A multiscale simulation study of carbon nanotube interactions with designed amphiphilic peptide helices†
E. Jayne
Wallace
,
Robert S. G.
D'Rozario
,
Beatriz Mendoza
Sanchez
and
Mark S. P.
Sansom
*
Department of Biochemistry, University of Oxford, South Parks Road, Oxford, OX1 3QU, UK. E-mail: mark.sansom@bioch.ox.ac.uk; Fax: +44 (0)1865 613238; Tel: +44 (0)1865 613306
First published on 9th April 2010
Abstract
The dispersion and manipulation of carbon nanotubes (CNTs) are of great importance if we are to utilise the unique properties of CNTs in a range of biological, electrical and mechanical applications. Recently, a designed amphiphilic peptide helix termed nano-1 has been shown to solubilise CNTs in aqueous solution. Furthermore, the peptide is capable of assembling these coated tubes into fibres. We use a multiscale molecular dynamics approach to study the adsorption profile of nano-1 on a CNT surface. We find that nano-1 interacts with a CNT in a preferred orientation, such that its hydrophobic surface is in contact with the tube. The adsorption profile is unchanged upon increasing the number of peptides on the CNT. Interestingly, when few peptides are adsorbed onto the CNT surface we find that the secondary structure of the peptide is unstable. However, the helical secondary structure is stabilised upon increasing the number of peptides on the CNT surface. This study sheds light on the adsorption of peptides on CNTs, and may be exploitable to enhance the selective solubilisation and manipulation of CNTs.
Introduction
There is great interest in exploring the novel properties of carbon nanotubes (CNTs) in electrical and mechanical devices.1 For example, single-walled CNTs have potential application in rectifying heterojunctions,2 field-effect transistors,3 nanowires,4 and high-strength, light-weight materials.5,6 Furthermore, the use of CNTs in biological applications such as artificial muscles (actuators),7 biomedical sensors8 and drug delivery vehicles9–12 is being investigated. However, manipulating CNTs into various structures is problematic due to their high affinity for one another, making it difficult to disperse them in an aqueous environment.13–15 Dissolution of CNTs has been facilitated by covalently attaching polar or charged groups to CNT surfaces. However, this can alter the inherent properties of the tube.16,17 Thus attention is now focussed on the noncovalent adsorption of molecules, primarily detergent,18–23 onto the surface of CNTs, thereby preserving the extended π networks of the tubes. Despite the ability of detergent to solubilise CNTs, these molecules do not promote higher-order structures or impart tissue selectivity in biomedical applications. Thus, there is much interest in exploring the potential uses of alternative biomolecular species in CNT solubilisation and manipulation, including peptides24 and DNA.25 Furthermore, from a more general bionanotechnological perspective there is a desire to understand CNT/peptide (and by extension CNT/protein) interactions at a fundamental level.In recent years Wang et al.24 discovered that peptides that are typically enriched in aromatic residues can display binding specificity for CNTs. In light of this, it has been suggested that biological molecules may be exploited to facilitate self-assembly of CNTs, owing to the accomplishment of biological systems in controlled self-assembly. A structural motif used by proteins to promote self-assembly is the amphiphilic α-helix.26 Considering this, Dieckmann et al.27–29 designed a 29-residue peptide denoted nano-1 (Fig. 1a and b) which folds into an amphiphilic α-helix in the presence of CNTs.
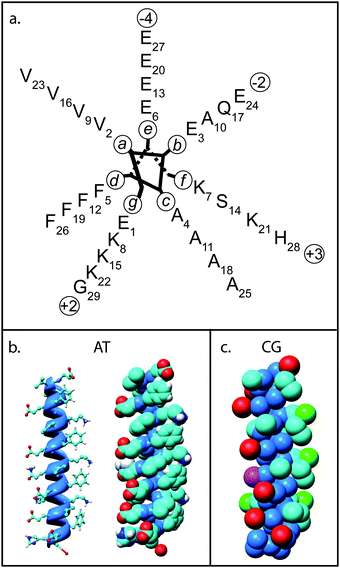 | ||
| Fig. 1 Model structure of nano-1. (a) Helical wheel diagram of nano-1 showing residue positions in the α-helix. The nano-1 sequence is Ac-E(VEAFEKK)(VAAFESK)(VQAFEKK)(VEAFEHG)-CONH2, where Ac indicates acetylation of the N-terminus, CONH2 indicates amidation of the C-terminus, and the parentheses denote heptad repeats. (b) Atomistic (AT) nano-1 highlighting the α-helical structure of the peptide (left) and in space filling format (right). Atoms are coloured using the CPK convention. (c) Coarse-grain (CG) nano-1, with particles coloured as follows: blue = “mixed polar/nonpolar particle”; cyan = “hydrophobic particle”; red/green = negative/positive “charged particle”; and pink = “polar particle”. | ||
Amphiphilic nano-1 has a nonpolar surface, comprising aliphatic valine (Val) and aromatic phenylalanine (Phe) residues, designed to noncovalently interact with CNTs. Nano-1 also has a polar surface, comprising glutamic acid and lysine, designed to interact with both the aqueous environment and with other peptides. As anticipated, Dieckmann and colleagues found that nano-1 effectively dispersed CNTs in aqueous solution.27 In addition, the hydrophilic surface of nano-1 and the peptide termini was found to promote assembly of these coated CNTs into fibres, with the CNTs aligned along the fibre axis. Crucially, it was found that the size and morphology of these fibres can be controlled by manipulating solution conditions that affect peptide–peptide interactions.29,30 Therefore, this approach to CNT solubilisation is not only appealing for electrical and mechanical devices, but also for drug delivery vehicles since peptides can be designed such that the CNT/peptide complex may target specific cells.
If we are to successfully exploit the use of peptides to solubilise CNTs it is important to have a greater understanding of their fundamental interactions with CNTs. Atomic force microscopy (AFM) and transmission electron microscopy (TEM) experiments have shed light on the structure of the CNT/nano-1 complex.29–31 However, these experiments were performed on dry samples and so may not reflect the adsorption profile in solution. A number of simulation studies address the interaction mechanism between a single peptide and a CNT. For example, Trzaskowski et al.32 have performed molecular dynamics simulations of two protein fragments, namely a β-hairpin and an α-helix, interacting with both the inner and outer surface of CNTs. They show that encapsulation of the peptide within a CNT can stabilise the peptide structure. Tomasio and Walsh33 recently performed molecular dynamics simulations of “strong-binder” and “non-binder” peptides, as identified by Wang et al.,24 interacting with a CNT. These simulations confirmed experimental observations of binding affinity, and showed that aromatic residues tend to be located close to the CNT surface. More recently, Fan et al.34 have confirmed this result via quantum mechanical modelling. Tomasio and Walsh35 further investigated the importance of the tryptophan aromatic residue in CNT adsorption. They mutated each tryptophan of the “stronger-binder” peptides with either tyrosine or phenylalanine. They find that the original tryptophan containing sequences bind more strongly to a CNT. Shen et al.36 have studied the adsorption of human serum albumin on CNTs via molecular dynamics. They find that the protein adsorbs onto the CNT in a stepwise manner, and that aromatic residues have a higher affinity to the CNT. Kang et al.37 later found that the protein SmtA is spontaneously encapsulated within a CNT, also making a stepwise conformational change in order to maximise its affinity to the CNT walls. During this conformational change, the β-sheet content of the protein is dramatically decreased.
Chiu et al.38 have performed fully atomistic molecular dynamics simulations of a single nano-1 peptide at various water/hydrophobic interfaces, including water/graphite and water/CNT. They found that the peptide retained approximately 40% of its α-helicity when interacting with graphite, whilst 65–95% of the peptide's α-helicity was retained when interacting with a single-walled (6,6) CNT of 8.1 Å in diameter. As with the simulations of Tomasio and Walsh,33 Chiu et al. also found that the aromatic residues tend to be located close to the CNT surface. To date, there are few simulation studies that address the adsorption of multiple peptides onto a CNT surface. Friling et al.39 have studied the adsorption of either one or two cyclic peptides on CNTs via molecular dynamics simulations. Chiu et al.40 have recently performed a simulation study of a (6,6) and an (8,8) CNT having saturated shells of nano-1 peptides. They observed that the peptides had α-helical conformations.
In this study we report on a systematic study of nano-1 interactions with CNTs, exploring the influence of nano-1/CNT ratio on CNT adsorption and peptide α-helical stability. We employ a multiscale molecular dynamics (MD) approach, incorporating both coarse-grained41–45 (CG) and atomistic (AT) levels of description. Multiscale simulations have been applied with some success to simulation of a number of biomolecular systems.46 The advantage of a multiscale approach is that whilst CG MD allows much larger length and longer time scales to be accessed than AT MD, an AT model enables detailed insight into the underlying intermolecular interactions. We show that nano-1 has a preferred orientation when adsorbed onto a CNT surface. Additionally, we show that the concentration of nano-1 clearly affects the stability of the secondary structure of the peptide.
Methods
Unless otherwise stated, all simulations were performed using the GROMACS 3.3 software package,47,48 at constant temperature (323 K), pressure (1 bar) and number of particles. Simulations were performed at 323 K since the CG force field has been validated at this temperature.41,49 In order to assess the robustness of our model, several AT simulations were also performed at 300 K. Unless stated otherwise, our AT simulations were performed with the GROMOS53A650 force field. However, as a further test of model robustness, several AT simulations were also performed with the OPLS51,52 force field (details of these OPLS simulations are in the ESI†). Molecular graphics images were produced using Chimera.53Atomistic model
We used PyMOL54 to construct an AT model of nano-1, basing the structure on an idealised α-helix with backbone dihedral angles of φ = −57° and ψ = −47°.55 We will focus on peptide–CNT and helix–helix interactions, hence for simplicity we model the acylated and amidated N- and C-termini of the peptide (which promote head-to-tail stacking of helices) by maintaining the termini in a neutral (i.e. uncharged) state. Note that unless explicitly stated, we applied α-helical restraints to the backbone of nano-1 in order to preserve its secondary structure. We did this by placing distance restraints between the carbonyl oxygen of residue i and nitrogen of residue i + 4.The simple point charge56 (SPC) model was used for water molecules. In order to ensure electrical neutrality counter ions were added. We simulate a finite (18,0) single-walled CNT that is 14 Å in diameter and 43 Å in length. Note that nano-1 has an extended length of approximately 40 Å, therefore the peptide is able to interact with the CNT along its entire length. We model the CNT using the sp2 hybridised carbon parameters (C) in the GROMOS53A6 force field. This is the same atom type as that used for the C-delta atom of the aromatic tryptophan amino acid. To assess the robustness of our CNT, we also modelled the tube using aromatic carbons (CR1) that are also found in the GROMOS53A6 force field. This did not give rise to an observable difference in CNT–peptide interactions.
For all simulations the CNT was restrained in the centre of the simulation box using a force constant of 100 kJ mol−1 Å−2. The initial box dimensions were set to 70 × 70 × 70 Å3. Long-range electrostatic interactions were calculated using the particle mesh Ewald method with a 12 Å cutoff for the real space calculation.57 A cutoff of 12 Å was used for the van der Waals interactions. The temperature of the peptide, CNT, water and ions was coupled separately using the Berendsen thermostat58 with a coupling constant τT = 0.1 ps. The pressure was coupled anisotropically using the Berendsen barostat with a coupling constant τP = 1 ps. The time step for integration was 2 fs, and the LINCS algorithm was used to constrain bond lengths.59
Coarse grain model
We converted the energy minimised AT structure of nano-1 into a CG representation. CG simulations were performed as described in Bond et al.,49 with CG parameters for lipid molecules and water as in Marrink et al.41 In the CG model, one particle comprises approximately four heavy (i.e. non-hydrogen) atoms. Particles interact via screened Lennard-Jones and Coulombic potentials, while harmonic terms maintain bond lengths and angles. This model leads to an ∼100 fold increase in speed with respect to AT simulations, therefore allowing the simulation of large systems for longer time scales. We have extended the CG model developed by Marrink et al.41 to enable simulation of the self-assembly of membrane proteins with lipid bilayers, proteins and detergent into mixed micelles,60,61 and CNTs with lipids and detergent.23,62 A closely related CG model has also been used to explore the interactions of fullerenes with lipid bilayer membranes.45 Hence, the CG methodology is appropriate to study the adsorption of nano-1 onto CNTs.The CG nano-1 structure is displayed in Fig. 1c. We use the same CG treatment of the amino acid side chains and backbone particles as in Bond et al.49 As with the AT representation of nano-1, we model the acetylated and amidated N- and C-termini of the peptide by maintaining the termini in a neutral state. Harmonic distance restraints that have an equilibrium length of 6 Å and a force constant of 10 kJ mol−1 Å−2 were applied between backbone particles to mimic secondary structure H-bonds in the AT structure.49
The model for our CG CNT is as in Wallace and Sansom,12,23,62 where we use an approximate 4 : 1 mapping of the carbon atoms so that the hexagonal symmetry of the CNT lattice is preserved. We use Marrink's41 apolar (C) particles to model the CNT particles. In this study we simulate single-walled CG CNTs based on a finite AT (18,0) CNT of 14 Å in diameter and 43 Å in length. See Wallace and Sansom12,23,62 for further information on the CG model. The CNT was restrained in the centre of the simulation box using a force constant of 10 kJ mol−1 Å−2. The initial box dimensions were set at 110 × 110 × 110 Å3.
Coarse grain to atomistic conversion
In AT simulations sampling is frequently incomplete due to the computational demands of long simulations. In view of this, we have developed a multiscale approach that enables us to obtain equilibrated AT CNT/peptide complexes. Initially we performed 2 µs CG self-assembly simulations since the reduced complexity of the model allows access to longer time scales than AT simulation. Upon equilibration, we performed a CG to AT conversion of the CNT/peptide complex. In order to do this we created a library of 1000 AT nano-1 structures. This library comprised 500 structures taken from nano-1 simulated in water, and also 500 nano-1 structures from simulations of nano-1 adsorbed onto a CNT surface. Each nano-1 structure in the library is then placed sequentially on top of a particular peptide in the CG CNT/peptide complex via least-square fitting. The root mean square deviation (rmsd) of the 2 structures is then computed. The library structure with the smallest rmsd is selected as the AT equivalent of the CG peptide. This process is repeated for all peptides in the complex. Placing an AT CNT where the CG tube had previously been located may result in steric conflicts between the peptides and CNT. Instead, we slowly “grow” the CNT by concentrically placing an increasingly larger diameter tube into a cavity that is centred on the position of the original CG CNT. Upon placement of each tube <1000 steps of steepest decent energy minimisation are performed, allowing relaxation of any steric conflicts. This process ceases when an (18,0) AT CNT has been placed into the position of the original CG CNT and energy minimisation has been performed. As with the exclusively AT simulations, α-helical restraints were applied to the backbone of nano-1 unless explicitly stated.Results and discussion
A single nano-1 peptide interacting with a CNT
To investigate the adsorption profile of nano-1 with a CNT, we place a CNT in the centre of a simulation box and randomly position a peptide within the box. Our CG and AT nano-1 peptides are α-helical since circular dichroism (CD) experiments show that nano-1 has an α-helical conformation when solubilising CNTs in aqueous solution.27 In the AT simulations we ensure peptide α-helicity using restraints. This allows us to study the binding profile as a function of number of α-helical peptides that are adsorbed onto the CNT surface. It should be noted that α-helical restraints allow some deviation from a “rigid” α-helix, enabling “coiled-coil” geometries. Nevertheless, below we also study the effect of removing these helical restraints.In total we perform ten 2 µs CG simulations and four 40 ns AT simulations of a single nano-1 peptide interacting with a CNT in aqueous solution. In all our CG simulations, the peptide molecule made contact with the CNT within 200 ns, and then remained in contact with the nanotube for the rest of the simulation.
Fig. 2a shows the normalised number of contacts between the CG CNT and each of the heptads of the CG peptide. Phe-rich heptad d makes the most contact with the CNT, as is illustrated in Fig. 2c. In our analogous AT simulations, the peptide was placed just over 12 Å away from the CNT. Encouragingly, the AT adsorption profile (Fig. 2b) is similar to that observed for the CG system, with heptad d making the most contact with the CNT. Hence, the Phe side chains are in close contact with the CNT, as found in previous simulation studies.33,38 Following heptad d, the heptad that makes the next largest contribution to the peptide/CNT contacts is the hydrophobic heptad a. In both the CG and AT models, the peptide–CNT interaction energies are of the same order of magnitude, with CG and AT interaction energies of −456 ± 46 kJ mol−1 and −306 ± 17 kJ mol−1 respectively (averaged over 20 ns).
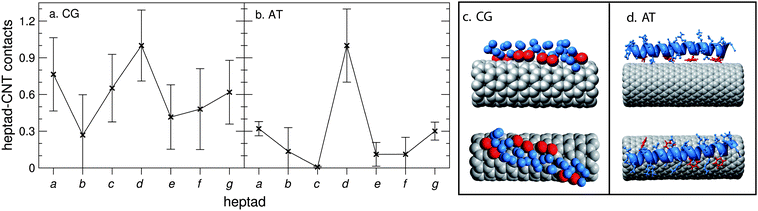 | ||
| Fig. 2 One nano-1 interacting with a CNT. Number of contacts between each heptad and the CNT for (a) CG and (b) AT simulations. We calculate a heptad–CNT contact as follows: the minimum distance between the CNT and each residue of a heptad is calculated. The residue and CNT are defined to be in contact if this minimum distance is less than 6 Å (a, CG simulations) or 3.5 Å (b, AT simulations). The data are normalised such that the heptad with the most CNT contacts is set equal to 1. In (a), nano-1 was placed randomly in the simulation box, and the data were averaged over 10 simulations, from 0.5 to 2 µs. In (b), nano-1 was placed just over 12 Å away from the CNT. The data were averaged over 4 simulations, from 30 to 40 ns. (c) CG and (d) AT snapshots showing nano-1 interacting with the CNT. In both (c) and (d) the peptide backbone is shown in blue, and the Phe side chains are shown in red. | ||
In Fig. 3 we calculate the contact angles between the Phe rings and the CNT surface for an AT CNT/peptide complex. The rings predominantly have a contact angle of 0 to 20°. This implies that the rings align approximately parallel with the CNT surface (Fig. 3b), which would enable π–π stacking.33 Note that the residues that comprise each heptad trace a slightly helical path along the peptide. Therefore, in order for all the Phe rings to align parallel with the CNT surface nano-1 adopts a more “coiled-coil” geometry38 (as observed in Fig. 3b). There is also a small frequency of rings that have a large contact angle with the CNT. This can be explained by examining the inset shown in Fig. 3a. These large contact angles originate primarily from Phe 1 (see Fig. 3c), the ring located at one end of the peptide. Large contact angles arise when nano-1 has a canonical α-helical geometry, with the ring at one end of the peptide (Phe 1) unable to maintain a parallel orientation with respect to the CNT surface.
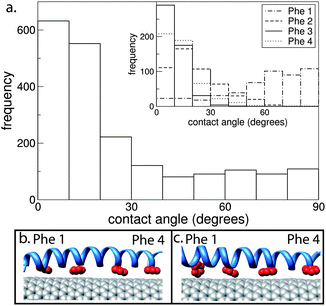 | ||
| Fig. 3 (a) Contact angle between the Phe rings and CNT for an AT simulation where one nano-1 is adsorbed onto a CNT. The contact angle was calculated between the normal vectors of the CNT surface and the plane of the Phe ring. Hence, an angle of 0° corresponds to the Phe ring aligning parallel with the CNT surface. Contact angles were calculated from 30 to 40 ns. The inset shows the contact angle for each individual Phe ring. (b) and (c) are snapshots of the Phe rings (red) interacting with the CNT. In (b) all Phe rings have contact angles between 0 and 20°. In (c) Phe 1 has a contact angle of 74°. | ||
Multiple nano-1 peptides interacting with a CNT
In Fig. 4a we increase the number of nano-1 peptides that are adsorbed onto the CNT surface in order to study the effect of peptide number on the adsorption profile. We performed simulations with various different initial configurations (Fig. 4c–e) in order to examine the robustness of these adsorption profiles. As can be observed, the adsorption profile for six CG peptides adsorbed onto the CNT surface is very similar to that observed for just one peptide (Fig. 2), with heptad d making the highest number of contacts with the CNT. Furthermore, the adsorption profile is unaffected upon a change in starting configuration. For the “circle Phe in” and “circle random” configurations the peptides adsorb onto the CNT with their N- and C-termini at opposite ends of the tube. The peptides remain in this parallel configuration for the rest of the simulation.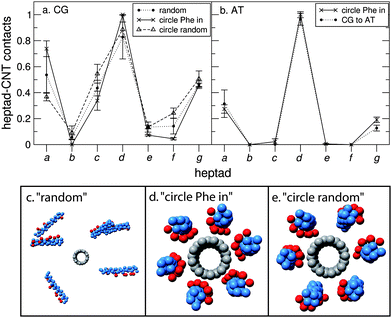 | ||
| Fig. 4 Six nano-1 interacting with a CNT. Number of contacts between each heptad and the CNT for (a) CG and (b) AT simulations. We calculate a heptad–CNT contact as described in Fig. 2. The data are normalised such that the heptad with the most CNT contacts is set equal to 1. In (a), contact plots are shown for three initial peptide configurations: “random” (c) where peptides are placed randomly in the simulation box (the minimum initial peptide–CNT distance was 20 Å); “circle Phe in” (d) where peptides are placed in a circle around the CNT with the Phe residues facing towards the tube; “circle random” (e) where peptides are placed randomly in a circle around the CNT. For the “circle Phe in” and “circle random” configurations the peptides are parallel such that their N- and C-termini are at opposite ends of the CNT. Two simulations were performed for each initial configuration. Data were averaged from 0.5 to 2 µs (circle Phe in) and 2.5 to 4 µs (“random” and “circle random”). In (b), contact plots are shown for two initial peptide configurations: “circle Phe in” and “CG to AT”. For the “CG to AT” protocol (described in the Methods section), the starting AT configuration was obtained after 2 µs of CG simulation. For both starting configurations the peptides are in a parallel arrangement around the CNT. Two simulations were performed for each initial configuration. Data were averaged from 30 to 40 ns (circle Phe in) and 10 to 20 ns (CG to AT). (c–e) CG snapshots of the different starting configurations implemented. | ||
For the “random” initial configuration the peptides do not necessarily adsorb onto the CNT in a parallel configuration. In one simulation (run 1, Fig. 5a–d), 5 out of 6 peptides initially formed a peptide bundle (Fig. 5b), with the hydrophobic Phe side chains pointing towards the centre of the bundle. This bundle encountered the CNT/peptide complex at approximately 100 ns (Fig. 5c). The bundle then proceeded to rearrange in order to encapsulate the CNT, with 5 out of 6 peptides adsorbed onto the CNT cylindrical outer surface in a parallel arrangement, and the remaining peptide (orange peptide in Fig. 5d) adsorbed onto the tube end. All of the peptides remained in contact with the CNT for the rest of the simulation (4 µs in length), suggesting that it is more energetically favourable for the peptides to interact with the CNT/peptide complex rather than be in the 5-peptide bundle.
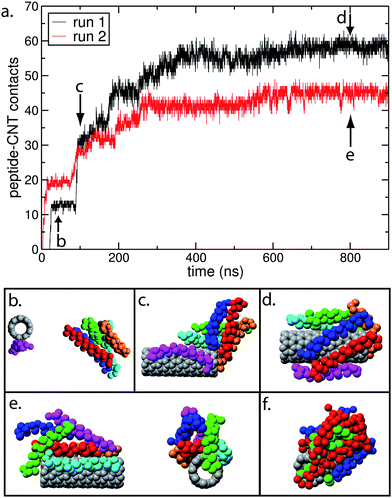 | ||
| Fig. 5 (a) Number of contacts between the peptides and the CNT as a function of time, for the CG “random” nano-1 simulations shown in Fig. 4. We calculate a peptide–CNT contact as follows: the minimum distance between the CNT and each peptide residue is calculated. The residue and CNT are defined to be in contact if this distance is less than 6 Å. (b) to (f) are snapshots of key events during the adsorption process for run 1 (b–d) and run 2 (e and f). In (b)–(e) the backbone of each peptide is coloured differently. In (f) the peptide backbone is red when in a parallel configuration, and blue when in an antiparallel configuration. The green spheres are the hydrophobic Phe side chains. | ||
In the other “random” configuration simulation (run 2, Fig. 5a, e and f), some peptides adsorb in an antiparallel configuration (Fig. 5f). The peptides that are in an antiparallel orientation experience side chain–side chain electrostatic repulsion, resulting in bundle formation (red peptides in Fig. 5f) on the CNT with the hydrophobic Phe residues buried in the centre of the bundle. Hence, there are fewer contacts for run 2 in Fig. 5a since several peptides in this bundle are not adsorbed directly onto the CNT surface.
We also performed AT simulations with six peptides placed around a CNT, where the hydrophobic surface of each peptide faces towards the tube (Fig. 4b). Once again, the CG and AT models produce very similar results, with the hydrophobic surface of the peptide contacting the CNT.
Next we wish to study AT CNT/nano-1 complexes that comprise a higher density of peptides. In order to do this, and to ensure well equilibrated AT systems, we have employed a multiscale approach. We initially performed 2 µs CG simulations, allowing equilibration of the CNT/nano-1 complexes. Next the CG to AT conversion was carried out as described in the Methods section. Fig. 6 shows snapshots of the CG complex prior to conversion, and the AT complex after conversion. As can be observed, the CG and AT structures are similar. The root mean square deviation (rmsd) between the Cα particles of the two structures is 1.8 Å. Additionally, the Phe side chains are located towards the CNT in both models. In order to validate this technique, we have plotted the adsorption profile for six peptides adsorbed onto a CNT (Fig. 4b), where the CNT/nano-1 complex was obtained via a CG to AT conversion. As can be seen, the profile is very similar to that obtained for the exclusively AT simulation.
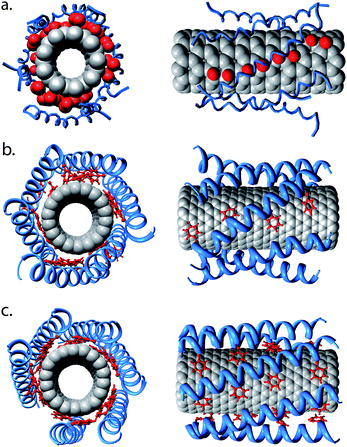 | ||
| Fig. 6 CG to AT conversion. (a) An equilibrated CG CNT/nano-1 complex, with 6 peptides interacting with the tube surface. The snapshot was taken after a 2 µs self-assembly simulation. (b) The AT complex resulting from conversion of the CG structure shown in (a). This structure has been energy minimised. The CG to AT protocol is described in the Methods section. (c) The CNT/nano-1 complex after a 20 ns simulation. The α-helical backbone is shown in blue, and the Phe side chains are shown in red. Note that in (b) and (c) the peptides have α-helical restraints. | ||
Peptide α-helical stability
We will now utilise this CG to AT methodology to examine the stability of the α-helical secondary structure of nano-1 as a function of number of peptides adsorbed onto the CNT surface. As mentioned above, in all prior AT simulations the peptide was restrained to remain α-helical. However, we will now remove the helical restraints to test for helical stability. For a single AT nano-1 peptide simulated in water in the absence of a CNT, we find that the peptide loses its α-helicity. In order to examine how robust this result is, we also performed analogous simulations with the GROMOS53A6 force field at the lower temperature of 300 K, and with the OPLS force field51,52 at 300 K and 323 K. Again, we find that the peptide loses its α-helicity (data shown in ESI, Fig. S1†). This behaviour is unsurprising since nano-1 is amphiphilic when α-helical and hence this conformation should be energetically unfavourable for a single peptide in water. Indeed, at low concentration of nano-1 in aqueous solution, CD shows that nano-1 is not α-helical.27In Fig. 7a we plot the percentage of α-helicity versus the number of peptides adsorbed onto the CNT surface. For all simulations, the initial AT complexes were obtained from a CG to AT conversion. After the conversion process, the complexes were allowed to relax for 20 ns before the α-helical restraints were removed. After removal of the restraints, all the complexes were simulated for a further 40 ns. In all cases, the peptides remained within the complex for the duration of the simulations. We measured the percentage of peptide α-helicity (evaluated using DSSP55) from 30 to 40 ns following the removal of the restraints.
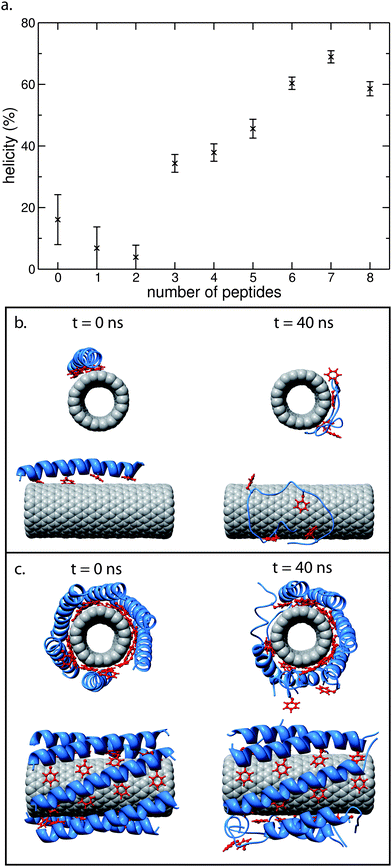 | ||
| Fig. 7 (a) AT nano-1 α-helicity as a function of number of adsorbed peptides onto the outer surface of a CNT. For all simulations, the initial AT configurations were obtained from CG to AT conversions. These conversions were based on equilibrated CG simulations of 2 µs in length. The AT CNT/peptide complexes were equilibrated for 20 ns prior to removal of the peptide α-helical restraints. This time period enabled all peptides to interact with the CNT, forming a stable CNT/peptide complex. Each complex was then simulated for a further 40 ns following the removal of the α-helical restraints, with the data shown in (a) averaged from 30 to 40 ns. Peptide α-helicity was evaluated using DSSP.55 Each data point in (a) is based on one simulation. (b) and (c) are snapshots taken at the start (0 ns) and end (40 ns) of the unrestrained simulations, for one peptide (b) and six peptides (c) in the CNT/peptide complex. Prior to removal of α-helical restraints, all peptides in the CNT/multi-peptide complexes are parallel such that their N- and C-termini are at opposite ends of the CNT. | ||
As for a single peptide simulated in water, when one peptide is adsorbed onto a CNT surface, the peptide loses its α-helical secondary structure within 40 ns. This loss of secondary structure is slightly less rapid for the peptide adsorbed onto the CNT (see ESI, Fig. S2†).
As observed in Fig. 7, the stability of nano-1 increases upon increasing the number of peptides on the CNT surface from one/two to seven peptides. This increase in stability is also seen clearly in the snapshots shown in Fig. 7b and c. Hence, nano-1 α-helicity is stabilised by inter-peptide interactions. In Fig. 8 we calculate the heptad–heptad contacts within a CNT/multi-peptide complex. In both the CG and AT simulations, heptads a and d, and heptads e and g make dominant contributions to the inter-peptide interactions. Note that nano-1 was designed specifically so that heptads e (negative) and g (positive) interact favourably.
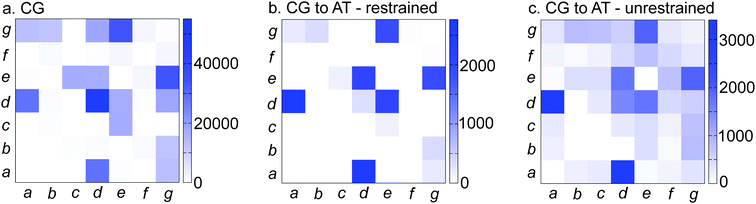 | ||
| Fig. 8 Heptad–heptad contacts within a CNT/6 nano-1 complex for (a) CG and (b and c) AT simulations. We calculate a heptad–heptad contact as follows: the minimum distance between two residues of different peptides is calculated. The residues are defined to be in contact if this minimum distance is less than 6 Å (a, CG simulations) or 3.5 Å (b and c, AT simulations). For the CG simulation (a), the peptides were initially placed in a circle around the CNT with the Phe residues facing towards the tube. Data were averaged from 0.5 to 2 µs. In (b), the initial AT configuration was obtained from a CG to AT conversion of the CG simulation in (a) at 2 µs. The AT CNT/peptide complex was equilibrated for 20 ns with peptide helical restraints. Data were averaged from 10–20 ns. For (c), the helical restraints in (b) were removed and the complex was then simulated for a further 40 ns. Data were averaged from 30–40 ns. In (a), (b) and (c) the peptides are parallel such that their N- and C-termini are at opposite ends of the CNT. | ||
In the CG simulations, heptad d also interacted with neighbouring d heptads. It should be noted that in Fig. 8, all particles of a residue are considered when calculating a potential contact. However, the interaction profiles are unchanged when only side chain particles are considered.
When the number of peptides on the CNT surface was increased to eight, this corresponds to the “saturation point” in the CG model, i.e. the point at which no more peptides can adsorb directly onto the CNT surface. However, in the AT model the saturation point is slightly lower (∼7) due to the larger conformational degree of freedom of the peptides. Hence, upon conversion from the CG to AT model when eight peptides are within the CNT/peptide complex, one peptide is subsequently pushed out from the inner shell of peptides surrounding the tube. This more disordered complex explains the loss of α-helicity seen in Fig. 7a. Note that we have extended several simulations in Fig. 7 up to 100 ns (see ESI, Fig. S3†). We find that the α-helical secondary structure of nano-1 decreases a little further in this period.
Conclusions
In conclusion, we have performed CG, AT and multiscale MD simulations to probe the adsorption profile of nano-1 with CNTs. The advantage of our approach is that the coarse-grained model allows access to larger length and longer time scales, whilst the AT-level model gives a more detailed description of interactions. By combining the two levels of description, one can perform AT simulations of well equilibrated larger systems obtained from CG simulations.We have shown that both our CG and AT models give rise to similar peptide–CNT adsorption profiles, with the hydrophobic Phe-rich heptad d making the most contacts with the CNT, as anticipated from the design of the peptide. This is in agreement with previous simulation studies.33,38 The adsorption profile is unchanged upon increasing the number of peptides on the CNT surface up to the saturation point.
Interestingly, we found that without application of α-helical restraints on a single AT nano-1 peptide adsorbed onto a CNT surface, the peptide loses almost all of its α-helicity. We systematically increased the number of peptides on the CNT surface up to the saturation point and found that the nano-1 α-helical secondary structure is stabilised by inter-peptide interactions. When nano-1 is in an α-helical conformation, the Phe and Val residues form the hydrophobic face of the helix, while the remaining residues form the hydrophilic face. Thus, nano-1 has a maximum amphiphilicity when in an α-helix. Hence, the loss of peptide secondary structure that we observe at low nano-1 concentration will reduce the amphiphilicity of the peptide, and therefore prevent solubilisation of the CNTs. This result complements experimental data that show that decreasing nano-1 concentration leads to a decrease in α-helical content.27 Furthermore, our finding that nano-1 α-helical conformation is stabilised when at high concentration on a CNT surface is in agreement with a recent simulation study of a (6,6) and an (8,8) CNT with a saturated shell of nano-1 peptides. In this study Chiu et al.40 found that nano-1 is also α-helical.
In an MD simulation study also by Chiu et al.,38 it was found that a single nano-1 peptide retains between 65 and 95% of it α-helicity when adsorbed onto a (6,6) CNT of 8.1 Å in diameter. However, the amount of α-helicity decreases when the peptide interacts with a planar sheet of graphite. Here we show that a single peptide adsorbed onto an (18,0) CNT surface loses almost all of its α-helicity. An explanation for this loss of α-helicity is that our CNTs are less highly curved than the tubes studied by Chiu et al.,38 thus the peptide is unable to match its hydrophobic face against the CNT surface. This issue of peptide α-helical stability on curved surfaces is an interesting issue which requires further study. It is possible that modifying the sequence of an amphipathic α-helix to include flexible regions, e.g.via glycine residues, may modulate stability on a curved surface. One can envisage that if a peptide is only able to maintain its secondary structure when adsorbed onto a CNT of a certain diameter, then it may be capable of selectively solubilising CNTs. We also note that in our simulations the CNT is open ended and so water can flow into the tube. This may conceivably modulate the energetics of CNT/peptide interactions.
In conclusion, findings reported here shed light on the adsorption profile of nano-1 with CNTs, and may guide future research on the selective solubilisation and targeting of CNTs to specific cells.
Acknowledgements
This work was supported via a grant from the EPSRC and BBSRC via the Bionanotechnology IRC, and via the James Martin 21st Century School.Notes and references
- R. H. Baughman, A. A. Zakhidov and W. A. de Heer, Science, 2002, 297, 787 CrossRef CAS.
- J. T. Hu, O. Y. Min, P. D. Yang and C. M. Lieber, Nature, 1999, 399, 48 CrossRef CAS.
- S. J. Tans, A. R. M. Verschueren and C. Dekker, Nature, 1998, 393, 49 CrossRef CAS.
- Y. Cui, Q. Wei, H. Park and C. M. Lieber, Science, 2001, 293, 1289 CrossRef CAS.
- B. Vigolo, A. Penicaud, C. Coulon, C. Sauder, R. Pailler, C. Journet, P. Bernier and P. Poulin, Science, 2000, 290, 1331 CrossRef CAS.
- A. B. Dalton, S. Collins, E. Munoz, J. Razal, V. H. Ebron, J. P. Ferraris, J. N. Coleman, B. G. Kim and R. H. Baughman, Nature, 2003, 423, 703 CrossRef CAS.
- R. H. Baughman, C. X. Cui, A. A. Zakhidov, Z. Iqbal, J. N. Barisci, G. M. Spinks, G. G. Wallace, A. Mazzoldi, D. De Rossi, A. Rinzler, O. Jaschinski, S. Roth and M. Kertesz, Science, 1999, 284, 1340 CrossRef CAS.
- R. J. Chen, S. Bangsaruntip, K. A. Drouvalakis, N. W. S. Kam, M. Shim, Y. Li, W. Kim, P. J. Utz and H. Dai, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 4984 CrossRef CAS.
- P. Cherukuri, S. M. Bachilo, S. H. Litovsky and R. B. Weisman, J. Am. Chem. Soc., 2004, 126, 15638 CrossRef CAS.
- X. Chen, A. Kis, A. Zettl and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 8218 CrossRef CAS.
- I. U. Vakarelski, S. C. Brown, K. Higashitani and B. M. Moudgil, Langmuir, 2007, 23, 10893 CrossRef CAS.
- E. J. Wallace and M. S. P. Sansom, Nano Lett., 2008, 8, 2751 CrossRef CAS.
- W. Zhao, C. H. Song and P. E. Pehrsson, J. Am. Chem. Soc., 2002, 124, 12418 CrossRef CAS.
- A. Star and J. F. Stoddart, Macromolecules, 2002, 35, 7516 CrossRef CAS.
- K. D. Ausman, R. Piner, O. Lourie, R. S. Ruoff and M. Korobov, J. Phys. Chem. B, 2000, 104, 8911 CrossRef CAS.
- J. L. Bahr, J. P. Yang, D. V. Kosynkin, M. J. Bronikowski, R. E. Smalley and J. M. Tour, J. Am. Chem. Soc., 2001, 123, 6536 CrossRef CAS.
- J. Chen, M. A. Hamon, H. Hu, Y. S. Chen, A. M. Rao, P. C. Eklund and R. C. Haddon, Science, 1998, 282, 95 CrossRef CAS.
- M. J. O'Connell, S. M. Bachilo, C. B. Huffman, V. C. Moore, M. S. Strano, E. H. Haroz, K. L. Rialon, P. J. Boul, W. H. Noon, C. Kittrell, J. P. Ma, R. H. Hauge, R. B. Weisman and R. E. Smalley, Science, 2002, 297, 593 CrossRef CAS.
- M. F. Islam, E. Rojas, D. M. Bergey, A. T. Johnson and A. G. Yodh, Nano Lett., 2003, 3, 269 CrossRef CAS.
- V. C. Moore, M. S. Strano, E. H. Haroz, R. H. Hauge, R. E. Smalley, J. Schmidt and Y. Talmon, Nano Lett., 2003, 3, 1379 CrossRef CAS.
- K. Yurekli, C. A. Mitchell and R. Krishnamoorti, J. Am. Chem. Soc., 2004, 126, 9902 CrossRef CAS.
- R. Qiao and P. C. Ke, J. Am. Chem. Soc., 2006, 128, 13656 CrossRef CAS.
- E. J. Wallace and M. S. P. Sansom, Nano Lett., 2007, 7, 1923 CrossRef CAS.
- S. Q. Wang, E. S. Humphreys, S. Y. Chung, D. F. Delduco, S. R. Lustig, H. Wang, K. N. Parker, N. W. Rizzo, S. Subramoney, Y. M. Chiang and A. Jagota, Nat. Mater., 2003, 2, 196 CrossRef CAS.
- R. R. Johnson, A. T. C. Johnson and M. L. Klein, Nano Lett., 2008, 8, 69 CrossRef CAS.
- W. D. Kohn, C. T. Mant and R. S. Hodges, J. Biol. Chem., 1997, 272, 2583 CrossRef CAS.
- G. R. Dieckmann, A. B. Dalton, P. A. Johnson, J. Razal, J. Chen, G. M. Giordano, E. Munoz, I. H. Musselman, R. H. Baughman and R. K. Draper, J. Am. Chem. Soc., 2003, 125, 1770 CrossRef CAS.
- V. Zorbas, A. L. Smith, H. Xie, A. Oritz-Acevedo, A. B. Dalton, G. R. Dieckmann, R. K. Draper, R. H. Baughmann and I. H. Musselman, J. Am. Chem. Soc., 2005, 127, 12323 CrossRef CAS.
- V. Zorbas, A. Oritz-Acevedo, A. B. Dalton, M. M. Yoshida, G. R. Dieckmann, R. K. Draper, R. H. Baughman, M. Jose-Yacaman and I. H. Musselman, J. Am. Chem. Soc., 2004, 126, 7222 CrossRef CAS.
- A. B. Dalton, A. Oritz-Acevedo, V. Zorbas, E. Brunner, W. M. Sampson, S. Collins, J. M. Razal, M. M. Yoshida, R. H. Baughman, R. K. Draper, I. H. Musselman, M. Jose-Yacaman and G. R. Dieckmann, Adv. Funct. Mater., 2004, 14, 1147 CrossRef CAS.
- H. Xie, A. Ortiz-Acevedo, V. Zorbas, R. H. Baughman, R. K. Draper, I. H. Musselman, A. B. Dalton and G. R. Dieckmann, J. Mater. Chem., 2005, 15, 1734 RSC.
- B. Trzaskowski, A. F. Jalbout and L. Adamowicz, Chem. Phys. Lett., 2006, 430, 97 CrossRef CAS.
- S. D. Tomásio and T. R. Walsh, Mol. Phys., 2007, 105, 221 CrossRef.
- W. J. Fan, J. Zeng and R. Q. Zhang, J. Chem. Theory Comput., 2009, 5, 2879 CrossRef CAS.
- S. M. Tomásio and T. R. Walsh, J. Phys. Chem. C, 2009, 113, 8778 CrossRef CAS.
- J. W. Shen, T. Wu, Q. Wang and Y. Kang, Biomaterials, 2008, 29, 3847 CrossRef CAS.
- Y. Kang, Y. C. Liu, Q. Wang, J. W. Shen, T. Wu and W. J. Guan, Biomaterials, 2009, 30, 2807 CrossRef CAS.
- C. Chiu, G. R. Dieckmann and S. O. Nielsen, J. Phys. Chem. B, 2008, 112, 16326 CrossRef CAS.
- S. R. Friling, R. Notman and T. R. Walsh, Nanoscale, 2010, 2, 98 RSC.
- C. C. Chiu, G. R. Dieckmann and S. O. Nielsen, Biopolymers, 2009, 92, 156 CrossRef CAS.
- S. J. Marrink, A. H. de Vries and A. E. Mark, J. Phys. Chem. B, 2004, 108, 750 CrossRef CAS.
- J. C. Shelley, M. Y. Shelley, R. C. Reeder, S. Bandyopadhyay and M. L. Klein, J. Phys. Chem. B, 2001, 105, 4464 CrossRef CAS.
- B. Smit, P. A. J. Hilbers, K. Esselink, L. A. M. Rupert, N. M. Vanos and A. G. Schlijper, Nature, 1990, 348, 624 CrossRef CAS.
- V. Tozzini, Curr. Opin. Struct. Biol., 2005, 15, 144 CrossRef CAS.
- J. Wong-Ekkabut, S. Baoukina, W. Triampo, I. M. Tang, D. P. Tieleman and L. Monticelli, Nat. Nanotechnol., 2008, 3, 363 CrossRef CAS.
- G. S. Ayton, W. G. Noid and G. A. Voth, Curr. Opin. Struct. Biol., 2007, 17, 192 CrossRef CAS.
- H. J. C. Berendsen, D. van der Spoel and R. van Drunen, Comput. Phys. Commun., 1995, 91, 43 CrossRef CAS.
- E. Lindahl, B. Hess and D. van der Spoel, J. Mol. Model, 2001, 7, 306 CAS.
- P. J. Bond, C. L. Wee and M. S. P. Sansom, Biochemistry, 2008, 47, 11321 CrossRef CAS.
- C. Oostenbrink, A. Villa, A. E. Mark and W. F. Van Gunsteren, J. Comput. Chem., 2004, 25, 1656 CrossRef CAS.
- W. L. Jorgensen, D. S. Maxwell and J. TiradoRives, J. Am. Chem. Soc., 1996, 118, 11225 CrossRef CAS.
- G. A. Kaminski, R. A. Friesner, J. Tirado-Rives and W. L. Jorgensen, J. Phys. Chem. B, 2001, 105, 6474 CrossRef CAS.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605 CrossRef CAS.
- W. L. DeLano, The PyMOL Molecular Graphics System, DeLano Scientific, Palo Alto, CA, USA, 2002 Search PubMed.
- W. Kabsch and C. Sander, Biopolymers, 1983, 22, 2577 CrossRef CAS.
- H. J. C. Berendsen, J. P. M. Postma, W. F. Van Gunsteren, J. Hermans, Interaction Models for Water in Relation to Protein Hydration, in Intermolecular Forces, Reidel, Dordrecht, The Netherlands, 1981, p. 331 Search PubMed.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089 CrossRef CAS.
- H. J. C. Berendsen, J. P. M. Postma, W. F. Van Gunsteren, A. Dinola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684 CrossRef CAS.
- B. Hess, H. Bekker, H. J. C. Berendsen and J. Fraaije, J. Comput. Chem., 1997, 18, 1463 CrossRef CAS.
- P. J. Bond and M. S. P. Sansom, J. Am. Chem. Soc., 2006, 128, 2697 CrossRef CAS.
- P. J. Bond, J. Holyoake, A. Ivetac, S. Khalid and M. S. P. Sansom, J. Struct. Biol., 2007, 157, 593 CrossRef CAS.
- E. J. Wallace and M. S. P. Sansom, Nanotechnology, 2009, 20, 045101 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Details of the OPLS simulations and peptide α-helicity. See DOI: 10.1039/b9nr00355j |
| This journal is © The Royal Society of Chemistry 2010 |