A hydrogel-based enzyme-loaded polymersome reactor
Hans-Peter M.
De Hoog
ab,
Isabel W. C. E.
Arends
*b,
Alan E.
Rowan
a,
Jeroen J. L. M.
Cornelissen†
*a and
Roeland J. M.
Nolte
a
aInstitute for Molecules and Materials, Radboud University Nijmegen, Toernooiveld 1, 6525, ED, Nijmegen, The Netherlands. E-mail: J.J.L.M.Cornelissen@tnw.utwente.nl; Fax: +31 53 489 4645; Tel: +31 53 489 4380
bDepartment of Biotechnology, Delft University of Technology, Julianalaan 136, 2628, DL, Delft, The Netherlands. E-mail: I.W.C.E.Arends@tudelft.nl
First published on 17th March 2010
Abstract
In this study we report the immobilization of enzyme-containing polymersomes into a macromolecular hydrogel. Whereas free enzyme shows progressive leakage from the hydrogel in a period of days, leakage of the polymersome-protected enzyme is virtually absent. The preparation of the hydrogel occurs under mild conditions and does not inhibit the activity of the encapsulated enzymes nor does it affect the structure of the polymersomes. The stability of the polymersome hydrogel architecture is demonstrated by the facile recycling of the polymersomes and their use in repeated reaction cycles. A ‘continuous-flow polymersome reactor’ is constructed in which substrate is added to the top of the reactor and product is collected at the bottom. This set-up allows the use of different enzymes and the processing of multiple substrates, as is demonstrated by the conversion of 2-methoxyphenyl acetate to tetraguaiacol in a reactor loaded with polymersome hydrogels containing the enzymes Candida antarctica lipase B (CALB) and glucose oxidase (GOx).
Introduction
Polymersomes are polymer capsules with a bilayer membrane, which are formed by the self-assembly of amphiphilic block copolymers.1 Their ability to function as nanoreactors by the encapsulation of enzymes has been demonstrated for different types of polymersomes and is often facilitated by the incorporation of membrane-spanning channel proteins, which facilitates the diffusion of substrates into their aqueous interior.2,3 These studies range from the impact encapsulation has on enzyme functionality to the construction of complex cascade reaction systems, including trigger-sensitive systems.3–6 In recent years, we have reported on the successful immobilization of various enzymes into polymersomes assembled from block copolymers of polystyrene–poly(isocyanoalanine-(2-thiophene-3-yl-ethyl amide)) (PS-PIAT). An interesting feature of these polymersomes is their intrinsic porosity to small organic substrates.7 The kinetics of cascade reactions catalyzed by the encapsulated enzymes has been studied and it was shown that upon encapsulation the enzymes could still function as highly enantioselective catalysts for the synthesis of chiral compounds.8,9The purpose of the present study is the study of a property of polymersomes that is often mentioned, but scarcely investigated, namely their recyclability. Although the membrane stability of polymersomes is enhanced compared to liposomes, the recyclability of the freely dispersed polymersomes may still be limited. In preliminary experiments we observed that recycling of CALB-loaded polymersomes resulted in a steady decrease of enzyme activity (40% loss after filtration 8 times) (unpublished data). We therefore decided to immobilize the polymersomes in a suitable matrix and to study the recyclability of this system as well as its ability to function in a reactor set-up. A prerequisite for the immobilization of polymersomes into a carrier matrix is that the inner aqueous compartment of the polymersomes is not affected. Thus, ideally the matrix should be constructed from high molecular weight polymers that are unable to penetrate the polymersome membrane. Obviously the morphology of the polymersomes should not be changed by the process and neither should the activity of the enzymes.
A viable option for immobilization was to use polysaccharide precursors to produce hydrogels. These compounds generally have a high molecular weight and easily trap water upon cross-linking, leading to gelation. A recent study reported the functionalization of hyaluronic acid (a polysaccharide consisting of disaccharide units of α-1,4-glucuronic acid and β-1,3-N-acetyl glucosamine; HA) with either azide or acetylene moieties and its subsequent gelation by the CuI-catalyzed Huisgen 1,3-dipolar cycloaddition reaction (more commonly referred to as ‘clicking’).10 It was decided to start from this precursor as HA is a natural product, which is readily available and the derivatization process is relatively easy. Most importantly, gelation by ‘clicking’ is tolerant to the use of biological compounds, such as enzymes.11 When successful the immobilization could facilitate the use of the polymersomes in a flow-reactor set-up, in which substrate is added to the top of the reactor and product is collected at the bottom, or in a batch-like process, in which the formed product is collected from the supernatant.
So far, no studies have been reported on the immobilization of polymersomes into a polymer or hydrogel matrix. Comparable architectures, such as nanoparticles,12,13 liposomes,14–16 and cells,17,18 have however been studied for immobilization. For cells, as well as for the immobilization of proteins or enzymes, the application is primarily focused on the controlled release of the trapped species,19–21 although cells and enzymes have also been studied with regard to their catalytic behaviour.22–26 Encapsulation of the enzyme in a polymersome and subsequent gelation of the polymersome solution will provide a gentle means of trapping the enzyme, as the polymersomes will prevent the release of the enzyme and the hydrogel the leakage of the polymersomes (Scheme 1).
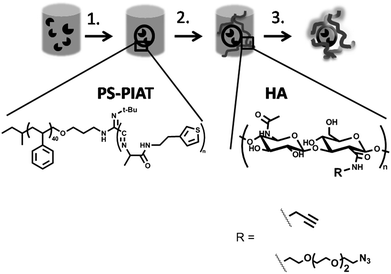 | ||
| Scheme 1 Procedure for the immobilization of enzyme-loaded polymersomes into a polysaccharide hydrogel matrix: 1. Addition of PS-PIAT polymer to the enzyme solution and subsequent filtration to yield enzyme-loaded polymersomes; 2. Addition of the enzyme-loaded polymersomes to a freeze-dried sample of the functionalized hyaluronic acid (HA); 3. Addition of the Cu-catalyst to create the hydrogel embedding the polymersomes. | ||
Results and discussion
Synthesis and characterization of enzyme-loaded polymersome hydrogel
Following a slight modification of a published procedure,10 HA (MW = 1200 kDa) was functionalized with an azide or an acetylene function on its glucuronic acid moieties by a peptide coupling with either the bifunctional oligoethylene linker 1-amino-3,6,9-trioxaundecan-11-azide (ATA), or propargyl amine (PA). The successful incorporation of the functionalities was demonstrated by IR (Fig. 1).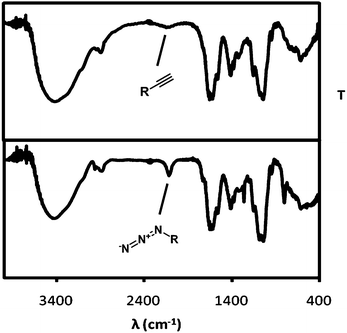 | ||
| Fig. 1 (Top) IR spectrum of hyaluronic acid (HA) provided with an acetylene function after reaction with propargylamine (PA). (Bottom) HA with an azide functionality obtained after reaction with 1-amino-3,6,9-trioxaundecan-11-azide (ATA). | ||
The ultimate test for the functionalization of the HA was its gelation upon addition of the click solution. A 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 solution of PA–HA and ATA–HA was prepared (8 mg ml−1) and gelation was induced by the addition of the Cu catalyst (2 mM; 0.1 monomer equivalents). The solution was found to gelate within 2–3 min, as was shown by inversion of the test tube (Fig. 2A). Transmission electron microscopy (TEM) on the gel matrix showed an opaque featureless structure with occasional electron-dense spots that are presumably crystallized salts (Fig. 2B). These could be removed by incubating the gel repeatedly with Milli-Q water. This method was easily extended to CALB-loaded polymersomes by dissolving the derivatized polysaccharides in the polymersome dispersion and adding the Cu-catalyst. The effective immobilization of the polymersomes in the gel was best visualized by TEM-imaging or confocal fluorescence microscopy (CFM). TEM showed dense spherical objects in the gelated samples with the expected diameters, but, in contrast to free polymersomes (Fig. 2C), with a somewhat blurred interface (Fig. 2D). This picture also shows that, within the experimental time-frame, the polymersomes remain intact when the polysaccharide derivatives are dissolved in the polymersome dispersion (complete dissolution of the polysaccharide was generally achieved overnight). The minor aggregation observed seems to be the result of the force enacted upon the polymersomes by the filtration procedure, as observed previously for non-gelated polymersome dispersions.9,27
:1 solution of PA–HA and ATA–HA was prepared (8 mg ml−1) and gelation was induced by the addition of the Cu catalyst (2 mM; 0.1 monomer equivalents). The solution was found to gelate within 2–3 min, as was shown by inversion of the test tube (Fig. 2A). Transmission electron microscopy (TEM) on the gel matrix showed an opaque featureless structure with occasional electron-dense spots that are presumably crystallized salts (Fig. 2B). These could be removed by incubating the gel repeatedly with Milli-Q water. This method was easily extended to CALB-loaded polymersomes by dissolving the derivatized polysaccharides in the polymersome dispersion and adding the Cu-catalyst. The effective immobilization of the polymersomes in the gel was best visualized by TEM-imaging or confocal fluorescence microscopy (CFM). TEM showed dense spherical objects in the gelated samples with the expected diameters, but, in contrast to free polymersomes (Fig. 2C), with a somewhat blurred interface (Fig. 2D). This picture also shows that, within the experimental time-frame, the polymersomes remain intact when the polysaccharide derivatives are dissolved in the polymersome dispersion (complete dissolution of the polysaccharide was generally achieved overnight). The minor aggregation observed seems to be the result of the force enacted upon the polymersomes by the filtration procedure, as observed previously for non-gelated polymersome dispersions.9,27
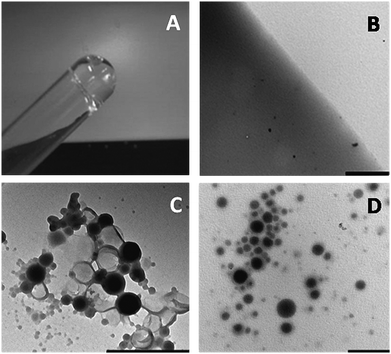 | ||
| Fig. 2 Photograph of the 1:1 solution of azide-functionalized HA and acetylene HA directly after gelation (A). TEM picture of the same hydrogel (B). TEM picture of CALB-loaded polymersomes deposited from solution (C). CALB-polymersomes immobilized in the HA hydrogel (D). Bars correspond to 1 μm. | ||
To prove the actual immobilization of the CALB-polymersomes, i.e. the restriction of their translational movement by trapping in the gel matrix, their mobility was studied with respect to time by CFM. To this end the fluorescence emanating from the polymersomes was detected at λem = 505 nm.8,28 Samples of the CALB-polymersomes, after and before gelation of the dissolved polysaccharide, were placed on a CFM glass slide and inspected at an elevation of 5 μm from the surface of the probe (Fig. 3). Whereas a solution of the polysaccharide and the polymersomes showed changes in the number and locations of the particles over a period of 15 min, the gelated solutions did not display such behaviour. The particles appeared to be situated at virtually fixed positions over a 15 min period, as a result of their trapping in the hydrogel matrix.
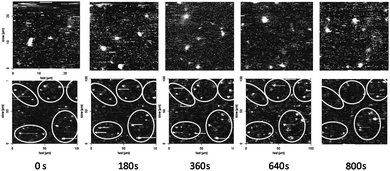 | ||
| Fig. 3 CFM images (λex = 480 nm, λem = 505 nm) of CALB-polymersomes taken at 3 min time-intervals in a HA solution before gelation (upper series) and after gelation (lower series). The width of the images in the lower series was increased from 25 to 100 μm to visualize a sufficient number of polymersomes. | ||
Leakage of polymersomes and enzymes from HA hydrogel
An interesting feature of hydrogels, in view of biological applications, is their ability to slowly release incorporated substances, for example drugs. In the present case, where we focus on biocatalytic applications, release of compounds is, however, undesirable. The release of the enzyme-loaded polymersomes from the hydrogel was studied and compared with the release of free enzyme (i.e. non-encapsulated) from the hydrogel in two ways: 1) the hydrogel, either containing CALB-polymersomes or free CALB, was incubated, directly after preparation, for 3 × 24 h with 0.5 ml of Milli-Q. The Milli-Q phase was then assayed for CALB activity by measuring the fluorescence increase at λem = 460 nm from the CALB-catalyzed hydrolysis of (non-fluorescent) DiFMU-octanoate to the fluorescent 6,8-difluoro-7-hydroxy-4-methylcoumarin;29 2) polymersomes encapsulating alexa-labeled chloroperoxidase from Caldariomyces fumago (CPO) or free alexa-labeled CPO were immobilized in the hydrogel and incubated with 0.5 ml Milli-Q.8 The fluorescence increase at λem = 633 nm of the Milli-Q phase was measured over time for 72 h.The CALB-activity in the Milli-Q fractions after incubation at different time intervals (t = 24 h, 48 and 72 h) showed a clear difference between the enzyme-containing polymersomes and the free enzyme (Fig. 4). Whereas the incubation solution showed clear enzymatic activity for the free enzyme, indicating leakage of the enzyme from the gel, the incubation solution for the polymersomes did not show any enzymatic activity at all. Apparently, the immobilization of the polymersomes is an efficient process and more successful than the immobilization into the hydrogel of the free enzyme.
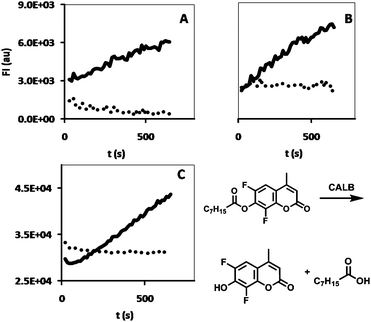 | ||
| Fig. 4 CALB-activity of incubation solutions collected after 24 h incubation time at (A) t = 24 h, (B) t = 48 h and (C) t = 72 h, for HA hydrogels containing CALB (black line) and HA hydrogels containing CALB-polymersomes (dots). The DiFMU assay reaction is shown at the bottom right. | ||
Although the hydrogel leakage experiments with the CALB-polymersomes and free CALB showed a clear difference, these experiments did not reveal how much enzyme had leaked out. To obtain a more quantitative picture, leakage from the hydrogel was studied for polymersomes containing alexa-663-labeled chloroperoxidase (CPO) and compared to the fluorescence intensity of the solutions before gelation (which was the same for the free enzyme and the CPO-polymersomes). Similarly to the CALB-polymersomes, a distinct difference existed between encapsulated and free enzyme (Fig. 5). After 72 h, 30% of the free enzyme had leaked out into the Milli-Q phase, whereas for the encapsulated enzyme this leakage amounted to only 7%. Already after the first 20 h, 5% of the CPO-polymersomes had leaked out, a value half that observed for the free enzyme (10% in 20 h), suggesting that the observed leakage possibly resulted from the available free enzyme, and not of enzyme encapsulated in the polymersomes. This may be due to the release of a small amount of labeled CPO from the polymersomes, as was also observed for freely dispersed CPO-polymersomes when the medium was changed from Milli-Q water to buffered solution.8 It could also mean that there was a minor amount of free CPO still present after preparation of the CPO-polymersomes. After 24 h there was no further significant leakage observed from the CPO-polymersomes, whereas for the free enzyme leakage continued at a similar rate.
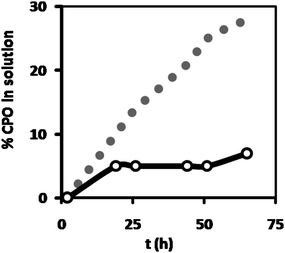 | ||
| Fig. 5 Percentage leakage of fluorescently labeled CPO from the HA hydrogel, incorporated as either free enzyme (grey dots) or encapsulated in polymersomes (open dots), as measured from the increase in fluorescence intensity of the supernatant Milli-Q added to the hydrogel after preparation. The fluorescence intensity of the solutions before gelation was taken as 100%. | ||
Upon the successful demonstration of the stability of the enzyme-loaded polymersomes in the hydrogel, the activity of CALB-polymersomes in the hydrogel was studied. Incubation of a 50 mg piece of the hydrogel with a 100 μl aliquot of a 20 μM DiFMU-solution clearly showed the highest activity to occur in the sample which had free enzyme entrapped in the hydrogel (Fig. 6). Although diminished, the conversion of the DiFMU by the enzyme-loaded polymersomes in the hydrogel was still significant. Clearly the enhanced stability comes with the price of activity loss. An explanation for this behaviour may be the rate-limiting diffusion of the substrate into the polymersome-loaded gel. The two gels were prepared with solutions that contained the same amount of enzyme in terms of activity, where it should be noted that it was previously demonstrated that in a non-gelated solution the activity of the enzyme is not hampered by its incorporation into the polymersomes.8 As such, it seems that the difference in activity must be related to the specific physical properties of the enzyme-loaded polymersome hydrogel. More detailed experiments are required, however, to substantiate this tentative explanation.
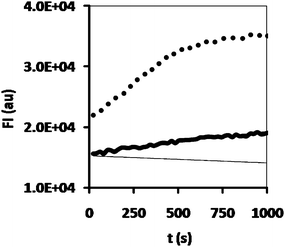 | ||
| Fig. 6 Hydrolysis of DiFMU octanoate by hydrogel samples containing CALB (dots), CALB-polymersomes (thick black line) or no enzyme at all (thin black line). | ||
Recycling of enzyme-loaded polymersome hydrogels
Although the enhanced stability of the enzyme-loaded polymersome hydrogel comes with a decreased intrinsic activity, this should not be a disadvantage if we can recycle the polymersome hydrogel after the reaction. The immobilized polymersomes were therefore tested for their recyclability. A 50 mg aliquot of gel was transferred to a 96-well plate and incubated with 100 μl of the 20 μM DiFMU-solution. The fluorescence increase was measured, the hydrogel was washed once with Milli-Q, and incubated again with substrate solution. This procedure was performed 8 times (Fig. 7). After the recycling procedure the supernatant of the last fraction was removed from the gel and tested for CALB activity.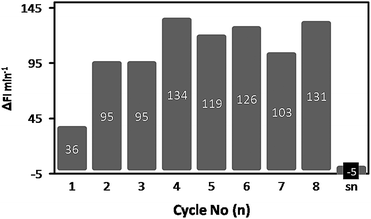 | ||
| Fig. 7 Recycling of the HA hydrogel containing CALB-loaded polymersomes. Each bar represents one cycle. The numbers on the bar indicate the CALB activity in FI min−1. The hydrogel was incubated with DiFMU and the increase in fluorescence at λem = 460 nm was measured, after which the hydrogel was washed once with Milli-Q and incubated again. This procedure was repeated 8 times. The supernatant of cycle 8 (sn) was transferred to an empty well and tested for its activity. | ||
Somewhat unexpectedly, there was a considerable increase in lipase activity between cycle 1 (36 FI min−1) and cycle 2 (95 FI min−1), after which the activity more or less remained constant at 118 ± 16 FI min−1, for at least 6 further cycles. As a minor amount of DMSO (1%) was used to solubilize the substrate, this could have caused the expansion of the pores of the gel and have resulted in an enhanced diffusion of the substrate into the hydrogel. Indeed it was observed in separate experiments that the presence of DMSO led to a modest swelling of the hydrogel, which could allow substrate to penetrate more deeply into the hydrogel structure and enable it to reach more active sites. Besides swelling, the diffusion of the substrate across the membrane into the polymersomes could also be facilitated by DMSO. Most important though, the trapping of the polymersomes in the hydrogel was still effective, as no CALB activity could be detected in the isolated supernatant after cycle 8, implying that no enzyme had leaked out during the recycling process and the observed activity resided in the hydrogel.
CALB-loaded polymersome hydrogel reactor
The absence of leakage from the polymersome hydrogel and the recycling of the system indicated that we could use it in a reactor setup. In a subsequent series of experiments a continuous-flow ‘enzyme-loaded polymersome reactor’ was constructed and DiFMU (3.5 μM) was added to the top of the reactor and fed through the reactor at a flow rate of 0.1 ml min−1 by the action of gravity. The flow-through was collected in a 96-well plate in fractions of 125 μl per well and the fluorescence intensity of the product was measured (Fig. 8). After collecting 6 fractions the substrate solution was replaced and Milli-Q was fed through the reactor. This process was repeated 5 times. The extent of the conversion was compared to that of the free, homogeneous enzyme, by taking a 125 μl sample of the substrate solution and incubating this solution with a CALB solution of high concentration (5 mg ml−1) and measuring the fluorescence intensity after 1 h incubation. This value was taken as 100%.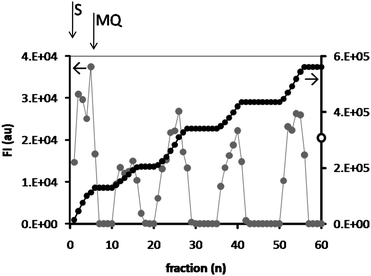 | ||
| Fig. 8 Conversion of DiFMU, measured by the fluorescence intensity of the product at λem = 460 nm, in a reactor constructed from an HA hydrogel containing CALB-polymersomes. Fractions of 125 μl each were collected and the fluorescence intensity was measured (grey dots, left axis). After 6 fractions the reactor was purged with Milli-Q. The black dots show the sum of the fluorescence intensities (right axis). For comparison, the fluorescence intensity of the substrate solution incubated with free CALB (FI = 3.1 × 105) is shown at the intersect with the right axis (open dot). | ||
The reactor experiments clearly demonstrated the feasibility of using the CALB-polymersome hydrogels in repeated cycles of product formation. It also showed that the system is very stable, especially when it is realized that no covalent bonds have been used to construct it, i.e., neither between the enzyme and the polymersome, nor between the polymersomes and the hydrogel. The average yield of product in each fraction was 5.4%, which was not further optimized but can be enhanced by increasing the residence time of the substrate in the column (see below). Although the highest conversions were obtained in the first 6 fractions (8.2% average yield with respect to the maximum possible conversion in one fraction), leakage of CALB-polymersomes from the hydrogel was not expected to have occurred. These would have caused a much higher fluorescence as a result of ongoing product formation in the wells. In addition, during subsequent cycles the activity of the reactor did not decrease further (4.9% average yield). An alternative explanation is therefore that the initial flow rate was somewhat lower than the final flow rate, which could have increased the conversion, because of the longer residence time of the substrate in the hydrogel.
Considering the product formation within each cycle, the general trend is that the product yield increases until Milli-Q is added to flush the column. This increase may result from the DMSO, which was shown to increase the rate of conversion of substrate in the recycling experiments, probably because it increases the pore size of the hydrogel (see Fig. 7). The same phenomenon seems to have occurred when substrate solution was flowed through the column.
To show that the hydrolysis of the substrate was the result of the action of CALB and not of spontaneous hydrolysis or autohydrolysis of DiFMU, the hydrogel was fed with the irreversible lipase inhibitor diethyl 4-nitrophenyl phosphate (DNP) in a 100:1 excess. Feeding the reactor with DiFMU before and after treatment with DNP revealed that, after feeding of DNP, the enzymatic activity decreased to 30% of the original value, demonstrating that the hydrolytic activity in the hydrogel resulted from the action of CALB (Fig. 9). Determining roughly the amount of p-nitrophenolate that was released from DNP by measuring its absorbance at λ = 420 nm, showed that approximately 10 nmol was set free. Because the total amount of CALB in the polymersomes in the hydrogel was estimated to be 2.4 nmol there seems to be a significant degree of autohydrolysis of the inhibitor.9 Indeed the half-life of the phosphate ester is half an hour in neutral water. As some time is needed for the inhibitor to penetrate through the gel, it seems therefore reasonable to assume that not all CALB has been deactivated by the inhibitor, explaining the residual 30% activity after DNP treatment.
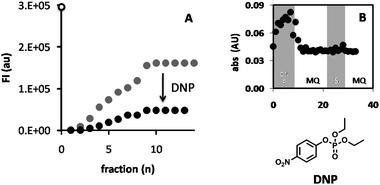 | ||
| Fig. 9 Conversion of DiFMU by feeding it through the polymersome hydrogel reactor, before (A; grey dots) and after (A; black dots) purging with the inhibitor DNP. The product was collected at the end of the reactor in a 96-well plate and the fluorescence at λem = 460 nm was measured. The fluorescence intensity of the free enzyme blank is shown at the intersect with the y-axis (FI = 3.1 × 105, open dot). Also shown (B) is the variation in absorbance at λ = 405 nm (indicating release of p-nitrophenol) of the fractions collected during the feeding of the reactor with DNP, Milli-Q (MQ), substrate, (S), and again Milli-Q (B). | ||
Stacked hydrogels of different enzyme-loaded polymersomes
The reactor procedure can easily be extended to other substrates and other enzymes as well. To demonstrate this, the substrate 2-methoxyphenyl acetate (2-MPA) was dissolved in water (1 mM) and fed through the reactor, along with H2O2 (2 mM). The hydrolyzed product, 2-methoxyphenol, was collected in a well plate and a peroxidase (chloroperoxidase; CPO) was added, initiating the formation of tetraguaiacol (3,3′-dimethoxy-4,4′-biphenylquinone; λmax = 470 nm) from 2-methoxyphenol with H2O2 as the oxidant.30,31 After 10 fractions the column was washed with Milli-Q water and the procedure was repeated. Similarly to the conversion of DiFMU, the 2-MPA was readily converted by feeding it through the reactor (Fig. 10), with an average yield in each fraction of 11%, compared to the free enzyme system. It was observed that the conversion of substrate was more or less constant in each fraction within the cycles, in particular when compared to the feeding of DiFMU through the reactor, where the product yield increased with each fraction until Milli-Q was added to flush the reactor. This is because at the concentration used, 2-MPA dissolves in water without the need for DMSO or another cosolvent, preserving the structure of the hydrogel.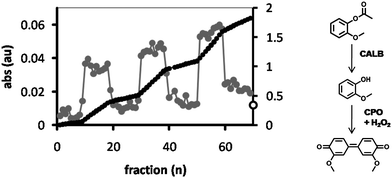 | ||
| Fig. 10 CPO-catalyzed formation of tetraguaiacol (λmax = 470 nm) from 2-methoxyphenol, which is generated from 2MPA by feeding it through a HA hydrogel containing CALB-polymersomes and collecting it in a 96-well plate. The product was collected in fractions of 125 μl (grey dots, left axis). After 10 fractions the reactor was purged with Milli-Q after which the procedure was repeated. The black dots show the sum of the absorbances (right axis). The open circle at the intersect of the right axis corresponds to the absorbance of the substrate solution after incubation with the free enzymes. | ||
In contrast to the feeding of DiFMU, the average absorption of the Milli-Q washings (i.e. the baseline) increased with each cycle, from 0.23% in the first cycle to 0.42% in the fourth cycle. A similar trend was observed for the 2-MPA fractions where the total yield in each cycle increased from 102% in the first cycle to 147% for the third cycle. This phenomenon seems to result from the presence of some substrate or product, which accumulates in the hydrogel and is not effectively washed out during the washing cycle and is gradually released. The same effect was seen when the absorbance at λ = 420 nm was monitored during the inhibition of the CALB-polymersome hydrogel with DNP (Fig. 9, small graph). After inhibition with DNP the addition of Milli-Q water caused an initial sudden increase of the absorbance from 0.059 to 0.070 (between fraction 9 and 10), which is most likely the result of the release of accumulated p-nitrophenolate.
Finally, in order to be able to perform multiple enzymatic transformations, the feasibility of stacking two hydrogels was studied, each containing polymersomes encapsulating different enzymes. (Fig. 11A). To this end, glucose oxidase (GOx), which catalyzes the oxidation of glucose to gluconolactone, giving H2O2 as a (by)product, was encapsulated in the polymersomes and the dispersion was gelated following the protocol described above. The hydrogel was placed directly on the CALB-polymersome hydrogel. A solution of glucose (2 mM) and 2-MPA (1 mM) was fed through the reactor, which was collected in a well-plate and detected by CPO. If successful, such a reactor can be provided with another hydrogel containing CPO-polymersomes. The reactor was continuously fed for a prolonged period of time without intermediate washings with Milli-Q, and it was observed that the rate of product formation was fairly constant over the applied period of time (Fig. 11B). With the objective of taking this set-up to its limit, a larger volume of substrate was fed through the reactor, e.g., 2 cycles of 6 ml each, and the product was collected in cuvettes, to directly quantify the product produced in the reactor by measuring its absorption at λ = 470 nm (Fig. 11C). Compared to the maximum yield realized by the enzymes in free solution at the same concentration of substrate and oxidant (7.5 nmol after 20 min), the conversion in the reactor was 36.9% for each fraction. This is significantly higher than the yields obtained with DiFMU or 2MPA and H2O2, and may be explained by the fact that the elution rate in the column was slower, which increased the residence time of the substrate in the column and hence the amount of substrate converted. However, when the amount of produced product was measured by its absorption (ε470 = 26600 M−1),30 the yield of the reaction was a mere 0.5%. This low efficiency, however, is not the result of the ineffectiveness of the reactor but rather the result of the low yield of the cascade reaction itself.
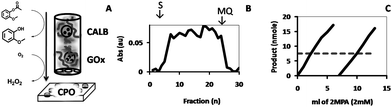 | ||
| Fig. 11 CPO-catalyzed formation of tetraguaiacol from 2-methoxyphenol and H2O2, which was obtained from 2MPA and glucose, respectively, by feeding these substrates through stacked HA hydrogels containing GOx and CALB (A). Shown are; the absorbance of the product fractions collected in a 96-well plate (B), the yield (nmol) determined for product fractions of larger volume collected in 1 ml cuvettes (C; straight lines) and the absorbance level of the substrate solution after incubation with the free system (C; dashed line). | ||
In a final series of experiments the stacked enzyme-loaded polymersome hydrogels of CALB and GOx were expanded with a hydrogel loaded with CPO-polymersomes. Unfortunately, feeding the reactor with the same mixture as described above did not lead to product formation. Variation of the thickness of the CPO-polymersome hydrogel to increase the residence time of the products to be converted did not change this. Most likely, this negative result is related to the kinetics of the 3-enzyme cascade reaction. The residence time of the 2-MPA should be sufficiently long for (final) product formation to occur, as a lag time is observed in the one-pot reaction. Furthermore, it may be necessary for CPO to be permanently present in the reaction mixture to drive the reaction to product formation. Integrating the CPO into the reactor removes it from the reaction mixture and, consequently, makes it ineffective.
Conclusions
In this study we have presented a proof-of-principle for an enzyme-loaded polymersome continuous-flow batch reactor, which is based on the immobilization of functional polymersomes in a HA hydrogel carrier. The reactor can be used for multiple cycles of product formation, which was demonstrated for the conversion of DiFMU-octanoate and for 2-methoxyphenyl acetate (2-MPA) by CALB. The stability of the reactor, which can be operated in multiple cycles of product formation, is largely due to the efficient trapping of the enzymes in the polymersomes, when compared to free enzymes, which progressively leak out over time. The set-up allows one to carry out tandem or cascade reactions, viz. by stacking hydrogels that contain polymersomes encapsulating different enzymes. Thus, HA hydrogels with CALB- and GOx-loaded polymersomes were stacked and the substrates 2-MPA and glucose were fed through the reactor. The products, H2O2 and 2-methoxyphenol were identified after collection by converting them to the strongly absorbing 3,3′-dimethoxy-4,4′-biphenylquinone with the help of the enzyme chloroperoxidase.Experimental
Chemicals
Hyaluronic acid sodium salt (Streptococcus equi; MW = 1.4–1.6 mDa),32 was bought from Sigma-Aldrich (BioChemika). Candida antarctica lipase B (E.C. 3.1.1.3; 1.0 U mg−1), glucose oxidase (E.C. 1.1.3.4; 200 U mg−1) from Aspergillus niger and chloroperoxidase (E.C. 1.11.1.10; 22371 U ml−1) from Caldariomyces fumago were purchased from Sigma-Aldrich (Biochemicka). 1-Amino-3,6,9-tri-oxaundecan-11-azide (ATA), propargylamine (PA), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide (EDC), N-hydroxysuccinimide (NHS), MES (2-(N-morpholino)ethanesulfonic acid), CuSO4, ascorbic acid, diethyl 4-nitrophenyl phosphate (DNP), 2-methoxyphenyl acetate (2-MPA), H2O2 and α-D-glucose were obtained from Sigma-Aldrich. Alexa fluor 633 succinimidyl ester and 6,8-difluoro-4-methylumbelliferyl (DiFMU) octanoate were bought from Molecular Probes (Invitrogen).Instrumentation
IR spectra were acquired with a Bruker Tensor 27 FT-IR spectrometer. Fluorescence was measured in 50 μl fluorescence cuvettes (Hellma) using a Perkin Elmer Luminescence Spectrometer LS50B or in a 96-well plate on a Wallac Victor 1420 multilabel counter. UV-vis spectra were recorded either on a Varian Cary 50 UV-Vis spectrophotometer or in a 96-well plate on the same Wallac Victor 1420 multilabel counter. For transmission electron microscopy (TEM) imaging a JEOL JEM-1010 instrument was used. Hydrogel samples for TEM were prepared by applying a small piece of this material on a carbon-coated copper grid (Electron Microscopy Sciences, Hatfield, PA) and by drying under a stream of argon for 30 min. No coating or staining was applied. CFM was carried out with a previously reported set-up.33Procedures
The hyaluronic acid was functionalized following a slight modification of a procedure published in the literature.34 The polysaccharide was dissolved in MES-buffer (pH = 5.2, 0.1 M) to a concentration of 4.0 mg ml−1, by heating the solution close to boiling overnight. After cooling, EDC and NHS were added followed by the respective amine, after which the solution pH was brought back to pH = 5.2 by the addition of HCl. The solution was allowed to stand for 24 h during which the pH dropped to pH = 3. The solution was subsequently transferred to a 10 K MWCO dialysis tube and dialyzed for 4 × 24 h against aqueous 0.1 M NaCl (1×) and distilled water (3×). CPO was labeled with alexa-663 succinimidyl ester (molecular probes) following the procedure supplied with the labeling kit. PS-PIAT was synthesized as reported previously.35A typical preparation for an enzyme-loaded hydrogel was conducted as follows: 200 μl of a 2.5 mg ml−1 stock solution of CALB in Milli-Q water was diluted to 2.5 ml and 0.5 ml of a 1.0 mg ml−1 PS-PIAT solution in THF was slowly added. The solution was incubated overnight and filtered 8 times to near-dryness over a 100 kDa MWCO Amicon ultra-free MC centrifugal filter (Millipore), while after each filtration step the sample was redispersed in Milli-Q to the original volume. The eventual volume of the polymersome dispersion was brought back to 1.0 ml and was added to 4.0 mg of HA-ATA and 4.0 mg of HA-PA (6 μM in total). After 8–10 h and occasional agitation complete dissolution of the polysaccharide had occurred. Gelation was induced by the addition of 10 μl of a solution containing 12.5 mg ml−1 CuSO4 (2 mM) and 35 mg ml−1 ascorbic acid (4 mM) to 0.5 ml of the HA polymersome solution, and occurred typically within 2–3 min as was observed by inverting the test tube. The solution was left for 12 h, the supernatant fluid on top was removed, and the gel was incubated for 48 h with Milli-Q. The supernatant solution was refreshed every 8 h. This procedure resulted in a clear gel.
The leakage experiments and the recycling of the hydrogel were performed as described in the main text. The enzyme concentration in the polymersomes was equal to the concentration of the free enzyme. For CALB this was estimated to be 0.2 mg ml−1, for CPO the concentrations were equalized by comparing the fluorescence of the samples. The enzyme concentration was calculated to be approximately 0.5 mg ml−1. For assaying CALB activity a 20 μM solution of DiFMU, which was prepared by the addition of 2 μl of DiFMU stock (1 mM) to 100 μl of Milli-Q, was used.
For the construction of the reactor the hydrogels containing the polymersomes were gently loaded into a 6 ml filtration column (pore size 20 μm, Biotage). Further experimental details are given in the main text.
References
- D. E. Discher and A. Eisenberg, Science, 2002, 297, 967 CrossRef CAS.
- S. F. M. van Dongen, H.-P. M. de Hoog, R. J. R. W. Peters, M. Nallani, R. J. M. Nolte and J. C. M. van Hest, Chem. Rev., 2009, 109, 6212 CrossRef CAS.
- C. LoPresti, H. Lomas, M. Massignani, T. Smart and G. Battaglia, J. Mater. Chem., 2009, 19, 3576 RSC.
- F. Axthelm, O. Casse, W. H. Koppenol, T. Nauser, W. Meier and C. G. Palivan, J. Phys. Chem. B, 2008, 112, 8211 CrossRef CAS.
- Q. Chen, H. Schönherr and G. J. Vancso, Small, 2009, 5, 1436 CrossRef CAS.
- M. Nallani, S. Benito, O. Onaca, A. Graff, M. Lindemann, M. Winterhalter, W. Meier and U. Schwaneberg, J. Biotechnol., 2006, 123, 50 CrossRef CAS.
- D. M. Vriezema, J. Hoogboom, K. Velonia, K. Takazawa, P. C. M. Christianen, J. C. Maan, A. E. Rowan and R. J. M. Nolte, Angew. Chem., Int. Ed., 2003, 42, 772 CrossRef CAS.
- H. M. de Hoog, M. Nallani, J. J. L. M. Cornelissen, A. E. Rowan, R. J. M. Nolte and I. W. C. E. Arends, Org. Biomol. Chem., 2009, 7, 4604 RSC.
- S. F. M. van Dongen, M. Nallani, J. J. L. M. Cornelissen, R. J. M. Nolte and J. C. M. van Hest, Chem. Eur. J., 2008, 15, 1107.
- V. Crescenzi, L. Cornelio, C. Di Meo, S. Nardecchia and R. Lamanna, Biomacromolecules, 2007, 8, 1844 CrossRef CAS.
- J.-F. Lutz, Angew. Chem., Int. Ed., 2007, 46, 1018 CrossRef CAS.
- G. Murtas, Y. Kuruma, P. Bianchini, A. Diaspro and P. L. Luisi, Biochem. Biophys. Res. Commun., 2007, 363, 12 CrossRef CAS.
- D. Hardison, H. U. Deepthike, W. Senevirathna, T. Pathirathne and M. Wells, J. Mater. Chem., 2008, 18, 5368 RSC.
- Y. Li and W. T. Yip, J. Am. Chem. Soc., 2005, 127, 12756 CrossRef CAS.
- S. A. Yamanaka, D. H. Charych, D. A. Loy and D. Y. Sasaki, Langmuir, 1997, 13, 5049 CrossRef CAS.
- T.-J. M. Luo, R. Soong, E. Lan, B. Dunn and C. Montemagno, Nat. Mater., 2005, 4, 220 CrossRef CAS.
- F. Khan, R. S. Tare, R. O. C. Oreffo and M. Bradley, Angew. Chem., Int. Ed., 2009, 48, 978 CrossRef CAS.
- L. Perlin, S. MacNeil and S. Rimmer, Chem. Commun., 2008, 5951 RSC.
- J. K. Tessmar and A. M. Gopferich, Adv. Drug Delivery Rev., 2007, 59, 274 CrossRef CAS.
- M. Ikeda, S. Ueno, S. Matsumoto, Y. Shimizu, H. Komatsu, K.-I. Kusumoto and I. Hamachi, Chem.–Eur. J., 2008, 14, 10808 CrossRef CAS.
- B. Wei, I. Cheng, K. Q. Luo and Y. Mi, Angew. Chem., Int. Ed., 2008, 47, 331 CrossRef CAS.
- H. Groger, E. Capan, A. Barthuber and K.-D. Vorlop, Org. Lett., 2001, 3, 1969 CrossRef CAS.
- A. Harada, K. Johnin, A. Kawamura and K. Kono, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 5942 CrossRef CAS.
- M. Jekel, A. Buhr, T. Willke and K.-D. Vorlop, Chem. Eng. Technol., 1998, 21, 275 CrossRef CAS.
- I. R. Wheeldon, J. W. Gallaway, S. C. Barton and S. Banta, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 15275 CrossRef CAS.
- J. Heo and R. M. Crooks, Anal. Chem., 2005, 77, 6843 CrossRef CAS.
- M. Nallani, R. Woestenenk, H. M. de Hoog, S. F. M. van Dongen, J. Boezeman, J. J. L. M. Cornelissen, R. J. M. Nolte and J. C. M. v. Hest, Small, 2009, 5, 1138 CAS.
- T. Liedl, H. Dietz, B. Yurke and F. Simmel, Small, 2007, 3, 1688 CrossRef CAS.
- Clustering of polymersomes is often observed in the specific prepartion procedure of PS-PIAT vesicles, e.g. see: M. Nallani, H. M. de Hoog, J. J. L. M. Cornelissen, A. R. A. Palmans, J. C. M. van Hest and R. J. M. Nolte, Biomacromolecules, 2007, 8, 3723 Search PubMed . In different systems also the addition of homopolymer imparts fusion and aggregation, see: T. P. Smart, C. Fernyhough, A. J. Ryan and G. Battaglia, Macromol. Rapid Commun., 2008, 29, 1855 CrossRef CAS.
- K. M. Manoj and L. P. Hager, Biochemistry, 2008, 47, 2997 CrossRef CAS.
- M.-Y. Lee, A. Srinivasan, B. Ku and J. S. Dordick, Biotechnol. Bioeng., 2003, 83, 20 CrossRef CAS.
- A. Shiedlin, R. Bigelow, W. Christopher, S. Arbabi, L. Yang, R. V. Maier, N. Wainwright, A. Childs and R. J. Miller, Biomacromolecules, 2004, 5, 2122 CrossRef CAS.
- M. Comellas-Aragones, H. Engelkamp, V. I. Claessen, N. A. J. M. Sommerdijk, A. E. Rowan, P. C. M. Christianen, J. C. Maan, B. J. M. Verduin, J. J. L. M. Cornelissen and R. J. M. Nolte, Nat. Nanotechnol., 2007, 2, 635 CrossRef CAS.
- V. Crescenzi, L. Cornelio, C. Di Meo, S. Nardecchia and R. Lamanna, Biomacromolecules, 2007, 8, 1844 CrossRef CAS.
- H. M. de Hoog, D. M. Vriezema, M. Nallani, S. Kuiper, J. J. L. M. Cornelissen, A. E. Rowan and R. J. M. Nolte, Soft Matter, 2008, 4, 1003 RSC.
Footnote |
| † Present address: Laboratory for Biomolecular Nanotechnology, MESA+ Institute, University of Twente, P.O. Box 217, 7500 AE, Enschede, The Netherlands. |
| This journal is © The Royal Society of Chemistry 2010 |