Facile ligand-exchange with polyvinylpyrrolidone and subsequent silica coating of hydrophobic upconverting β-NaYF4:Yb3+/Er3+ nanoparticles†
Noah J. J.
Johnson
,
Neralagatta M.
Sangeetha
,
John-Christopher
Boyer
and
Frank C. J. M.
van Veggel
*
Department of Chemistry, University of Victoria, P.O. Box 3065, Victoria, British Columbia, Canada V8W 3V6. E-mail: fvv@uvic.ca
First published on 4th March 2010
Abstract
A facile ligand-exchange strategy with a water-soluble polymer, i.e. polyvinylpyrrolidone (PVP), to replace the surface passivating oleate ligands on the β-NaYF4 nanoparticle surface is reported. Highly monodisperse oleate-stabilized β-NaYF4 nanoparticles were synthesized and the oleates were exchanged with a commercially available PVP allowing the phase transfer of these nanoparticles. The exchanged nanoparticles are readily dispersible in water and other polar solvents. To show the effectiveness of the exchange reaction we used the affinity of the PVP chains to silica and coated the nanoparticles with a uniform, thin silica shell. The PVP exchanged and silica-coated nanoparticles show longer colloidal stability and no surfactant related problems as compared to the reverse microemulsion-based silica-coated nanoparticles, which show a high tendency to aggregate, when removed from the microemulsion. The optical properties of the ligand-exchanged nanoparticles dispersed in water were compared with that of the oleate-stabilized nanoparticles in organic solvents. A decrease in the upconversion emission intensity and a different relative ratio of the green and red upconverted light were observed for the particles dispersed in water after ligand-exchange. PVP is a highly biocompatible polymer and is reported to have a longer blood circulation time and very low accumulation in vital organs, two highly desired properties for in vivo studies. This ligand-exchange strategy opens a new pathway to study the use of β-NaYF4 for biological applications in vivo.
Introduction
Inorganic nanoparticles have widely been studied for their use towards biological applications. Nanoparticles based on quantum dots and metals such as gold and silver show unique size dependent optical properties and their use as bio-tools has been studied extensively.1–4 Recently, there is an increased interest towards the use of lanthanide-based nanoparticles. This can be attributed to the use of heavy metals and toxicity related to some of the quantum dots5,6 and sensitive size/shape dependent optical properties of gold and silver nanoparticles which overlap with the biological tissue fluorescence in the visible region. Trivalent lanthanide ions have unique luminescent properties due to the shielding of the valence 4f electrons by the filled 5s and 5p orbitals. The f–f transitions of the trivalent lanthanide ions result in sharp, narrow spectral bands and are highly specific to the ion. Different lanthanide-doped luminescent materials have been reported in the literature,7–13 among which, lanthanide-doped β-NaYF4 (hexagonal) upconverting nanoparticles (UCNPs) are being studied extensively for biological applications because of their highly efficient upconversion luminescence.14 When co-doped with Yb3+ ions, lanthanide-doped NaYF4 yields upconversion emission ranging from visible to NIR wavelengths for excitation with a low energy NIR light of 980 nm. This selectivity of the emission wavelength using a single excitation source, higher penetration depth, and low auto-fluorescence from the biological tissues at NIR wavelengths has resulted in an increased interest in this class of nanoparticles.Synthesis of near-monodisperse β-NaYF4 usually involves the use of high boiling, non-aqueous solvents and oleate as the coordinating ligand. The resulting nanoparticles are hydrophobic and form stable colloidal dispersions in non-polar organic solvents. In order to use these nanoparticles for biological applications they have to be rendered water-dispersible via phase-transfer. Different strategies have been reported for the phase transfer of β-NaYF4 hydrophobic nanoparticles from organic to aqueous phase, viz. silica coating in reverse microemulsion,15 coating with an amphiphilic polymer shell,16 and ligand exchange.17 Silica coating within a reverse microemulsion leads to silica-coated nanoparticles and allows for the facile phase transfer of the nanoparticles to aqueous solvents. Coating with amphiphilic polymer shells involves the use of polymers with both hydrophobic and hydrophilic segments in their backbone. On mixing the nanoparticles protected with a hydrophobic ligand with the amphiphilic polymer, the hydrophobic ligand intercalates with the hydrophobic segment of the polymer and the hydrophilic part of the polymer sticks out into water, rendering the nanoparticles dispersible in water. In the case of ligand exchange, the hydrophobic ligands on the surface of the nanoparticles are replaced with hydrophilic small molecules or polymer chains, resulting in water-dispersible nanoparticles. This strategy has largely been studied with different silanes18 and water-soluble polymers. Polymers with functional groups or heteroatoms which can coordinate to the surface of the nanoparticle are being explored widely over simple molecules, as they yield highly stable dispersions of nanoparticles in water after ligand exchange, because of their bulky framework. The ligand exchange with polymers has largely been focused on poly(acrylic acid) (PAA)19 and phosphine oxide-PEG based polymers20,21 and not much attention has been paid to other water-soluble polymers such as PVP that can coordinate to the nanoparticle surface.
PVP is a biocompatible, non-toxic, water-soluble polymer used in different pharmaceutical applications.22,23 The ability of the polymer chains to stabilize the surface of nanoparticle during growth is termed as the protective value of the polymer and PVP stands out with a high protective value.24 PVP chains coordinate to different nanoparticle surfaces through their carbonyl groups and have been used in the synthesis of metal25,26 and magnetic nanoparticles.27 When PVP is used as a coordinating ligand to synthesize NaYF4, the obtained nanoparticles have the α-phase and are polyhedral in shape with particle sizes greater than 30 nm.28 Recently Wang et al. reported on the β-phase NaYF4 stabilized by PVP using a hydrothermal method.29 The synthesized particles were on an average 40 nm in size and polydisperse, other hydrothermal methods reported in the literature have also reported on particle sizes greater than 50 nm and mostly polydisperse.30–33 On the other hand, β-phase NaYF4 obtained in high boiling solvents with oleates as stabilizing ligand are monodisperse (size dispersion <5%) and particle sizes less than 20 nm can easily be obtained. As smaller size and monodispersity play a vital role in determining their suitability for biological applications, we report on the preparation of PVP-stabilized upconverting β-NaYF4via ligand exchange by replacing the hydrophobic oleate ligands. The particles thus obtained are highly monodisperse and spherical in shape with particle size around 20 nm. The use of these particles for biological applications (in vivo) would be particularly advantageous as PVP is reported to have good antifouling properties,34 longer blood circulation time than the widely used poly(ethylene glycol) (PEG), minimum tissue distribution, and low accumulation in vital organs.35 Moreover, to show that the exchange was successful we used the affinity of PVP to silica36 and coated the PVP-stabilized nanoparticles with a thin silica shell. By growing a silica shell, these PVP-stabilized nanoparticles can be used as targeted probes as the silica shell can easily be functionalized by co-hydrolysis of functional silanes. Coating of silica shell on nanoparticles is focused on obtaining thin shell growth and non-aggregated particles as aggregation would be detrimental for in vivo studies.
Silica coating on oleate-stabilized β-NaYF4 nanoparticles reported in literature has largely been done by the reverse microemulsion route, wherein the silica coating is done in water-in-oil emulsions and the particles stabilized by surfactants during growth. This one-step strategy could be tuned to obtain silica shells of varying thickness.37 Although this synthetic route is simple, there is a tendency for the silica-coated particles to aggregate which leads to necking between the silica beads when they are removed from the emulsions.38 Graf et al. showed thin silica coatings on citrate-stabilized gold nanoparticles using PVP to stabilize the nanoparticles from aggregation in ethanol.36 The latter methodology has not been investigated for oleate-stabilized hydrophobic nanoparticles for growing thin silica shells. Here, we show that this method of silica coating can be extended to oleate-stabilized β-NaYF4 nanoparticles after ligand-exchange with PVP. We used a commercially available PVP for ligand-exchange with oleate-stabilized β-NaYF4 nanoparticles. Dispersions of PVP-stabilized nanoparticles obtained in this way have very good colloidal stability in a wide range of solvents and can readily be coated with a thin silica shell. Moreover, upconversion luminescence studies on the PVP-stabilized nanoparticles and silica-coated nanoparticles were done and compared with the oleate-stabilized NaYF4 nanoparticles. To the best of our knowledge this is the first report on oleate-stabilized β-NaYF4 nanoparticles being successfully exchanged with PVP to grow a uniform silica shell. We believe that this facile strategy of PVP exchange and silica coating can be extended to other oleate-stabilized nanoparticles having affinity to PVP chains.
Experimental section
Materials
All lanthanide chlorides (YCl3·6H2O, YbCl3·6H2O, ErCl3·6H2O) (>99.99%), oleic acid (90%), 1-octadecene (90%), ammonium fluoride (99.99%), tetraethyl orthosilicate (TEOS) (99.99%), polyvinylpyrrolidone 10 kg mol−1 (PVP-10) and Igepal CO-520 were purchased from Sigma-Aldrich. Sodium hydroxide and dichloromethane (DCM) were obtained from ACP chemicals. Ethyl ether (anhydrous), dimethylformamide (DMF), toluene, cyclohexane and ammonium hydroxide (30 wt% in water) were from Caledon laboratories. All chemicals were used as received without further purification.Synthesis of oleate-stabilized β-NaYF4:Yb3+/Er3+ nanoparticles
The synthesis of β-NaYF4 was done following a reported procedure with some modifications.39 In a typical synthesis, 1 mmol of the lanthanide chlorides (0.78 mmol YCl3·6H2O, 0.20 mmol YbCl3·6H2O and 0.02 mmol ErCl3·6H2O) were taken in a 100 ml 3-necked flask and 15 ml of octadecene and 6 ml of oleic acid were added. The flask was then heated to 150 °C under vacuum and held at this temperature for 30 min to get a homogenous solution. Subsequently, the flask was cooled down to room temperature and 10 ml methanol solution containing 4 mmol NH4F and 2.5 mmol NaOH was added dropwise. The resulting solution was stirred at room temperature for 2 h and heated slowly to 70 °C until all the methanol evaporated. The reaction vessel was then brought under a gentle flow of argon and heated up to 300 °C and held at that temperature for 90 min. The flask was then cooled to room temperature and the nanoparticles were precipitated by adding ethanol (15 ml), centrifuged (Beckman Coulter Spinchron 15 - rotor F0630), and washed with ethanol. The isolated oleate-stabilized nanoparticles were stored as 1 wt% dispersion in toluene.Ligand exchange of oleate-stabilized β-NaYF4:Yb3+/Er3+ with PVP
A dispersion of oleate-stabilized nanoparticles in toluene (0.2 ml) was taken in a 100 ml round bottom flask and diluted with 5 ml of 1:1 DMF–DCM. To this, 75 mg of PVP was added and refluxed at 100 °C for 6 h. The reaction mixture was then added dropwise into ethyl ether (60 ml) to precipitate the polymer-stabilized nanoparticles. The precipitate was washed once with ethyl ether and centrifuged at 4500 rpm for 5 min. The precipitate was transferred to 6.5 ml of ethanol to yield a stable dispersion of the PVP-stabilized NaYF4 nanoparticles. The precipitate could also be dispersed in several other solvents like water, chloroform, dichloromethane, DMF, and DMSO to yield optically transparent dispersions.Silica coating on PVP-stabilized NaYF4:Yb3+/Er3+ nanoparticles
Ethanol dispersion of phase-transferred nanoparticles (6.5 ml) was taken in a 15 ml vial and 0.28 ml of ammonium hydroxide (30 wt% in water) was added, followed by 65 μl of 10 vol% TEOS in ethanol solution. The vial was sealed and stirred for 15 h at room temperature. The silica-coated nanoparticles were isolated by centrifugation at 12000 rpm for 15 min and washed with ethanol. The collected silica beads were dispersed in distilled water.Silica coating on hydrophobic nanoparticles through microemulsion route
Microemulsion-based silica coating was done by following a reported procedure.40 Typically, ∼7 mg of the oleate-stabilized nanoparticles in cyclohexane was mixed with 6 ml cyclohexane and 0.1 ml of Igepal CO-520. To this, 0.4 ml Igepal CO-520 and 0.08 ml ammonium hydroxide (30 wt% in water) were added and the flask was sonicated until a transparent emulsion was obtained. TEOS (40 μl) was added and stirred for 2 days. The nanoparticles were precipitated by adding methanol and washed several times with ethanol–water, centrifuged at 4500 rpm for 5 min and the collected silica beads were dispersed in distilled water.Characterization of β-NaYF4:Yb3+/Er3+ nanoparticles
TEM images of the nanoparticles were obtained from a Hitachi H-7000 microscope operating at 75 kV. The TEM grids (Formar Carbon on 300 mesh Cu) were coated with the nanoparticles by drop casting a dilute dispersion of the nanoparticles (hexane dispersions of hydrophobic nanoparticles and ethanol dispersions of the hydrophilic nanoparticles) and air dried before imaging. The size distribution was calculated for an ensemble of ∼100 nanoparticles.The X-ray diffraction (XRD) patterns of the nanoparticles were acquired using a Rigaku Miniflex X-ray diffractometer with a Cr source (Kαλ = 2.2890 Å) operating at 30 kV and 15 mA. The XRD patterns were collected at a sampling width of 0.05° (2θ) and scan speed of 1° min−1.
FT-IR spectra were obtained from a Perkin Elmer FT-IR spectrometer 1000 with a resolution of 2 cm−1 and averaged over four scans. The samples were taken as dry powder and pelletized with KBr and placed on the sample holder.
Optical measurements were done on the UCNPs using an Edinburgh instruments FLS920 fluorimeter The luminescence spectra were collected using a red-sensitive Peltier-cooled Hamamatsu R955 PMT. A 980 nm laser diode (JDS Uniphase type 63-00342) coupled to a 100 μm core fiber was used as the excitation source. The output was collimated using a fiber coupler and a long band-pass filter (850 nm) was used on the excitation side and a short band-pass filter (800 nm) on the collecting end of the detector to remove the scattered excitation light. The optical measurements were done on toluene and hexane dispersions of oleate-stabilized nanoparticles and water dispersions of the hydrophilic nanoparticles (PVP-stabilized and silica-coated) taken in a 1 cm path-length quartz cuvette. All upconversion emission spectra were measured at a resolution of 1 nm, using the same laser power density (150 W cm−2) for excitation.
Results and discussion
The lanthanide chlorides, sodium hydroxide and ammonium fluoride were heated to 300 °C in a mixture of octadecene and oleic acid and subsequently aged for 90 min at that temperature to obtain β-NaYF4. The initially formed kinetic product, α-NaYF4 is transformed to the more stable product, β-NaYF4 upon aging at a high temperature.30,41 The β-NaYF4 nanoparticles co-doped with Yb3+(20%) and Er3+(2%) thus obtained were characterized using TEM and X-ray powder diffraction. The TEM image (Fig. 1A) of the synthesized oleate-stabilized NaYF4 nanoparticles show that the particles are spherical with a narrow size distribution (21.0 ± 0.5 nm). The particles on the TEM grids show a long range ordering into hexagonal, closed-packed structure because of their narrow size distribution and are regularly spaced (∼3.3 nm) by the surface stabilizing oleate ligand. The XRD pattern (Fig. 2A) shows that pure β-NaYF4 nanoparticles were obtained and the peaks match well with the standard spectral lines of β-NaYF4 (JCPDS #16-0334). The average crystallite size determined from X-ray diffraction data (20 nm), is in good agreement with the size determined from TEM analysis. The surface oleate ligands allow for the formation of stable, optically transparent dispersions of these nanoparticles in non-polar organic solvents such as toluene, hexane, and chloroform. | ||
| Fig. 1 TEM images of NaYF4:Yb3+/Er3+ (A) oleate-stabilized, (B) ligand-exchanged with PVP. | ||
 | ||
| Fig. 2 Powder X-ray diffraction pattern of NaYF4:Yb3+/Er3+ (A) oleate-stabilized, (B) ligand-exchanged with PVP and, (C) the corresponding reference pattern. | ||
Ligand exchange of the hydrophobic nanoparticles with PVP
The ligand exchange step allows for almost complete exchange of the oleate ligands with a hydrophilic ligand and opens the pathway to disperse them in aqueous media and their use in different biological applications. Amphiphilic PVP was selected as it has been shown to coordinate well with NaYF4 nanoparticles.28 A toluene dispersion of oleate-stabilized nanoparticles was mixed with PVP dissolved in a mixture of DCM and DMF to form a clear dispersion. This mixture was then refluxed at 100 °C to exchange the oleates with PVP. The exchange reaction is believed to be driven by mass action as the excess PVP in the medium slowly replaces the surface oleate ligand and the high temperature helps in the dynamic solvation of the ligands during exchange.42 DCM used in the reaction helps in the precipitation of PVP-stabilized nanoparticles into ethyl ether and the oleates remain in the supernatant, which allows for their easy removal after exchange. After exchange, the precipitate was dried and dispersed in different solvents to yield stable, optically clear dispersions (Fig. 3). The beam sharpness in all photographs demonstrates that these dispersions have little or no scatter of the excitation and emission light. Excitation of the organic layer after the exchange showed no observable intensity, suggesting that the exchange is quantitative. | ||
| Fig. 3 TEM images of silica-coated NaYF4:Yb3+/Er3+ nanoparticles, (A and B) PVP-exchanged and subsequently coated, (C) from reverse microemulsion before washing, (D) from reverse microemulsion showing aggregation and necking after washing to remove the excess surfactants. | ||
The TEM images (Fig. 1B) of the PVP-stabilized nanoparticles have the same morphology after exchange but did not show any long range ordering on the TEM grids as observed for the oleate-stabilized nanoparticles. This indicates that the oleate ligands responsible for the packing of the hydrophobic nanoparticles had been successfully replaced by PVP chains. The XRD pattern (Fig. 2B) shows that the PVP-stabilized nanoparticles retain their phase after exchange. Comparison of the FT-IR spectra of the ligand-exchanged NPs with that of the oleate-stabilized nanoparticles indicated that the oleates were exchanged with PVP. The FT-IR spectrum of the oleate-stabilized nanoparticles (Fig. 4A) shows the carboxylate stretching at 1566 and 1465 cm−1. On the other hand, the FT-IR spectra of the exchanged nanoparticles (Fig. 4B) shows a strong carbonyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) stretching peak at 1651 cm−1 and a broad peak at 3500 cm−1 due to the adsorbed water molecules. The appearance of the carbonyl stretching peak and the absence of the carboxylate stretching peaks indicates that the exchange of oleates with PVP was successful.
O) stretching peak at 1651 cm−1 and a broad peak at 3500 cm−1 due to the adsorbed water molecules. The appearance of the carbonyl stretching peak and the absence of the carboxylate stretching peaks indicates that the exchange of oleates with PVP was successful.
 | ||
| Fig. 4 Colloidal dispersion of PVP-stabilized nanoparticles in different solvents (0.5 wt%) and their total fluorescence under 980 nm laser excitation (same power density). (A) chloroform, (B) DCM, (C) ethanol, (D) DMSO, (E) DMF and (F) water. | ||
Silica coating on PVP-stabilized β-NaYF4:Yb3+/Er3+ nanoparticles
The affinity of PVP-stabilized nanoparticles to silica is well documented.36 In order to show that the exchange reaction with PVP was successful and aggregation of the nanoparticle did not occur during the exchange reaction, we used the affinity of the PVP chains to silica and grew a shell of silica around the nanoparticles. The PVP-stabilized nanoparticles were dispersed in ethanol and the silica shells were grown by a Stöber-like synthesis. PVP on the surface of the nanoparticles allows for their stability in ethanol and their affinity to silica allows for uniform silica shell growth on the nanoparticle surface. The silica-coated nanoparticles thus obtained have an average shell thickness of 9 nm (Fig. 5A–B) and there were few silica-coated nanoparticles existing as dimers which can be attributed to the multidentate coordination of the PVP, which leads to some cross-linking between the nanoparticles during exchange. The surface of the silica shells are rough, probably due to the silica nucleated PVP chains attached to the surface.43,44 The excess PVP which is not coordinated to the nanoparticle surface acted as nucleation sites for silica growth, leading to the formation of some empty silica beads. This silica being less dense than the silica-coated nanoparticles was easily removed by centrifugation at the end of the reaction. The FT-IR spectra of the silica-coated nanoparticles (Fig. 4C) shows the Si–O–Si stretching band at 1097 cm−1 and a small peak at 1651 cm−1 indicating the presence of some remnant PVP chains in the silica beads. | ||
| Fig. 5 FT-IR spectra of β-NaYF4:Yb3+/Er3+ (A) oleate-stabilized nanoparticles, (B) ligand-exchanged with PVP, (C) silica-coated on the ligand-exchanged nanoparticles. | ||
The reverse microemulsion route was used to coat the hydrophobic nanoparticles with a thin shell to compare with the silica coating on the PVP-stabilized nanoparticles. The thin silica shell obtained by the reverse microemulsion route also had an average shell thickness of 9 nm. These silica-coated nanoparticles have the same morphology when compared to the silica-coated PVP-stabilized nanoparticles (Fig. 5C). The excess surfactant used in the microemulsion route was hard to remove even by successive centrifugation and washing steps and the removal of the surfactants led to much higher necking between the silica beads (Fig. 5D). The lower colloidal stability and aggregation tendency of thin silica shells grown in reverse microemulsion has been attributed to the dynamic nature of the reverse micelles and the presence of excess surfactants, by earlier reports.38 Our experimental studies also show the same features with respect to the silica-coated nanoparticles grown in reverse micelles. Recently Liu et al. have reported that the silica-coated NaYF4 particles from reverse micelles were stable for one day,45 which implies that they have a tendency to aggregate once removed from the stabilizing micelles which kept them apart during growth. Nanoparticles that were silica-coated by reverse emulsion were mostly aggregated within one day after washing and storing as a dispersion in water (Fig. S3C and D). On the other hand, the PVP-exchanged nanoparticles coated with silica were individual with no aggregation after washing and storing for two days (Fig. S1). Some aggregation was noticeable after 5 days (Fig. S2A) and only after a week most of them were aggregated (Fig. S2B). These results clearly show that the silica-coated nanoparticles made via PVP-stabilized precursor nanoparticles show less tendency to aggregate than the silica-coated nanoparticles obtained via the reverse emulsion method when all excess surfactant has been removed.
Most of the biological studies require sterile samples, and our attempts to filter the silica nanoparticles synthesized in reverse microemulsion through a 0.2 μm sterile syringe filter showed that they could not be filtered. This might be due to the excess surfactants clogging the pores and the aggregation resulting from the necking between the silica beads. On the other hand, the PVP-exchanged and subsequently silica-coated nanoparticles could easily be filtered through 0.2 μm sterile syringe filters.
Optical properties of β-NaYF4:Yb3+/Er3+ nanoparticles
The colloidal dispersion of the oleate-stabilized nanoparticles in organic solvents showed bright green luminescence (Inset Fig. 6) when excited with a 980 nm laser diode as observed by naked eye. The upconversion luminescence spectra (Fig. 6) obtained from hexane and toluene dispersions show the characteristic green and red upconverted light with green being more intense than the red. The peaks at 520 and 540 nm are attributed to the transitions from the 2H11/2, 4S3/2 levels to the ground state (4I15/2), respectively. The peak at 654 nm corresponds to the transition from the 4F9/2 to 4I15/2 energy levels of the Er3+ doped in the nanoparticle matrix. | ||
| Fig. 6 Upconversion emission spectra of oleate-stabilized β-NaYF4:Yb3+/Er3+ nanoparticles (λex = 980 nm) (A) in hexane, (B) in toluene, Inset: upconversion emission from the colloidal dispersion under 980 nm diode excitation. | ||
The ligand-exchanged nanoparticles dispersed in water showed decreased upconversion luminescence when compared with the oleate-stabilized nanoparticles dispersed in organic solvents. The high phonon energy of O–H groups in the water molecules can easily quench the upconversion luminescence by enhancing the non-radiative relaxation of the excited photons in Er3+ energy levels which account for the green and red emissions. The quenching of lanthanide excited states by O–H vibrations is well studied and has been reported to be highly effective compared to the C–H vibrational quenching of the excited states.46 Hydrophilic upconverting nanoparticles obtained by other pathways have also been reported to show decreased luminescence intensity when dispersed in water.16,47 Yi et al. used a core-shell nanoparticle approach with an undoped NaYF4 as a shell so that the emitting ions are shielded from the O–H vibrations.16 Yet there was some quenching of upconversion emission when these core-shell nanoparticles coated with amphiphilic PAA were dispersed in water. This shows that the upconversion emission is highly dependent on the environment or the solvent used.
The PVP-exchanged nanoparticles dispersed in water showed a different ratio of the green and red upconverted emission when compared with the oleate-stabilized nanoparticles dispersed in organic solvents. While green upconverted emission was dominant in oleate-stabilized nanoparticles, red upconverted emission was dominant in PVP-stabilized nanoparticles dispersed in water (Fig. 7A). Typical upconversion emission mechanism for β-NaYF4:Yb3+/Er3+ is shown in Fig. 8. In the presence of O–H vibrations, the non-radiative relaxation of (2H11/2, 4S3/2) to 4F9/2 level and 4I11/2 to 4I13/2 are increased. The non-radiative relaxation of (2H11/2, 4S3/2) to 4F9/2 quenches the green emission and populates the red emitting level. In addition to that, as the probability of 4I11/2 to 4I13/2 non-radiative relaxation increases, the ions populating the 4I13/2 can absorb another photon of 980 nm and eventually populate the red emitting level (4F9/2). Thus, an increase in the non-radiative relaxation pathways in the presence of water enhances the red emission compared to the green. This effect has been observed on the upconverting nanoparticles in presence of water.47–49 The upconversion spectra of the silica-coated nanoparticles in water shows that the coating was partially effective in shielding the high energy O–H vibrations as relatively equal amounts of green and red upconversion emission were observed (Fig. 7B).
 | ||
| Fig. 7 Upconversion emission spectra of β-NaYF4:Yb3+/Er3+ nanoparticles (λex = 980 nm) (A) PVP-stabilized in water, (B) PVP-stabilized and subsequently silica-coated nanoparticles in water. | ||
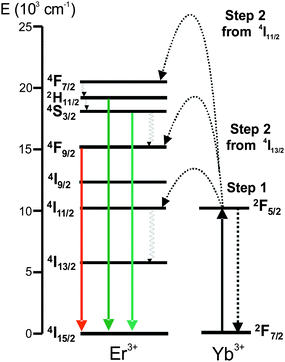 | ||
| Fig. 8 Upconversion emission mechanism of β-NaYF4:Yb3+/Er3+ (λex = 980 nm). | ||
Conclusions
Highly monodisperse β-NaYF4:Yb3+/Er3+ UCNPs capped with oleates were successfully phase transferred from organic solvent to water after ligand-exchange with a commercially available polymer, PVP. These PVP-stabilized nanoparticles were used to grow thin silica shells to show the success of the PVP exchange reaction. The PVP-stabilized nanoparticles were individually coated with a silica shell of ∼9 nm showing that nanoparticle aggregation did not happen during the ligand exchange step. These nanoparticles show decreased upconversion emission in aqueous medium and the intensity of the red upconversion emission increases relative to the green due to increased non-radiative relaxation of Er3+ excitation levels in the presence of –OH phonons. This methodology of phase transfer and silica coating opens a different pathway for using these nanoparticles in biological studies, as the silica aggregation and excess surfactant related problems as seen in the reverse emulsion-based silica shells are avoided. This strategy is currently being investigated with terminally functionalized PVP, allowing for direct bioconjugation of the exchanged nanoparticles and the results will be reported in a future publication.Acknowledgements
Natural Science and Engineering Research Council (NSERC), the Canada Foundation for Innovation (CFI), and the British Columbia Knowledge Development Fund (BCKDF) of Canada are acknowledged for support.References
- S. H. Liu and M. Y. Han, Adv. Funct. Mater., 2005, 15, 961–967 CrossRef CAS.
- S. Santra, H. S. Yang, P. H. Holloway, J. T. Stanley and R. A. Mericle, J. Am. Chem. Soc., 2005, 127, 1656–1657 CrossRef CAS.
- A. Schroedter and H. Weller, Angew. Chem., Int. Ed., 2002, 41, 3218–3221 CrossRef CAS.
- R. E. Bailey, J. B. Strausburg and S. M. Nie, J. Nanosci. Nanotechnol., 2004, 4, 569–574 CrossRef.
- A. Hoshino, K. Fujioka, T. Oku, M. Suga, Y. F. Sasaki, T. Ohta, M. Yasuhara, K. Suzuki and K. Yamamoto, Nano Lett., 2004, 4, 2163–2169 CrossRef CAS.
- K. B. Male, B. Lachance, S. Hrapovic, G. Sunahara and J. H. T. Luong, Anal. Chem., 2008, 80, 5487–5493 CrossRef CAS.
- F. Meiser, C. Cortez and F. Caruso, Angew. Chem., Int. Ed., 2004, 43, 5954–5957 CrossRef CAS.
- J. L. Bridot, A. C. Faure, S. Laurent, C. Riviere, C. Billotey, B. Hiba, M. Janier, V. Josserand, J. L. Coll, L. Vander Elst, R. Muller, S. Roux, P. Perriat and O. Tillement, J. Am. Chem. Soc., 2007, 129, 5076–5084 CrossRef CAS.
- J. W. Stouwdam, G. A. Hebbink, J. Huskens and F. C. J. M. van Veggel, Chem. Mater., 2003, 15, 4604–4616 CrossRef CAS.
- F. Wang, X. J. Xue and X. G. Liu, Angew. Chem., Int. Ed., 2008, 47, 906–909 CrossRef CAS.
- C. A. Traina and J. Schwartz, Langmuir, 2007, 23, 9158–9161 CrossRef CAS.
- Z. G. Yan and C. H. Yan, J. Mater. Chem., 2008, 18, 5046–5059 RSC.
- S. Sivakumar, P. R. Diamente and F. C. J. M. van Veggel, Chem.–Eur. J., 2006, 12, 5878–5884 CrossRef CAS.
- K. W. Krämer, D. Biner, G. Frei, H. U. Güdel, M. P. Hehlen and S. R. Lüthi, Chem. Mater., 2004, 16, 1244–1251 CrossRef.
- Z. Q. Li, Y. Zhang and S. Jiang, Adv. Mater., 2008, 20, 4765–4769 CrossRef CAS.
- G. S. Yi and G. M. Chow, Chem. Mater., 2007, 19, 341–343 CrossRef CAS.
- M. Nyk, R. Kumar, T. Y. Ohulchanskyy, E. J. Bergey and P. N. Prasad, Nano Lett., 2008, 8, 3834–3838 CrossRef CAS.
- R. De Palma, S. Peeters, M. J. Van Bael, H. Van den Rul, K. Bonroy, W. Laureyn, J. Mullens, G. Borghs and G. Maes, Chem. Mater., 2007, 19, 1821–1831 CrossRef CAS.
- W. Lin, K. Fritz, G. Guerin, G. R. Bardajee, S. Hinds, V. Sukhovatkin, E. H. Sargent, G. D. Scholes and M. A. Winnik, Langmuir, 2008, 24, 8215–8219 CrossRef CAS.
- H. Bin Na, I. S. Lee, H. Seo, Y. Il Park, J. H. Lee, S. W. Kim and T. Hyeon, Chem. Commun., 2007, 5167–5169 RSC.
- S. W. Kim, S. Kim, J. B. Tracy, A. Jasanoff and M. G. Bawendi, J. Am. Chem. Soc., 2005, 127, 4556–4557 CrossRef CAS.
- D. Le Garrec, S. Gori, L. Luo, D. Lessard, D. C. Smith, M. A. Yessine, M. Ranger and J. C. Leroux, J. Controlled Release, 2004, 99, 83–101 CrossRef CAS.
- O. Rabin, J. M. Perez, J. Grimm, G. Wojtkiewicz and R. Weissleder, Nat. Mater., 2006, 5, 118–122 CrossRef CAS.
- L. D. Pachon and G. Rothenberg, Appl. Organomet. Chem., 2008, 22, 288–299 CrossRef.
- Y. G. Sun and Y. N. Xia, Science, 2002, 298, 2176–2179 CrossRef CAS.
- Y. G. Sun, Y. D. Yin, B. T. Mayers, T. Herricks and Y. N. Xia, Chem. Mater., 2002, 14, 4736–4745 CrossRef CAS.
- X. Y. Lu, M. Niu, R. R. Qiao and M. Y. Gao, J. Phys. Chem. B, 2008, 112, 14390–14394 CrossRef CAS.
- Z. Q. Li and Y. Zhang, Angew. Chem., Int. Ed., 2006, 45, 7732–7735 CrossRef CAS.
- M. Wang, C. C. Mi, J. L. Liu, X. L. Wu, Y. X. Zhang, W. Hou, F. Li and S. K. Xu, J. Alloys Compd., 2009, 485, L24–L27 CrossRef CAS.
- C. X. Li, J. Yang, Z. W. Quan, P. P. Yang, D. Y. Kong and J. Lin, Chem. Mater., 2007, 19, 4933–4942 CrossRef CAS.
- X. Liang, X. Wang, J. Zhuang, Q. Peng and Y. D. Li, Adv. Funct. Mater., 2007, 17, 2757–2765 CrossRef CAS.
- L. Y. Wang and Y. D. Li, Chem. Mater., 2007, 19, 727–734 CrossRef CAS.
- J. H. Zeng, J. Su, Z. H. Li, R. X. Yan and Y. D. Li, Adv. Mater., 2005, 17, 2119–2123 CrossRef CAS.
- S. Robinson and P. A. Williams, Langmuir, 2002, 18, 8743–8748 CrossRef CAS.
- Y. Kaneda, Y. Tsutsumi, Y. Yoshioka, H. Kamada, Y. Yamamoto, H. Kodaira, S. Tsunoda, T. Okamoto, Y. Mukai, H. Shibata, S. Nakagawa and T. Mayumi, Biomaterials, 2004, 25, 3259–3266 CrossRef CAS.
- C. Graf, D. L. J. Vossen, A. Imhof and A. van Blaaderen, Langmuir, 2003, 19, 6693–6700 CrossRef CAS.
- D. K. Yi, S. S. Lee, G. C. Papaefthymiou and J. Y. Ying, Chem. Mater., 2006, 18, 614–619 CrossRef CAS.
- N. R. Jana, C. Earhart and J. Y. Ying, Chem. Mater., 2007, 19, 5074–5082 CrossRef CAS.
- Z. Q. Li and Y. Zhang, Nanotechnology, 2008, 19, 345606 CrossRef.
- R. A. Jalil and Y. Zhang, Biomaterials, 2008, 29, 4122–4128 CrossRef.
- G. S. Yi and G. M. Chow, Adv. Funct. Mater., 2006, 16, 2324–2329 CrossRef CAS.
- T. R. Zhang, J. P. Ge, Y. P. Hu and Y. D. Yin, Nano Lett., 2007, 7, 3203–3207 CrossRef CAS.
- I. Pastoriza-Santos, J. Perez-Juste and L. M. Liz-Marzan, Chem. Mater., 2006, 18, 2465–2467 CrossRef CAS.
- Y. Wang, Z. Y. Tang, X. R. Liang, L. M. Liz-Marzan and N. A. Kotov, Nano Lett., 2004, 4, 225–231 CrossRef CAS.
- C. Liu, H. Wang, X. Li and D. Chen, J. Mater. Chem., 2009, 19, 3546–3553 RSC.
- G. A. Hebbink, J. W. Stouwdam, D. N. Reinhoudt and F. C. J. M. van Veggel, Adv. Mater., 2002, 14, 1147–1150 CrossRef CAS.
- R. Naccache, F. Vetrone, V. Mahalingam, L. A. Cuccia and J. A. Capobianco, Chem. Mater., 2009, 21, 717–723 CrossRef CAS.
- J. W. Zhao, Y. J. Sun, X. G. Kong, L. J. Tian, Y. Wang, L. P. Tu, J. L. Zhao and H. Zhang, J. Phys. Chem. B, 2008, 112, 15666–15672 CrossRef CAS.
- H. Schäfer, P. Ptacek, K. Kömpe and M. Haase, Chem. Mater., 2007, 19, 1396–1400 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S3. See DOI: 10.1039/b9nr00379g |
| This journal is © The Royal Society of Chemistry 2010 |