.
- P. Ionita, A. Caragheorgheopol, B. C. Gilbert and V. Chechik, J. Phys. Chem. B, 2005, 109, 3734–3742 CrossRef CAS.
- P. Ionita, A. Volkov, G. Jeschke and V. Chechik, Anal. Chem., 2008, 80, 95–106 CrossRef CAS.
- G. Jeschke, ChemPhysChem, 2002, 3, 927–932 CrossRef CAS.
- V. K. Khlestkin, J. F. Polienko, M. A. Voinov, A. I. Smirnov and V. Chechik, Langmuir, 2008, 24, 609–612 CrossRef CAS.
- V. V. Khramtsov and L. B. Volodarsky, Spin Labeling: The Next Millenium, ed. L. J. Berliner, Plenum Press, New York, 1998, vol. 14, p. 109 Search PubMed.
| This journal is © The Royal Society of Chemistry 2010 |