Deciphering ionic current signatures of DNA transport through a nanopore
Aleksei
Aksimentiev
*
Department of Physics, Beckman Institute for Advanced Science and Technology, University of Illinois at Urbana-ChampaignE-mail: aksiment@illinois.edu
First published on 2nd February 2010
Abstract
Within just a decade from the pioneering work demonstrating the utility of nanopores for molecular sensing, nanopores have emerged as versatile systems for single-molecule manipulation and analysis. In a typical setup, a gradient of the electrostatic potential captures charged solutes from the solution and forces them to move through a single nanopore, across an otherwise impermeable membrane. The ionic current blockades resulting from the presence of a solute in a nanopore can reveal the type of the solute, for example, the nucleotide makeup of a DNA strand. Despite great success, the microscopic mechanisms underlying the functionality of such stochastic sensors remain largely unknown, as it is not currently possible to characterize the microscopic conformations of single biomolecules directly in a nanopore and thereby unequivocally establish the causal relationship between the observables and the microscopic events. Such a relationship can be determined using molecular dynamics—a computational method that can accurately predict the time evolution of a molecular system starting from a given microscopic state. This article describes recent applications of this method to the process of DNA transport through biological and synthetic nanopores.
 Aleksei Aksimentiev | Aleksei Aksimentiev is Professor of physics at the University of Illinois at Urbana-Champaign. He received his PhD in chemistry from the Institute of Physical Chemistry, Poland, after completing a Master's degree in physics at the Ivan Franko Lviv State University in Ukraine. After postdoctoral training at Mitsui Chemicals, Japan, he joined the Theoretical and Computational Biophysics Group in Urbana as a postdoctoral research associate. He became a faculty member of the physics department at Illinois in 2005. His research interests include systems that combine biological macromolecules and man-made nanostructures, membrane proteins, and molecular machinery of DNA replication. |
1 Introduction
Imagine pulling a thread through a needle's eye, just a nanometre in diameter, a thousand times per minute, in the dark. That is precisely what so-called nanopore technology1 enables. Fig. 1 illustrates the basic principle. A nanometre-thin membrane with a single nanopore in it separates an aqueous solution into two compartments, connected through the nanopore. A transmembrane voltage bias drives charged solutes, such as biomolecules and dissociated salt ions through the nanopore, from one compartment to the other. The specific distribution of the electric field, which is high near and inside the nanopore but negligible away from the membrane, performs the hard task of navigating the thread (biomolecule) through the needle eye (nanopore), enabling automatic reloading of the pore with a new biomolecule as soon as another one exits the pore (hence is the throughput). The steric constraints of the nanopore ensure that only one molecule is loaded at a time.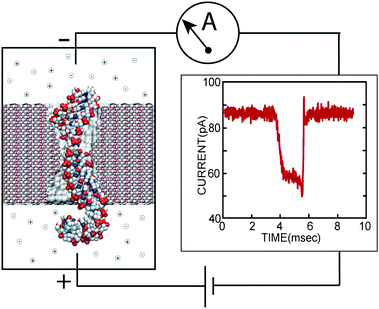 | ||
| Fig. 1 Experimental setup of a single molecule nanopore experiment. Driven by the electrical field between two electrodes, a biomolecule transits the pore in a thin, synthetic membrane (left), inducing a transient blockade of the ionic current (right), measured by the amplifier. | ||
What are the merits of such a remarkable technology? For a period that can be as brief as a few nanoseconds2,3 or as long as a few seconds,4,5 a fragment of or an entire (small) biomolecule can be confined to a volume of just several cubic nanometres. Thus, the nanopore confinement enables properties of a confined molecule to be examined, tens of atoms at a time. In particular, single-file transport of a linear, unfolded polymer sequentially exposes its fragments to the examination volume—a process ideal for protein or nucleic acid sequencing. Although various methods to perform the examination have been proposed and realized, including transverse tunneling current recordings,6,7 capacitance measurements8,9 and fluorescent readout,10 the most useful method to date is also the one most easily realizable in practice: measurement of the ionic current co-passing through the pore along with the analyte molecule. In the absence of an analyte, the geometry and the fixed charge of a nanopore determines its ionic conductance. The presence of an analyte alters the nanopore conductance, which is registered by an electric amplifier. In a way, the modus operandi of a nanopore detector is similar to that of a Coulter counter—a device routinely used in biology labs to count cells by measuring the ionic current blockades they produce passing though a micron-size aperture.
The first experimental realization of a molecular Coulter counter was done using the alamethicin channel,11 where the noise spectrum revealed permeation of poly(ethylene glycol) molecules. About the same time, the idea of using nanopores to sequence DNA was articulated by Deamer, Brandon and, independently, Church.12 In 1996, Kasianowicz and co-workers jump-started the field by demonstrating a sequence-dependence of the ionic current blockades produced by transport of RNA molecules through the α-hemolysin channel.13 Since then, nanopores have been employed to detect nucleotide content of DNA and RNA molecules,14–20 to measure the type and the concentration of small analytes,21–24 to detect proteins,25–33 to identify the stereoisoforms of a common drug,34 to determine the mass spectrum of a polymer mixture,35 to exert forces on biomolecules,4,5,36–39 and to sort proteins.40,41 The largest technology drive beyond the explosion of publications in the nanopore field is the possible application of nanopores in DNA sequencing,42–44 with major biotech companies already eyeing nanopores as prospective generation IV sequencing platforms. Various spin-off applications such as general purpose stochastic sensors,45–47 single-molecule manipulators,4,36,37,48 ion filters,49–53 nanofluidic electronics,54–57etc. ensures that the nanopore field will be flourishing long after the DNA sequencing goals are met.
Apart from interest in using nanopores for various nano(bio)technology applications, transport of solutes through nanopores presents a rich and non-trivial physics problem. In fact, the microscopic mechanism underlying the basic nanopore functionality—ionic current blockades—remains largely unknown, as it is not currently possible to characterize the microscopic conformations of single biomolecules directly in a nanopore and thereby unequivocally establish the causal relationship between the ionic current measurements and the underlying microscopic events. Many unanswered questions pertain to the mechanism of a solute's capture, the role of hydrodynamic interactions, solvation forces, and entropic forces of confinement, the electro-osmotic effect, mechanical deformation of solutes, atomic-scale friction, interaction of inorganic surfaces with biomolecules, sorption/desorption of solutes to/from the nanopore walls, spontaneous closure of biological nanopores, noise in nanoelectrofluidic systems, etc. Nanopore translocation experiments present a unique opportunity to test these physical models of diverse nanoscale phenomena at the single-molecule level.
The high throughput and the relatively simple setup of the nanopore experiments come with a price: the ionic current is often the only observable that can be directly measured. Microscopic simulations, in particular all-atom molecular dynamics (MD), can be used to interpret the results of a nanopore experiment as they can not only describe the dynamics of a polymer in a nanopore, but also predict the associated ionic current values. Below we discuss the capability of the MD method in answering the following questions:
• For a pore of fixed geometry and charge, what is the magnitude of the ionic current and how does it depend on electrolyte concentration and make-up?
• For a solute of a known structure, a pore of known geometry and an electrolyte of known concentration, what permeation pathway is the most likely and what is the permeation rate?
Once a method is available to answer these questions, it will become possible to reconstruct the molecular events underlying measured ionic current blockades. This reconstruction method will enable further optimization of the nanopore structure, solvent properties and external conditions (transmembrane bias, temperature, etc.) to control the permeation mechanism and increase the sensitivity of ionic current blockades to the underlying molecular events.
2 Biological and solid-state nanopores
Nanopores are ubiquitous in nature. Equipped with voltage-driven levers, selective gates, and electrochemical motors, nanometre-size pores in lipid membranes transport biomolecules in and out of living cells and between the intracellular organelles. Viruses use nanopores to infect susceptible cells. Bacteria employ nanopores for horizontal gene transfer. Cells divide, change their shapes and move in space in part due to the transport of biomolecules through nanometre-cross section porous filaments, i.e., microtubules and flagella, that allows the filaments to grow or contract.Nanopores are ubiquitous in engineering. They can be deployed in a variety of applications, ranging from high-performance lightweight materials, thermal insulators, and catalytic membranes to hydrogen gas storage, protein filters and drug delivery systems. Here, we focus on a particular type of nanopores that can serve as models for biological systems and that can be employed for various applications in bionanotechnology. Incorporated or produced in a nanometre-thick biological or synthetic membrane, such pores have diameters from one to tens of nanometres.
Until recently, bacterial toxin α-hemolysin (Fig. 2a) was the system of choice for nanopore translocation experiments, as this protein easily forms a water-filled pore in a lipid bilayer membrane58,59 and retains such conformation for extended periods of time, up to several days.60 The utility of the α-hemolysin pore as a stochastic sensor can be enhanced by placing a molecular adapter near the constriction of the pore.21 Through genetic engineering, specific sites61,62 or even entire proteins63,64 can be incorporated into the nanopore's structure, further increasing the sensor's sensitivity. To be detected, the analytes do not have to permeate the pore. Indeed, a toggling-type auxiliary molecule can transduce the binding of an analyte at the periphery of the channel into a pattern of conductance states, which can be recognized using a support vector machine.27
 | ||
| Fig. 2 Biological and solid-state nanopores. (a, b) All-atom model of the α-hemolysin (a) and MspA (b) channels suspended in a lipid bilayer membrane.65 (c) TEM image of a nanopore in a Si3N4 membrane.66 (d) Transmission electron microscopy (TEM) image of a metal-oxide-semiconductor capacitor membrane.8 TEM images courtesy of Gregory Timp. | ||
Another biological pore that was recently shown to have extraordinary potential for molecular sensing is MspA—a porin protein from the outer membrane of Mycobacterium smegmatis, Fig. 2b. The X-ray structure of MspA67 revealed its unusual architecture: eight β-strand monomers form a goblet-like structure, with the inner diameter of the constriction ∼1 nm and inner diameter of the widest part ∼4.5 nm. This particular shape of the MspA pore makes it an ideal platform for stochastic sensing applications.47 The main difficulty with using an MspA pore for ionic current recordings was its propensity for gating, which produces ionic current blockades similar to blockades resulting from transport of solutes. Recently an interdisciplinary team of researchers succeeded in genetically modifying the MspA pore to produce a channel pore that remains open indefinitely in bilayer recording experiments.68
The main advantage of the α-hemolysin and MspA systems is that they always form nanopores of the same three-dimensional structure, which are known to atomic-level detail.67,69 An obvious disadvantage is the mechanical fragility of a lipid bilayer membrane. Furthermore, a rather tedious procedure is required to incorporate a single biological pore in a lipid bilayer membrane. Experimental work70,71 is underway to decrease the mechanical instability of the lipid bilayer, which is the main drawback of this system.
Recent advances in semiconductor nanotechnology allow pores to be fabricated with nanometre-size diameters (Fig. 2c) using highly focused ion72,73 and electron66,74–76 beams (reviewed in ref. 77). Such pores are mechanically stable, do not change their shapes after extensive use, can operate at various temperatures and pH conditions,78,79 and can withstand high electric fields.80 The pores can also be made of any size,74,75,81 can be arranged into arrays,76 and can be functionalized with various organic molecules82 including oligonucleotides.83,84 The main disadvantage of the solid-state pores is that, even when manufactured using the same procedure, all pores will have slightly different shapes and surface charges, which affect their transport properties. Coating the nanopores with an atomic layer of alumina was shown to reduce some surface effects.85
Nanopores in metal-oxide-semiconductor membranes (Fig. 2d) are similar to those in silicon-based insulators, but have the additional advantage of allowing direct measurement of the electrostatic field of transported solutes.8,86 Typical metal-oxide-semiconductor membranes used in nanopore experiments have three layers: a layer of SiO2 separating two layers of doped crystalline and polycrystalline silicon. The SiO2 layer can be made as thin as 7 Å.87 Single or multiple-layer structures can be manufactured using standard semiconductor fabrication procedures as well. Nanopores in such membranes can be produced using a highly focused electron beam8,86 or through a sequence of alternating etching and atomic layer deposition procedures. The latter method can produce large nanopore arrays and is suitable for mass production.
3 Theoretical and computational models of nanopore transport
The process of polymer transport through a nanopore has attracted the attention of many theoreticians who brought to the field expertise from diverse areas of physics. Consequently, the spectrum of methods used to model this process is broad, and a comprehensive review is beyond the scope of this article. Below, we briefly describe the most popular methods.Continuum, transport and polymer physics models
The first theoretical models of polymer transport aimed at providing a theoretical account of the measurements (reviewed in refs. 88 and 89). The three most popular approaches were based on transport equations, scaling concepts of polymer physics, and continuum mechanics. In the first approach, a Fokker–Planck equation or its equivalent is used to describe polymer flux through a nanopore. The same type of models can also predict ionic current.90–92 Concepts of semiconductor physics have been employed to described ion transport and the effective force on DNA in narrow pores.93,94 The second approach relies on scaling laws of polymer physics95 and is most often used to predict the dependence of the polymer transport time on the polymer length and transmembrane bias. Thus, by describing DNA as an ideal polymer chain, the free-energy barrier for the DNA translocation,96,97 the average translocation time as a function of the pore dimensions and the length of DNA,98,99 and the total polymer flux induced by the external electrical field100 could be computed. Mathematical models describing the diffusion of polymers through pores have been used to investigate the origin of the measured non-Gaussian distributions of DNA passage times.101–103 The third class of models treat polymer transport through nanopores at the level of continuum mechanics. Such models were instrumental in elucidating the role of hydrodynamic interactions in nanopore transport.104–106 A clear advantage of these approaches is that they describe the translocation process in statistical terms and thus can be compared with an ensemble average determined by experiment. A disadvantage is that these models are built assuming certain types of microscopic interactions between the polymer and the nanopore, solvent, and itself and cannot easily incorporate atom-scale information.All-atom molecular dynamics
At the other end of the spectrum of approaches is MD, which is a computational method that allows the microscopic dynamics of a molecular system to be modeled at atomic resolution. In this method, a molecular system is approximated by an ensemble of virtual atoms interacting with each other according to the molecular force field,107 which has been developed and calibrated to quantitatively reproduce the physical properties of a simulated system. The trajectory of each atom is obtained by integrating Newton's equation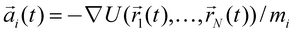 using the Verlet algorithm,108 where
using the Verlet algorithm,108 where 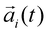 ,
, 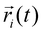 and mi are the acceleration, coordinates, and mass of atom i, respectively, and N is the total number of atoms in the system. For MD simulations of biomolecules, the molecular force field, e.g.,
and mi are the acceleration, coordinates, and mass of atom i, respectively, and N is the total number of atoms in the system. For MD simulations of biomolecules, the molecular force field, e.g., 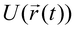 , is derived from quantum-mechanics calculations. Parameters of the molecular force field thus obtained are further refined to reproduce the physical properties of biomolecules in water.109,110
, is derived from quantum-mechanics calculations. Parameters of the molecular force field thus obtained are further refined to reproduce the physical properties of biomolecules in water.109,110
To model the transport of biomolecules through nanopores, an all-atom model of the experimental system is constructed using known crystallographic or other structures of the biomolecules, lipids, and inorganic materials. To model the nanopores in synthetic membranes, the force field describing the synthetic component is transcribed in the form of potential functions used by the biomolecular force fields, such as AMBER110 and CHARMM.109 Adapting the methodology developed for simulation of membrane proteins,65,90,115–124 a simulation of the electric field-driven translocation of biomolecules through nanopores can be accomplished.66,112Fig. 3 illustrates the process of building atom-scale models of biological65 and synthetic112 nanopores (reviewed in refs. 111 and 125).
 | ||
| Fig. 3 Atom-scale models of solid-state (top row) and biological (bottom row) nanopores. To build a microscopic model of a solid-state nanopore, a unit cell (a) of Si3N4 or SiO2 crystal is replicated in 3D to produce a solid-state membrane (b). A nanometre-size pore is produced by removing atoms according to the desired shape (c). An optional step is to melt and resolidify the membrane material to obtain an amorphous surface (not shown).111 Next, a fragment of DNA is placed near the nanopore's entrance (d), water molecules are added to fill the volume of the simulated system (e), selected water molecules are replaced with the ions of the electrolyte according to the specified concentration (f). Similar steps are required to build a microscopic model of a biological nanopore: an X-ray structure of the pore (g) is combined with the nucleic acid in the desired conformation (h) and embedded in a lipid bilayer membrane (i). The resulting system is solvated (j) and ionized (k). The procedures are described in detail in refs. 112 and 65,113 and online tutorials 114. | ||
The main advantage of the MD method is its atomic resolution and the possibility of direct comparison between simulation and experiment.65,79,80,126 The main disadvantage of the method is its timescale, which is currently limited to several microseconds.
Coarse-grained, Brownian dynamics and Monte Carlo simulations
Occupying a niche between all-atom and continuum models, this class of approaches relies on a less detailed description of the system than all-atom MD, while preserving enough detail to incorporate some structural features of a nanopore and a polymer. In coarse-grained (CG) MD,127,128 the polymer, nanopore and, sometimes, solvent are described as beads, each representing several atoms. In addition to reducing the number of degrees of freedom, a CG representation increases the timescale of an MD simulation by allowing for larger integration time steps. Additional speed-up originates from effective smoothing of the interbead interactions.129 In Brownian dynamics or Langevin models,130–133 the solvent effects enter as stochastic forces that apply to beads according to the underlying dynamical model.107 Monte Carlo simulations134–137 are often used to find a solution to the polymer physics models of nanopore transport. The timescale of these methods permits simulation of a polymer capture by the pore and complete permeation trajectories, in some cases allowing the distribution of the permeation times to be reconstructed.132 However, the computational efficiency comes at the price of accuracy, as all atomic-level details of the nanopore and polymer structure are washed out or blurred in this approach. Accordingly, these methods are much less successful in predicting the ionic current blockades and, generally, do not account for electro-kinetic effects.Multiscale approaches
Escaping the overidealization of a continuum approach and beating the timescale and lengthscale limits of a particle-based model, multiscale simulations typically combine a detailed description of the system in the vicinity of the nanopore with a coarser description of the system everywhere else. These types of models only recently have been applied to study polymer transport through nanopores. Thus, a CG MD model of dsDNA and a solid-state nanopore was coupled to a Lattice-Boltzmann (LB) model of an electrolyte138,139 to study the so-called fast translocation regime140 that is thought to be governed by hydrodynamic interactions. In another study, a continuum electrostatic model was coupled to LB hydrodynamics to predict ionic current blockades.141 A multiscale model combining all-atom MD and a continuum electrostatic solver was used to predict electric signatures resulting from DNA transport through a metal-oxide-semiconductor capacitor.8,142 This class of models has the ability to provide answers to almost all relevant questions of the field; however, it is also the least developed one.4 MD simulations of nanopore experiments
The indirect nature of information furnished by the nanopore translocation experiments creates the opportunity for molecular modeling to play a major role in the research process. Successful modeling approaches must not only accurately describe the process of polymer transport through a nanopore but also predict, with a quantitative accuracy, the associated ionic current blockades—a capability required for direct comparison between simulation and experiment. The all-atom MD method is ideally suited to interpret the nanopore experiments, as it can directly relate the conformation of a molecular system to the ionic current.65,112,126 The fundamental problem, however, is that the timescale of processes that can be investigated using this method is limited to several microseconds, whereas a typical translocation process takes milliseconds or more. Even using the newest generation of special purpose supercomputers that promise to push the simulation time into the submillisecond regime,143 a full account of the measurements cannot be obtained by the MD approach alone, as the permeation process is highly stochastic and must be characterized in statistical terms, which requires simulation of a large number of permeation events.While coarse-grained and multiscale computational approaches that preserve the accuracy of an all-atom MD simulation at a fraction of the computational cost are not yet available, all-atom MD simulations can be used to provide valuable information about a select class of translocation phenomena. For example, a typical polymer translocation event involves capture of the polymer by the pore and subsequent translocation. The capture process is difficult to characterize by all-atom MD, as diffusion governs DNA arrival at the pore's mouth and the subsequent search for the conformation that enables translocation. Hence, in a typical MD simulation of polymer transport, the polymer is placed in a conformation that enables translocation at the very beginning of the simulation. Such an approach is justified if the study's main focus is on the translocation events, not the capture. Furthermore, in some systems (for example, α-hemolysin, Fig. 3i), the translocation process is too slow for the all-atom MD even when the simulation is initiated from a conformation that is expected to occur during the translocation. Is this case, a viable approach is to take advantage of the thousandfold difference in the timescales of ion and polymer transport and characterize in statistical terms the ensemble of conformations a polymer can adopt inside the nanopore and compute (using MD) the corresponding distribution of the ionic current.
Below we describe the use of all-atom MD simulations for deciphering ionic current traces produced by polymer transport through biological and solid-state nanopores. Molecules of single- and double-stranded DNA (ssDNA and dsDNA) are used as model polymers, representing the case when the polymer's persistence length is much smaller (ssDNA) and much larger (dsDNA) than the membrane's thickness. The task of predicting the ionic current traces can be split into four parts: (i) Predicting ionic current through an open pore; (ii) predicting a typical translocation trajectory of a polymer through the pore; (iii) predicting ionic current blockades produced by the polymer in the nanopore; (iv) taking into account the electro-osmotic effect that couples the force experienced by the polymer in a nanopore to the ionic current.
Open pore current
The task of predicting the ionic current through a nanometre-size pore can be far from trivial: computing the conductance of even the simplest biological ion channels remains a major challenge.122,144 Luckily, nanopores used for DNA translocation experiments are considerably wider than the pores of biological ion channels. Not only does predicting the conductance of a larger-diameter pore require less computation, the simulations are more tolerant of the imperfections of the molecular force field.118In addition to the force field, other factors can influence the bulk conductivity of electrolyte in an MD simulation, for example, the type and the parameters of the thermostat107 used to maintain the temperature of the system constant, the use of rigid or flexible bonds in the water model.147 One should be particularly careful when determining the relative contribution of the cationic and anionic species to the total current, because the entire simulated system can randomly drift in space65 as a result of the particle-mesh Ewald electrostatics148 and/or Langevin thermostat.
 | ||
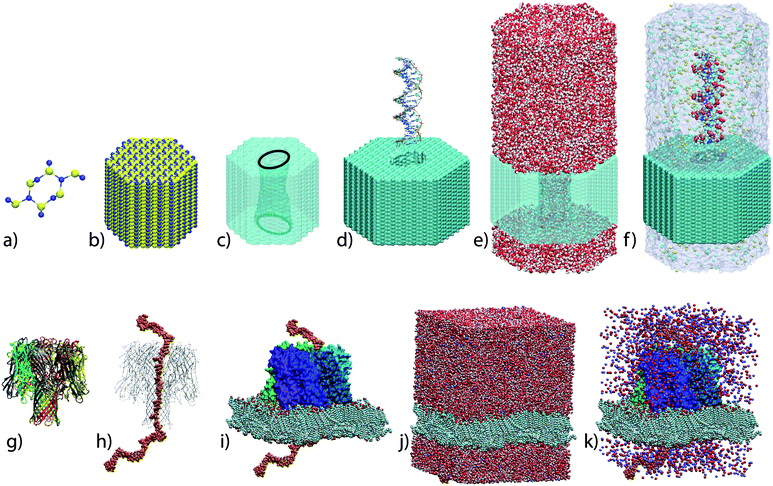
| Fig. 4 MD simulation of ionic current. (a) The microscopic model of a nanopore system is subject to an external electric field. Under the action of the field, water and ions rearrange, focusing the electric field to the vicinity of the membrane. (b) The resulting distribution of the electrostatic potential. A transmembrane bias of 0.6 V was imposed in this simulation. (c) Instantaneous currents sampled at 1 (black) and 10 (red) ps. The noise decreases as a square root of the sampling rate. (d) Integrated currents at different voltage biases. A linear cumulative current trace indicates a steady state conductance. (e) The simulated current–voltage curve of α-hemolysin.65 | ||
Driven by the effective electrostatic potential, ions of both types permeate through the nanopore in opposite directions. The instantaneous value of the current can be determined by summing up local displacements of individual ions between consecutive frames of an MD trajectory,65,150,151 which are usually recorded with the rate of one frame per 1 to 50 ps. The obtained instantaneous currents are very noisy. Hence, determining the value of the open pore current requires averaging over an extended period of time. In general, the standard error in determination of the current decreases as a square root of the number of ion permeation events, or the square root of the simulation time in the steady state regime. The statistical nature of the current evaluation makes predictions of the ionic current less computationally expensive for high-concentration electrolytes and at high transmembrane bias potentials.
Although the first MD simulations of an open pore current were performed using solid-state nanopores,112,150 it was the simulation of the α-hemolysin channel that demonstrated the ability of the MD method to quantitatively predict the ionic current.65 α-hemolysin was an ideal protein for this study because of its relatively large conductance and the known X-ray structure. The simulation of ionic current through an α-hemolysin channel was carried out at different values of the transmembrane voltage, yielding the current–voltage (I–V) curve, Fig. 4e. The simulations revealed features of a real channel, such a slight asymmetry of the I–V dependence and moderate selectivity to potassium ions. A major advancement reported in that study was the ability of the MD method to predict the ionic current at the voltage-bias conditions accessible to experiment, enabling direct verification of the simulation results. In the case of α-hemolysin, the agreement between the simulated and measured I–V curves was quantitative. Moreover, changing the protonation states of seven histidine residues near the pore constriction in an MD simulation was observed to alter the ionic current, which suggests that spontaneous protonation–deprotonation of these residues could be the source of the experimentally observed pH-dependent noise in the open pore current recordings.152,153 In addition to α-hemolysin, the MD method was applied to characterize the ionic conductance of MscS,154 Kv1.2,144 and OmpF.146,147
Determining the accuracy of the MD method in the case of solid-state nanopores is more difficult, as the structure of these pores is not known to atomic detail and, therefore, direct comparison to experiment is not possible. Nevertheless, all simulations carried out to date showed a qualitative agreement of the nanopore conductance values obtained in simulation and experiment.4,112,126 The most recent study of Si3N4126 and SiO2155 nanopores demonstrated high sensitivity of the open pore currents to atomic-scale roughness of the nanopore surface. The slow ion binding kinetics in Si3N4 pores requires long (10–50 ns) simulation times to reach a steady-state conductance regime, in particular in the case of large-area (15–20-nm membrane thickness) pores.
To illustrate the current capabilities of the MD method, let us estimate the supercomputer and real-life time required to simulate ionic conductance of an MspA channel,67Fig. 2a. The molecular dynamics code NAMD156 can apply thousands of central processing units (CPUs) to a single MD run. The runtime performance of the code scales linearly with the number of CPUs available, reaching the limit of scalability at about 50 to 100 atoms/per CPU, depending on the hardware architecture. A typical simulation system incorporating one copy of MspA, a lipid bilayer membrane, water and ions amounts to about 350![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) 000 atoms. Using 1024 CPUs in one run, a 100 ns simulation of the MspA system requires about 10 days to complete. The simulation time required to compute the ionic current I to desired accuracy δ = ΔI/I = 0.4(Iτ)−1/2, assuming that the standard deviation of the current is proportional to the square root of the number of elementary charges passing through the channel (which is the case for MD simulations). I and τ in the above formula have units of nA and ns, respectively. Determining the MspA open current (I0 = 450 pA) at a 200 mV bias to 10% accuracy will require a 35.5 ns MD simulation, and will take about 3 days to complete. The improved communication network of a new generation of supercomputers is expected to reduce the actual time required for a 100 ns simulation of an MspA system to about 1 day by increasing the number of CPUs that can be applied in one run. Using a special-purpose supercomputer,143 a 1 μs simulation of the MspA system could be accomplished in less than one day.
000 atoms. Using 1024 CPUs in one run, a 100 ns simulation of the MspA system requires about 10 days to complete. The simulation time required to compute the ionic current I to desired accuracy δ = ΔI/I = 0.4(Iτ)−1/2, assuming that the standard deviation of the current is proportional to the square root of the number of elementary charges passing through the channel (which is the case for MD simulations). I and τ in the above formula have units of nA and ns, respectively. Determining the MspA open current (I0 = 450 pA) at a 200 mV bias to 10% accuracy will require a 35.5 ns MD simulation, and will take about 3 days to complete. The improved communication network of a new generation of supercomputers is expected to reduce the actual time required for a 100 ns simulation of an MspA system to about 1 day by increasing the number of CPUs that can be applied in one run. Using a special-purpose supercomputer,143 a 1 μs simulation of the MspA system could be accomplished in less than one day.
DNA transport
Because the setup of an MD simulation can faithfully reproduce the experimental conditions, no special arrangements are required, in principle, to simulate DNA transport through a nanopore: driven by the gradient of the electrostatic potential, DNA, like any other charged solute, will find its way through a nanopore. The only limitation of this approach is the timescale of an MD simulation. Several methods have been developed to simulate DNA permeation when the experimental timescale of DNA transport greatly exceeds the range on conventional MD. | ||
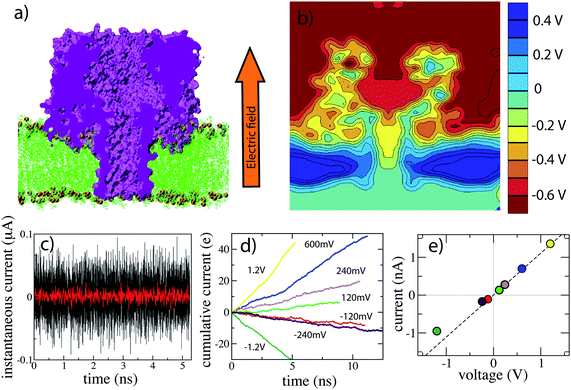
| Fig. 5 MD simulation of DNA translocation. (a–c) Electric field-driven transport of ssDNA through α-hemolysin. This proof-of-principle MD trajectory was obtained by applying a very high transmembrane bias of 15 V; protein and lipids were restrained. A more realistic trajectory can be obtained using the G-SMD method.163 (d–h) Transport of dsDNA transport through a Si3N4 pore. The DNA was observed to interact strongly with the surface of the solid-state membrane.112 (i–m) DNA hairpin permeation through a Si3N4 pore. The duplex part of the hairpin stretches under the influence of the electric field (panels j–l), which enables the translocation.126 | ||
In the case of relatively wide (>4 nm in diameter) solid-state nanopores, the translocation rate of dsDNA is of the order of one base pair per 30 ns at a 200 mV bias3,85 and increases with the bias.157 Similar translocation rates were observed in MD simulations.126 Furthermore, as solid-state membranes can withstand voltage biases of 1 V and higher, the translocation rates can be directly compared between simulation and experiment.79,158 The translocation rate of DNA through narrow (<4 nm in diameter) pores is determined by the DNA's interaction with the nanopore surface.101,112,157,159 In particular, nucleobases of ssDNA were observed to bind to Si3N4 surfaces with high affinity, often stalling the translocation in an MD simulation.112 The phosphate groups of dsDNA were observed to bind to the surfaces of crystalline Si3N4 and amorphous SiO2 solid as well (unpublished). As neither the accuracy of the force field that describes the interactions of a solid-state membrane with DNA in an MD simulation nor the structure and chemical composition of the nanopore surface used in actual experiment are known, direct quantitative comparison between simulation and experiment is, most of the time, not possible.
One noticeable exception is simulations and experiments pertaining to the so-called voltage threshold for DNA translocation,79,80 which is observed when molecules of DNA are forced to permeate through nanopores smaller in diameter than the translocating molecules. At a low transmembrane bias, DNA transport through such nanopores is sterically forbidden. However, if the force of the electric field in a nanopore is high enough to stretch the DNA molecule (reducing its cross section), the permeation can take place. This voltage threshold should not be confused with the entropic barrier for polymer transport.97 The voltage threshold for dsDNA translocation was first observed in MD simulations of Si3N4 pores and subsequently demonstrated in experiment.79,80 It is possible to trap a dsDNA molecule in a nanopore by switching off the electric field from a value above the translocation threshold before the DNA escapes the pore.158 The voltage threshold phenomena were also observed in experiments using DNA hairpins126,160 and DNA-restriction enzyme assemblies.4,161
Stretching a dsDNA molecule in a nanopore requires a high transmembrane bias. Therefore, subsequent translocation of short (up to 1000 base pairs) dsDNA fragments proceeds too fast to be detected as an ionic current blockade in experiments. Hence, the fact of DNA translocation (or its absence) is established by biochemical analysis of the solution samples taken from both sides of the membrane before and after the translocation experiment.80 In the absence of reliable experimental data on the ionic current blockades, the only quantity that can be directly compared between simulation and experiment is the value of the voltage threshold. Thus far, the simulations and experiments have produced consistent values of the voltage threshold, which is quite remarkable given that the only information about experiment used in the MD simulations are a TEM image of the pore, like the one shown in Fig. 2c, and the nucleotide sequence of DNA.
Transport of ssDNA through biological pores typically proceeds much slower than transport of dsDNA though solid-state pores, most likely because the constriction of the biological pore (α-hemolysin or MspA) is of the same size as the diameter of a single DNA strand. For example, the transport rate of DNA homopolymers through α-hemolysin is of the order of one nucleotide per μs at a 120 mV bias.15 This is consistent with the following MD result: at a 240 mV bias, DNA maintains the same position in the pore of α-hemolysin during a 150 ns simulation (unpublished). It is also clear that the utility of a conventional MD approach for characterizing the ssDNA translocation rate is severely limited.
A typical method to accelerate the rate of ssDNA transport in an MD simulation is to increase the transmembrane bias. Often, scaling the bias by a factor of ten can yield the desired results,112,113 however the dependence of the DNA transport on the bias is often nonlinear.162 In experiment, a solid-state membrane can withstand a transmembrane bias up to 5 V,79 whereas a lipid bilayer ruptures at about ±300 mV. Electroporation of a lipid bilayer in an MD simulation is observed at voltage biases of 2 V and higher, which could also depend on the overall duration of the MD run. The electroporation leads to ion leakage, which subsequently distorts the driving potential and thereby results in a simulation of an unrealistic permeation event. Grid-steered MD (G-SMD)163 is recommended for simulations at transmembrane biases higher than the threshold for electroporation (see below).
Due to these limitations, conventional SMD is seldom used to investigate polymer transport through nanopores. Nethertheless, this approach is preferred to the electric field-driven simulation when the force applied to a DNA polymer in a nanopore needs to be controlled or measured. Thus, using conventional SMD, it was discovered that a pathway (stretching or unzipping) a DNA hairpin undertakes to permeate through a nanopore that is smaller in diameter than its duplex part depends on the nanopore diameter.160 The SMD method was employed to determine the order the bonds between the restriction enzyme and DNA break in a nanopore force spectroscopy measurement.161 The G-SMD method (see next) could have been used to obtain these results as well.
Using this method, multiple complete permeation events of ssDNA through α-hemolysin could be simulated.163 Despite the dramatic acceleration of the permeation process, the results of the simulations were in good agreement with experiment: the ratio of permeation rates for (dA)58, (dC)58, and (dAdC)29 strands matched experiment. The ratio of the translocation rates of a poly(dA)58 strand in two possible global orientations (lead by 3′- or 5′-end) was found to be in quantitative agreement with experiment.113,163 The G-SMD method can be used to simulate protein transport through α-hemolysin as well as unzipping of DNA hairpins.163 The main deficiency of the method is that it cannot predict the absolute translocation times. Another imperfection of the approach is disproportionate scaling of the driving and stochastic forces, which may overestimate the effect of nanopore–DNA interactions on the translocation kinetics. Despite that, the method can already be applied to study slow (μs/DNA nucleotide) permeation processes. Work is underway to enable on-the-fly recalculations of the steering potential, making the simulation scheme self-consistent, and to better describe the stochastic forces that affect the permeation process.
Ionic current blockades
Transient reductions of the ionic current are recorded in almost all polymer translocation experiments. The next two sections discuss the physical effects that determine the level of the ionic current blockade and describe the use of the MD method to relate the microscopic conformations of a DNA polymer to the resulting departures of the nanopore current from the open pore level.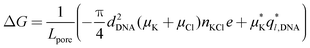 | (1) |
In addition to bulk ion concentration, MD simulations have determined that the depth and the sign of a nanopore's conductance change critically depend on the DNA conformation and the surface properties of the nanopore. Fig. 6a–c illustrates an MD simulation of dsDNA permeation through a 3.0 nm diameter pore in a 10 nm thick Si3N4 membrane driven by a 1.3 V bias. The KCl concentration in this simulation was 0.1 M. Fig. 6d shows the simulated ionic current. As the leading edge of dsDNA approaches the pore constriction, the current is reduced more than tenfold. About that time, the leading base pair adheres to the surface of the pore while the rest of the molecule continues to move through it. At the end of the simulation, dsDNA forms a loop in the exit compartment, piercing the nanopore constriction twice, whereas the ionic current returns to the open-pore level. This MD simulation highlights the importance of knowing the conformation of a DNA molecule in a nanopore for proper interpretation of ionic current blockades.
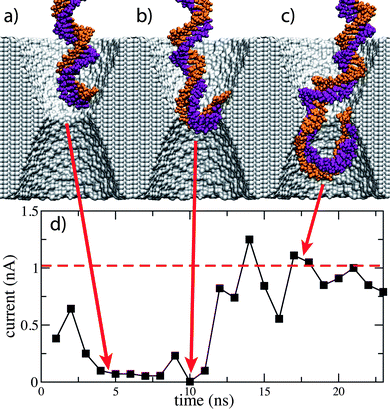 | ||
| Fig. 6 The effect of dsDNA conformation on ionic current blockades. (a–c) Snapshots from an MD simulation of dsDNA translocation through a 3.0 nm diameter pore in Si3N4. In this simulation, the Si3N4 membrane is 10 nm thick; the transmembrane bias is 1.3 V, the KCl concentration is 0.1 M. (d) The simulated ionic current. The ionic current returns to an open pore value while dsDNA is still in the pore. | ||
To reproduce the ionic current blockades recorded during the DNA trap experiments,158 MD simulations of a long dsDNA fragment in a 2.6 nm diameter nanopore were carried out using two models of the nanopore surface: crystalline Si3N4112 and annealed amorphous SiO2 solid.155,173 The two surfaces were previously found to have different affinity to the ions: the Si3N4 surface adsorbs both K+ and, to a lesser extent, Cl−ions,126 whereas the annealed SiO2 surface does not adsorb ions.155 In experiments, the presence of dsDNA in a nanopore was observed to reduce the nanopore current to 50–70% of the open pore level in a 0.1 M KCl electrolyte.158 The simulated ionic current blockades in the Si3N4 pore were ∼60% of the open pore level, whereas in the annealed SiO2 pore155 the current was enhanced to 130%. As both nanopore materials used in the simulations had neutral surfaces, the simulations demonstrate that atomic-scale roughness can play a major role in determining the nanopore current.
Hairpin DNA molecules are exceptionally attractive subjects for nanopore experiments because their double-helical portion can be trapped in a pore of the appropriate diameter while the remaining portion is threaded through the constriction, providing ample time to carry out the ionic current measurement.18,36,160,174–176 The arrested state does not last forever because the duplex part of the hairpin can thermally denature, allowing the hairpin to complete the translocation. In the case of α-hemolysin, the kinetics of duplex disruption can be used to discriminate molecules of different sequences.175 Contrary to α-hemolysin, in the case of solid-state nanopores, there are several plausible scenarios for how translocation of hairpin DNA could occur: a hairpin molecule could enter the pore in the coil-first orientation and complete its translocation by either mechanical unzipping or stretching of the duplex part, or a hairpin molecule could enter in the loop-first orientation and complete the translocation by stretching the duplex, as shown in Fig. 5i–m. In the DNA hairpin experiments,126,160 well-defined and recurring levels of ionic current below and above the open pore level I0 were observed within one translocation event, varying from 0.1 I0 to 2.0 I0 at 1 M KCl. Through MD simulations, some of the experimentally observed ionic current levels could be associated with the particular conformations of the hairpin DNA–nanopore system.126 Thus, the deepest blockades were observed when the loop of the DNA hairpin blocked the pore constriction, while the highest current levels (>2I0) developed when a large portion of the DNA hairpin accumulated in the exit chamber of the nanopore.
Fig. 7 illustrates the non-trivial dependence of the ionic current on the DNA conformation. During this particular trajectory, the ionic current changed from a 35% blockade to a 220% enhancement as hairpin DNA moved through the pore constriction. The plots of the ion concentrations demonstrate the complexity of the problem. In the absence of DNA, Fig. 7a, the local concentrations of K+ and Cl− ions are not only much lower in the nanopore than in the bulk but also asymmetric with respect to the pore constriction because of the applied bias (the nanopore itself is symmetric and electrically neutral). The presence of DNA in the nanopore, Fig. 7b and c increases the local concentration of both K+ and Cl− ions, but only a particular combination of the excluded volume and the enhanced ion concentrations results in ionic current enhancement. Predicting this effect based on eqn (1) is not possible, because the ion density, the DNA conformation and the distribution of the electrostatic potential are highly non-uniform. This example supports the notion that despite being computationally expensive, MD simulation is the only practical means to deal with the complexity of the problem.
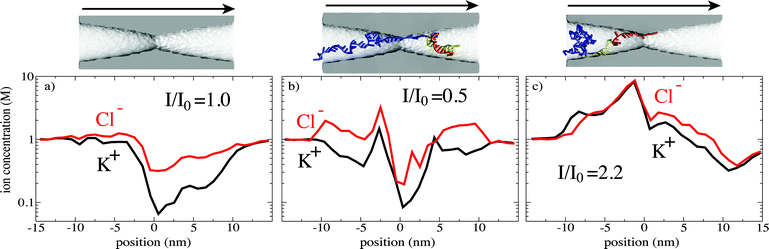 | ||
| Fig. 7 Influence of the local ion concentration on the nanopore current. The ion concentration profiles are shown for (a) an empty (no DNA) pore, (b) an ionic current blockade, and (c) an ionic current enhancement. The pore images are faithfully aligned with the position axes of the corresponding plots. The arrows indicate the direction of the applied electric field. The bulk ion concentration was 1 M. Other simulation conditions are described in ref. 126. | ||
Despite extensive efforts, ionic current blockades that are specific to the nucleotide sequence of a DNA molecule have not been reported yet from experiments employing solid-state nanopores. MD simulations suggest112 that a critical condition for obtaining such sequence-specific data from both ss- and dsDNA is the ability to restrain the DNA conformation for the duration of the ionic current measurement.158 Moving the same fragment of DNA back and forth through the pore9 is another possible strategy to beat the conformational noise.
MD simulations of ionic current blockades in α-hemolysin have been tempered by a rather small conductance of the channel in the blocked state (10–20 pA). A set of simulations was performed at a higher bias (1.2 V) to answer the question of an experimentalist (A. Meller, Boston U.) who challenged the author to predict how a DNA strand's global orientation affects its transport and ionic current blockades in α-hemolysin. In this blind test, no experimental data were given. The simulations of two α-hemolysin systems with DNA threaded in two opposite directions showed that DNA nucleotides in a narrow (<1.5 nm in diameter pore) tend to tilt toward the 5′-end, which was linked to the observed differences in the DNA translocation rate and the ionic current blockades.113 While these simulations provided a qualitatively correct picture, the absolute values of the currents were considerably higher than what one could expected from a linear scaling of the pore conductance with the voltage. The most recent simulations of the ionic current blockades in α-hemolysin were carried out at an experimentally accessible bias of 240 mV, yielding the ionic current at 15.6 ± 8.3 pA, a value consistent with experiment.113 From a simulation point of view, MspA is a much easier system to model, because its genetically engineered variants show an ion conductance that is stable and higher than that of α-hemolysin in both open and blocked states.68
The electro-kinetic effect
Quantitative characterization of the force experienced by a polymer in a nanopore is critical for predicting the polymer's translocation time. The notion of an effective charge q* can conveniently relate the force F experienced by a charged polymer in a nanopore to the applied electric field E: F = q*E, where q* is generally smaller than the nominal charge of the polymer. However, the value of the effective charge alone does not specify the physical origin of the polymer's charge reduction. Thus, the force required to stall dsDNA transport through a 10 nm diameter Si3N4 nanopore was directly measured by trapping one end of the DNA molecule with optical tweezers while the other end was subject to an electric field in a nanopore.37 The obtained dependence of the force on the transmembrane bias indicated a 75% reduction of the dsDNA charge—a value that seemed to be in perfect agreement with the Manning ion condensation theory.177 However the applicability of this theory to describe the force experienced by a charged polymer in an electric field was under question.178For a problem like that, MD simulations can serve not only as a microscope revealing atom-scale processes, but also as a probe measuring the forces involved. To determine the origin and the magnitude of the DNA's effective charge, a fragment of dsDNA was threaded through a nanopore and subject to a harmonic constraint so that displacement of the DNA in an electric field revealed the effective force. Using the setup shown in Fig. 8, the effective reduction of the DNA's charge was found to be produced by the hydrodynamic drag of the electro-osmotic flow created by the ionic current.179 A similar conclusion was reached independently by two other groups,39,180 however, only the all-atom MD study179 could fully describe the microscopic events giving rise to the effective screening and reproduce the results of the measurements without any adjustable parameters. Furthermore, the simulations determined that the effective force obeys the following dependence:178F = ξμE, where ξ and μ are the friction coefficient and the electrophoretic mobility of DNA, respectively (see Fig. 8b). This result enables determining the effective force on DNA in a nanopore without a laborious direct force measurement, as ξ and μ can be deduced from DNA escape113,174,181 and translocation13–15,157 experiments.
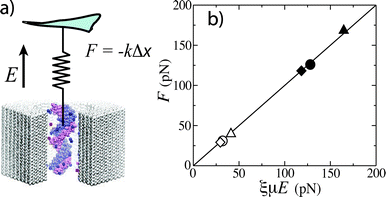 | ||
| Fig. 8 MD simulations of the effective force. (a) Setup of the simulations. A virtual spring limits displacement of the DNA fragment in an external electric field E. The effective force F is the product of the spring constant k and the DNA displacement Δx. (b) The simulated effective force F versus the product of the electrophoretic mobility μ, the friction coefficient ξ and the applied field E; μ and ξ were determined from independent MD simulations. The symbols indicate the results obtained for three different pores and two values of the applied field.179 | ||
It is now clear that electro-osmotic flow plays an important role in determining the timescale of a polymer translocation. The feedback loop is far from trivial, as the force experienced by a DNA fragment determines its conformation, which in turn affects the ionic current, which in turn affects the effective force. As the pattern of the flow depends on many factors, such as the surface properties of the nanopore, the local concentration of ionic species, and the electrolyte viscosity, MD simulations may be the only feasible approach to characterize the transport of flexible DNA molecules, such as ssDNA or DNA hairpins. Although the effect of the electro-osmotic flow on ssDNA transport through biological nanopores is not known, it is expected to have a considerable influence on the DNA capture rate.182,183
5 Outlook
Clearly, molecular simulations can play a major role in the development of the nanopore research field. The simulations can be used both to interpret the results of the actual measurements and to carry out nanopore translocation experiments in silico. Particularly attractive are simulations of engineered protein nanopores, whose experimental characterization is very laborious. Due to the timescale limit of the simulation methods and the force field inaccuracy, computational experiments are unlikely to eliminate the need for real experiments in the near future, although as the field matures it can be expected to follow the trend observed in other branches of technology, for example, in the aerospace industry, where computer simulations have already taken over from experiments.It is also clear that the MD method alone cannot address all the questions posed by experiment, even if the range of an MD simulation reaches a millisecond. Due to the local character of the translocation processes, the lengthscale problem can be tractable using a multiscale approach. Modeling the processive transport of a long (longer than the membrane thickness) polymer will require the ability to dynamically switch between the coarse and the finer representations of the same polymer fragment during the simulation. Although such a reduced description will not solve the timescale problem (the timescale will still be limited by the simulation of the highest-resolution fragment), it will dramatically improve the throughput, enabling statistical description of the translocation process.
The ability of brute-force MD to resolve ionic current blockades is limited by the time scale of the method and by the molecular force field. In the case of biological nanopores, the force-field bias can emerge as a systematic error caused by the difference between the simulated and measured bulk electrolyte conductance, systematic errors arising from the description of the interactions between the protein and the electrolyte, and yet unknown effects of the force field on microsecond dynamics of protein nanopores. Here, an important factor for future development will be the availability of experimental methods that can reliably characterize conformation dynamics of a protein at the microsecond time scale so that the force field accuracy can be unequivocally tested and refined. Stochastic gating of the nanopore current might be used to calibrate the protein force field.
In the case of solid-state nanopores, conformational fluctuations are negligible within the timescale of an MD simulation. In this case, extensive investments into development of an accurate force field to describe interactions between inorganic membranes and biomolecules can bring only modest improvement in the overall predicting power of the MD method because the atomic structure or even typical chemical make-up of a solid-state nanopore is not known and can dramatically vary from one sample to another (single-wall carbon nanotubes are a notable exception). Thus, the progress toward improving the accuracy of the simulations is limited by the advancements in the experimental methodology to characterize the atomic structure of the nanopores. Another possibility is to use solid-state nanopores coated with a molecular layer of a well-characterized chemical compound.82–84 Such nanopores will not only make molecular simulations more reliable, but also will provide the experimentalists with a means to manufacture solid-state nanopores that have reproducible properties.
Despite all the challenges and the shortcomings, microscopic simulations of the nanopore transport will remain a very active and exciting research field within the next decade, with the $1000 genome technology coming about in 2014, which may or may not utilize nanopores for DNA sequence detection. Having accomplished the $1000 genome goal, researchers will continue to explore nanopores for protein sequence detection, post-translational modification analysis and as a general tool to probe nanoscale interactions.
Acknowledgements
A. A. would like to thank Jeff Comer, Binquan Luan, David Wells, Gregory Timp, Klaus Schulten, Eduardo Cruz Chu, Jean-Pierre Leburton and Maria Gracheva for years of fruitful collaboration. This work was supported by grants from the National Institute of Health (Nos. R01-HG003713, R01-HG005115 and PHS 5 P41-RR05969), National Science Foundation (PHY0822613), and the Petroleum Research Fund (48352-G6). The authors gladly acknowledge supercomputer time provided by the Texas Advanced Computing Center, Pittsburgh Supercomputer Center and the National Center for Supercomputing Applications via Large Resources Allocation grant No. MCA05S028.References
- C. Dekker, Solid-state nanopores, Nat. Nanotechnol., 2007, 2, 209–215 CrossRef CAS.
- P. Chen, J. Gu, E. Brandin, Y.-R. Kim, Q. Wang and D. Branton, Probing single DNA molecule transpore using fabricated nanopores, Nano Lett., 2004, 4, 2293–2298 CrossRef CAS.
- A. J. Storm, J. H. Chen, H. W. Zandbergen and C. Dekker, Translocation of double-strand DNA through a silicon oxide nanopore, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2005, 71, 051903–051913 CrossRef CAS.
- Q. Zhao, G. Sigalov, V. Dimitrov, B. Dorvel, U. Mirsaidov, S. Sligar, A. Aksimentiev and G. Timp, Detecting SNPs using a synthetic nanopore, Nano Lett., 2007, 7, 1680–1685 CrossRef CAS.
- B. Hornblower, A. Coombs, R. D. Whitaker, A. Kolomeisky, S. J. Picone, A. Meller and M. Akeson, Single-molecule analysis of DNA-protein complexes using nanopores, Nat. Mater., 2007, 4, 315–317 CAS.
- J. Lagerqvist, M. Zwolak and M. D. Ventra, Fast DNA Sequencing via Transverse Electronic Transport, Nano Lett., 2006, 6, 779–782 CrossRef CAS.
- M. Zwolak and M. D. Ventra, Electronic signature of dna nucleotides via transverse transport, Nano Lett., 2005, 5, 421–424 CrossRef CAS.
- M. E. Gracheva, A. Xiong, J.-P. Leburton, A. Aksimentiev, K. Schulten and G. Timp, Simulation of the electric response of DNA translocation through a semiconductor nanopore-capacitor, Nanotechnology, 2006, 17, 622–633 CrossRef CAS.
- G. Sigalov, J. Comer, G. Timp and A. Aksimentiev, Detection of DNA sequence using an alternating electric field in a nanopore capacitor, Nano Lett., 2008, 8, 56–63 CrossRef CAS.
- G. V. Soni and A. Meller, Progress toward ultrafast DNA sequencing using solid-state nanopores, Clin. Chem., 2007, 53, 1996–2001 CrossRef CAS.
- S. M. Bezrukov, I. Vodyanoy and V. A. Parsegian, Counting polymers moving through a single ion channel, Nature, 1994, 370, 279–281 CrossRef CAS.
- G. Church, Genomes for all, Sci. Am., 2006, 294, 46–54 CrossRef CAS.
- J. J. Kasianowicz, E. Brandin, D. Branton and D. W. Deamer, Characterization of individual polynucleotide molecules using a membrane channel, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 13770–13773 CrossRef CAS.
- M. Akeson, D. Branton, J. J. Kasianowicz, E. Brandin and D. W. Deamer, Microsecond time-scale discrimination among polycytidylic acid, polyadenylic acid, and polyuridylic acid as homopolymers or as segments within singe RNA molecules, Biophys. J., 1999, 77, 3227–3233 CrossRef CAS.
- A. Meller, L. Nivon, E. Brandin, J. Golovchenko and D. Branton, Rapid nanopore discrimination between single polynucleotide molecules, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 1079–1084 CrossRef CAS.
- W. Vercoutere, S. Winters-Hilt, H. Olsen, D. Deamer, D. Haussler and M. Akeson, Rapid discrimination among individual DNA hairpin molecules at single-nucleotide resolution using an ion channel, Nat. Biotechnol., 2001, 19, 248–252 CrossRef CAS.
- W. A. Vercoutere, S. Winters-Hilt, V. S. DeGuzman, D. Deamer, S. E. Ridino, J. T. Rodgers, H. E. Olsen, A. Marziali and M. Akeson, Discrimination among individual Watson–Crick base pairs at the termini of single DNA hairpin molecules, Nucleic Acids Res., 2003, 31, 1311–1318 CrossRef CAS.
- N. Ashkenasy, J. Sánchez-Quesada, H. Bayley and M. R. Ghadiri, Recognizing a single base in an individual DNA strand: A step toward DNA sequencing in nanopores, Angew. Chem., Int. Ed., 2005, 44, 1401–1404 CrossRef CAS.
- Y. Wang, A. J. Rader, I. Bahar and R. L. Jernigan, Global ribosome motions revealed with elastic network model, J. Struct. Biol., 2004, 147, 302–314 CrossRef CAS.
- S. Iqbal, D. Akin and R. Bashir, Solid-state nanopore channels with DNA selectivity, Nat. Nanotechnol., 2007, 2, 243–248 CrossRef CAS.
- Q. Gu, O. Braha, S. Conlan, S. Cheley and H. Bayley, Stochastic sensing of organic analytes by a pore-forming protein containing a molecular adapter, Nature, 1999, 398, 686–690 CrossRef CAS.
- O. Braha, L. Q. Gu, L. Zhou, X. Lu, S. Cheley and H. Bayley, Simultaneous stochastic sensing of divalent metal ions, Nat. Biotechnol., 2000, 18, 1005–1007 CrossRef CAS.
- S. M. Bezrukov, Ion Channels as Molecular Coulter Counters to Probe Metabolite Transport, J. Membr. Biol., 2000, 174, 1–13 CrossRef CAS.
- P. Merzlyak, M.-F. Capistrano, A. Valeva, J. Kasianowicz and O. Krasilnikov, Conductance and ion selectivity of a mesoscopic protein nanopore probed with cysteine scanning mutagenesis, Biophys. J., 2005, 89, 3059–3070 CrossRef CAS.
- L. Movileanu, S. Howorka, O. Braha and H. Bayley, Detecting protein analytes that modulate transmembrane movement of a polymer chain within a single protein pore, Nat. Biotechnol., 2000, 18, 1091–1095 CrossRef CAS.
- J. J. Kasianowicz, S. E. Henrickson, H. H. Weetall and B. Robertson, Simultaneous multianalyte detection with a nanometer-scale pore, Anal. Chem., 2001, 73, 2268–2272 CrossRef CAS.
- S. Winters-Hilt and M. Akeson, Nanopore cheminformatics, DNA Cell Biol., 2004, 23, 675–683 CrossRef CAS.
- T. Sutherland, Y.-T. Long, R.-I. Stefureac, I. Bediako-Amoa, H.-B. Kraatz and J. Lee, Structure of peptides investigated by nanopore analysis, Nano Lett., 2004, 4, 1273–1277 CrossRef CAS.
- L. Movileanu, J.S., J. P. Schmittschmitt and H. Bayley, Interactions of peptides with a protein pore, Biophys. J., 2005, 89, 1030–1045 CrossRef CAS.
- A. Han, G. Scharmann, G. Mondin, R. Bitterli, N. Hegelbach, N. F. de Rooij and U. Staufer, Sensing protein molecules using nanofabricated pores, Appl. Phys. Lett., 2006, 88, 093901–3 CrossRef.
- M. Pastoriza-Gallego, G. Oukhaled, J. Mathe, B. Thiebot, J. Betton, L. Auvray and J. Pelta, Urea denaturation of α-hemolysin pore inserted in planar lipid bilayer detected by single nanopore recording: Loss of structural asymmetry, FEBS Lett., 2007, 581, 3371–3376 CrossRef CAS.
- G. Oukhaled, J. Mathe, A. Biance, L. Bacri, J. Betton, D. Lairez, J. Pelta and L. Auvray, Unfolding of proteins and long transient conformations detected by single nanopore recording, Phys. Rev. Lett., 2007, 98, 158101–4 CrossRef CAS.
- S. Benner, R. Chen, N. Wilson, R. Abu-Shumays, N. Hurt, K. Lieberman, D. Deamer, W. Dunbar and M. Akeson, Sequence-specific detection of individual DNA polymerase complexes in real time using a nanopore, Nat. Nanotechnol., 2007, 2, 718–724 CrossRef CAS.
- X. F. Kang, S. Cheley, X. Y. Guan and H. Bayley, Stochastic detection of enantiomers, J. Am. Chem. Soc., 2006, 128, 10684–10685 CrossRef CAS.
- J. W. F. Robertson, C. G. Rodrigues, V. M. Stanford, K. A. Rubinson, O. V. Krasilnikov and J. J. Kasianowicz, Single-molecule mass spectrometry in solution using a solitary nanopore, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 8207–8211 CrossRef CAS.
- C. Tropini and A. Marziali, Multi-nanopore force Spectroscopy for DNA analysis, Biophys. J., 2007, 92, 1632–1637 CrossRef CAS.
- U. Keyser, B. Koeleman, S. Dorp, D. Krapf, R. Smeets, S. Lemay, N. Dekker and C. Dekker, Direct force measurements on DNA in a solid-state nanopore, Nat. Phys., 2006, 2, 473–477 CrossRef CAS.
- E. H. Trepagnier, A. Radenovic, D. Sivak, P.G. and J. Liphardt, Controlling DNA capture and propagation through artificial nanopores, Nano Lett., 2007, 7, 2824–2830 CrossRef CAS.
- S. van Dorp, U.F.K., N. H. Dekker, C. Dekker and S. G. Lemay, Origin of the electrophoretic force on dna in solid-state nanopores, Nat. Phys., 2009, 5, 347–351 CrossRef CAS.
- C. C. Striemer, T. R. Gaborski, J. L. McGrath and P. M. Fauchet, Charge- and size-based separation of macromolecules using ultrathin silicon membranes, Nature, 2007, 445, 749–753 CrossRef CAS.
- A. Wolfe, M. Mohammad, S. Cheley, H. Bayley and L. Movileanu, Catalyzing the translocation of polypeptides through attractive interactions, J. Am. Chem. Soc., 2007, 129, 14034–14041 CrossRef CAS.
- D. Branton and et al, The potential and challenges of nanopore sequencing, Nat. Biotechnol., 2008, 26, 1146–1153 CrossRef CAS.
- H. Bayley, Sequencing single molecules of DNA, Curr. Opin. Chem. Biol., 2006, 10, 628–637 CrossRef CAS.
- R. F. Service, The race for the $1000 genome, Science, 2006, 311, 1544–1546 CrossRef CAS.
- H. Bayley and P. S. Cremer, Stochastic sensors inspired by biology, Nature, 2001, 413, 226–230 CrossRef CAS.
- J. Schmidt, Stochastic sensors, J. Mater. Chem., 2005, 15, 831–840 RSC.
- J. Kasianowicz, J. W. F. Robertson, E. R. Chan, J. E. Reiner and V. M. Stanford, Nanoscopic porous sensors, Annu. Rev. Anal. Chem., 2008, 1, 737–766 Search PubMed.
- N. A. Wilson, R. Abu-Shumays, B.G., H. Wang, K. Lieberman, M.A. and W. Dunbar, Electronic control of dna polymerase binding and unbinding to single dna molecules, ACS Nano, 2009, 3, 995–1003 CrossRef CAS.
- Z. S. Siwy, Ion current rectification in nanopores and nanotubes with broken symmetry revisited, Adv. Funct. Mater., 2006, 16, 735 CrossRef CAS.
- M. Gracheva and J.-P. Leburton, Electrolytic charge inversion at the liquid-solid interface in a nanopore in a doped semiconductor membrane, Nanotechnology, 2007, 18, 145704–145710 CrossRef.
- J. Vidal, M. Gracheva and J.-P. Leburton, Electrically tunable solid-state silicon nanopore ion filter, Nanoscale Res. Lett., 2007, 2, 61–68 Search PubMed.
- M. Gracheva and J.-P. Leburton, Simulation of electrically tunable semiconductor nanopores for ion current/single bio-molecule manipulation, J. Comput. Electron., 2008, 7, 6–9 Search PubMed.
- M. Gracheva, D. Melnikov and J.-P. Leburton, Multilayered semiconductor membranes for nanopore ionic conductance modulation, ACS Nano, 2008, 2, 2349–2355 CrossRef CAS.
- C. R. Martin and Z. S. Siwy, Learning nature's way: Biosensing with synthetic nanopores, Science, 2007, 317, 331 CrossRef CAS.
- I. Vlassiouk and Z. S. Siwy, Nanofluidic diode, Nano Lett., 2007, 7, 552 CrossRef CAS.
- L.-J. Cheng and L. Guo, Rectified ion transport through concentration gradient in homogeneous silica nanochannels, Nano Lett., 2007, 7, 3165–3171 CrossRef CAS.
- R. Karnik, C. Duan, K. Castelino, H. Daiguji and A. Majumdar, Rectification of ionic current in a nanofluidic diode, Nano Lett., 2007, 7, 547–551 CrossRef CAS.
- S. Bhakdi and J. Tranum-Jensen, Alpha-toxin of staphylococcus aureus, Microbiol. Rev., 1991, 55, 733–751 CAS.
- E. Gouaux, α-hemolysin from staphylococcus aureus: An archetype of β-barrel, channel-forming toxins, J. Struct. Biol., 1998, 121, 110–122 CrossRef CAS.
- G. Menestrina, Ionic channels formed by staphylococcus aureus α-toxin: Voltage-dependent inhibition by divalent and trivalent cations, J. Membr. Biol., 1986, 90, 177–190 CrossRef CAS.
- S. Howorka, L. Movileanu, X. Lu, M. Magnon, S. Cheley, O. Braha and H. Bayley, A protein pore with a single polymer chain tethered within the lumen, J. Am. Chem. Soc., 2000, 122, 2411–2416 CrossRef CAS.
- S. Howorka and H. Bayley, Probing distance and electrical potential within a protein pore with tethered DNA, Biophys. J., 2002, 83, 3202–3210 CrossRef CAS.
- Y. Jung, S. Cheley, O. Braha and H. Bayley, The internal cavity of the staphylococcal α-hemolysin pore accommodates approximately 175 exogenous amino acid residues, Biochemistry, 2005, 44, 8919–29 CrossRef CAS.
- S. Cheley, H. Xie and H. Bayley, A genetically encoded pore for the stochastic detection of a protein kinase, ChemBioChem, 2006, 7, 1923–1927 CrossRef CAS.
- A. Aksimentiev and K. Schulten, Imaging alpha-hemolysin with molecular dynamics: Ionic conductance, osmotic permeability and the electrostatic potential map, Biophys. J., 2005, 88, 3745–3761 CrossRef CAS.
- J. B. Heng, C. Ho, T. Kim, R. Timp, A. Aksimentiev, Y. V. Grinkova, S. Sligar, K. Schulten and G. Timp, Sizing DNA using a nanometer-diameter pore, Biophys. J., 2004, 87, 2905–2911 CrossRef CAS.
- M. Faller, M. Niederweis and G. Schultz, The structure of a mycobacterial outer membrane channel, Science, 2004, 303, 1189–1192 CrossRef CAS.
- T. Butler, M. Pavlenok, I. Derrington, M. Niederweis and J. Gundlach, Single-molecule DNA detection with an engineered MspA protein nanopore, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 20647 CrossRef CAS.
- L. Song, M. R. Hobaugh, C. Shustak, S. Cheley, H. Bayley and J. E. Gouaux, Structure of staphylococcal α-hemolysin, a heptameric transmembrane pore, Science, 1996, 274, 1859–1865 CrossRef CAS.
- D. Shenoy, W. Barger, A. Singh, R. Panchal, M. Misakian, V. Stanford and J. Kasianowicz, Functional reconstitution of protein ion channels into planar polymerizable phospholipid membranes, Nano Lett., 2005, 5, 1181–1185 CrossRef CAS.
- X.-f. Kang, S. Cheley, A. Rice-Ficht and H. Bayley, A storable encapsulated bilayer chip containing a single protein nanopore, J. Am. Chem. Soc., 2007, 129, 4701–4705 CrossRef CAS.
- J. Li, D. Stein, C. McMullan, D. Branton, M. J. Aziz and J. A. Golovchenko, Ion-beam sculpting at nanometre length scales, Nature, 2001, 412, 166–169 CrossRef CAS.
- J. Li, M. Gershow, D. Stein, E. Brandin and J. A. Golovchenko, DNA molecules and configurations in a solid-state nanopore microscope, Nat. Mater., 2003, 2, 611–615 CrossRef CAS.
- A. J. Storm, J. H. Chen, X. S. Ling, H. W. Zandergen and C. Dekker, Fabrication of solid-state nanopore with single-nanometre precision, Nat. Mater., 2003, 2, 537–540 CrossRef CAS.
- C. Ho, R. Qiao, A. Chatterjee, R. J. Timp, N. R. Aluru and G. Timp, Electrolytic transport through a synthetic nanometer-diameter pore, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10445–14450 CrossRef CAS.
- D. Kim, J. Park, J. Shin, P. Kim, G. Lim and S. Shoji, An extended gate FET-based biosensor integrated with a Si microfluidic channel for detection of protein complexes, Sens. Actuators, B, 2006, 117, 488–494 CrossRef.
- M. Rhee and M. A. Burns, Nanopore sequencing technology: nanopore preparations, Trends Biotechnol., 2007, 25, 174–181 CrossRef CAS.
- D. Fologea, M. Gershow, B. Ledden, D. S. McNabb, J. A. Golovchenko and J. Li, Detecting single stranded DNA with a solid state nanopore, Nano Lett., 2005, 5, 1905–1909 CrossRef CAS.
- J. B. Heng, A. Aksimentiev, C. Ho, P. Marks, Y. V. Grinkova, S. Sligar, K. Schulten and G. Timp, The electromechanics of DNA in a synthetic nanopore, Biophys. J., 2006, 90, 1098–1106 CAS.
- J. B. Heng, A. Aksimentiev, C. Ho, P. Marks, Y. V. Grinkova, S. Sligar, K. Schulten and G. Timp, Stretching DNA using an electric field in a synthetic nanopore, Nano Lett., 2005, 5, 1883–1888 CrossRef CAS.
- Q. Cai, B. Ledden, E. Krueger, J. A. Golovchenko and J. Li, Nanopore sculpting with noble gas ions, J. Appl. Phys., 2006, 100, 024914 CrossRef.
- M. Wanunu and A. Meller, Chemically modified solid-state nanopores, Nano Lett., 2007, 7, 1580–1585 CrossRef CAS.
- J. Nilsson, J. R. I. Lee, T. V. Ratto and S. E. Letant, Localized functionalization of single nanopores, Adv. Mater., 2006, 18, 427–431 CrossRef CAS.
- S. M. Iqbal, D. Akin and R. Bashir, Solid-state nanopore channels with DNA selectivity, Nat. Nanotechnol., 2007, 2, 243–248 CrossRef CAS.
- P. Chen, T. Mitsui, D. Farmer, J. Golovchenko, R. Gordon and D. Branton, Atomic layer deposition to fine-tune the surface properties and diameters of fabricated nanopores, Nano Lett., 2004, 4, 1333–1337 CrossRef CAS.
- J. B. Heng, A. Aksimentiev, C. Ho, V. Dimitrov, T. Sorsch, J. Miner, W. Mansfield, K. Schulten and G. Timp, Beyond the gene chip, AT&T Bell Lab. Tech. J., 2005, 10, 5–22 Search PubMed.
- G. Timp et al., Progress toward 10 nm CMOS devices, Tech. Dig. - Int. Electron Devices Meet., 1998, IEEE, pp. 615–618 DOI:10.1109/IEDM.1998.746433.
- A. Meller, Dynamics of polynucleotide transport through nanometre-scale pores, J. Phys.: Condens. Matter, 2003, 15, R581–R607 CrossRef CAS.
- M. Muthukumar, Mechanism of DNA transport through pores, Annu. Rev. Biophys. Biomol. Struct., 2007, 36, 435–450 CrossRef CAS.
- W. Im and B. Roux, Ions and counterions in a biological channel: a molecular dynamics study of OmpF porin from Escherichia coli in an explicit membrane with 1 M KCl aqueous salt solution, J. Mol. Biol., 2002, 319, 1177–1197 CrossRef CAS.
- S. Y. Noskov, W. Im and B. Roux, Ion permeation through the α-hemolysin channel: Theoretical studies based on Brownian Dynamics and Poisson-Nernst-Plank electrodiffusion theory, Biophys. J., 2004, 87, 2299–2309 CrossRef CAS.
- M. Saraniti, S. Aboud and R. Eisenderg, The simulation of ionic charge transport in biological ion channels: an introduction to numerical methods, Rev. Comput. Chem., 2006, 22, 229–294 Search PubMed.
- D. J. Bonthuis, J. Zhang, B. Hornblower, J. Mathe, B. I. Shklovskii and A. Meller, Self-energy-limited ion transport in subnanometer channels, Phys. Rev. Lett., 2006, 97, 128104 CrossRef.
- J. Zhang and B. I. Shklovskii, Effective charge and free energy of DNA inside an ion channel, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2007, 75, 021906 CrossRef.
- P. G. de Gennes, Scaling concepts in polymer physics, 1979, Cornell University Press, Ithaca, NY Search PubMed.
- S.-S. Sung and X.-W. Wu, Molecular dynamics simulations of synthetic peptide folding, Proteins: Struct., Funct., Genet., 1996, 25, 202–214 CrossRef CAS.
- M. Muthukumar, Polymer translocation through a hole, J. Chem. Phys., 1999, 111, 10371–10374 CrossRef CAS.
- E. Slonkina and A. B. Kolomeisky, Polymer translocation through a long nanopore, J. Chem. Phys., 2003, 118, 7112–7118 CrossRef CAS.
- M. Muthukumar, Polymer escape through a nanopore, J. Chem. Phys., 2003, 118, 5174–5184 CrossRef CAS.
- T. Ambjörnsson, S. P. Apell, Z. Konkoli, E. A. D. Marzio and J. J. Kasianowicz, Charged polymer membrane translocation, Biophys. J., 2002, 117, 4063–4073 CAS.
- D. K. Lubensky and D. R. Nelson, Driven polymer translocation through a narrow pore, Biophys. J., 1999, 77, 1824–1838 CrossRef CAS.
- R. Metzler and J. Klafter, When translocation dynamics becomed anomalous, Biophys. J., 2003, 85, 2776–2779 CrossRef CAS.
- O. Flomenbom and J. Klafter, Translocation of a single-stranded DNA through a conformationally changing nanopore, Biophys. J., 2004, 86, 3576–3584 CrossRef CAS.
- C. Wong and M. Muthukumar, Polymer capture by electro-osmotic flow of oppositely charged nanopores, J. Chem. Phys., 2007, 126, 164903 CrossRef CAS.
- S. Ghosal, Electrophoresis of a polyelectrolyte through a nanopore, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2006, 74, 041901 CrossRef.
- S. Ghosal, Effect of salt concentration on the electrophoretic speed of a polyelectrolyte through a nanopore, Phys. Rev. Lett., 2007, 98, 238104 CrossRef.
- M. P. Allen and D. J. Tildesley, Computer Simulation of Liquids, 1987, Oxford University Press Search PubMed.
- L. Verlet, Computer ‘experiments’ on classical fluids: I. Thermodynamical properties of Lennard-Jones molecules, Phys. Rev., 1967, 159, 98–103 CrossRef CAS.
- A. MacKerell, Jr. and et al, All-atom empirical potential for molecular modeling and dynamics studies of proteins, J. Phys. Chem. B, 1998, 102, 3586–3616 CrossRef CAS.
- W. D. Cornell, P. Cieplak, C. I. Bayly, I. R. Gould, K. M. Merz, Jr., D. M. Ferguson, D. C. Spellmeyer, T. Fox, J. W. Caldwell and P. A. Kollman, A second generation force field for the simulation of proteins, nucleic acids, and organic molecules, J. Am. Chem. Soc., 1995, 117, 5179–5197 CrossRef CAS.
- A. Aksimentiev, R. Brunner, E. R. Cruz-Chu, J. Comer and K. Schulten, Modeling transport through synthetic nanopores, IEEE Nanotechnol. Mag., 2009, 3, 20–28 Search PubMed.
- A. Aksimentiev, J. B. Heng, G. Timp and K. Schulten, Microscopic kinetics of DNA translocation through synthetic nanopores, Biophys. J., 2004, 87, 2086–2097 CrossRef CAS.
- J. Mathé, A. Aksimentiev, D. R. Nelson, K. Schulten and A. Meller, Orientation discrimination of single stranded DNA inside the α-hemolysin membrane channel, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 12377–12382 CrossRef CAS.
- http://tbgl.physics.illinois.edu/tutorials/ .
- B. L. de Groot and H. Grubmüller, Water permeation across biological membranes: Mechanism and dynamics of aquaporin-1 and GlpF, Science, 2001, 294, 2353–2357 CrossRef CAS.
- E. Tajkhorshid, P. Nollert, M. Ø. Jensen, L. J. W. Miercke, J. O'Connell, R. M. Stroud and K. Schulten, Control of the selectivity of the aquaporin water channel family by global orientational tuning, Science, 2002, 296, 525–530 CrossRef CAS.
- B. Ilan, E. Tajkhorshid, K. Schulten and G. A. Voth, The mechanism of proton exclusion in aquaporin channels, Proteins: Struct., Funct., Bioinf., 2004, 55, 223–228 Search PubMed.
- S. Bernèche and B. Roux, Energetics of ion conduction through the K+ channel, Nature, 2001, 414, 73–77 CrossRef CAS.
- C. Domene, P. J. Bond, and M. S. P. Sansom, Membrane protein simulations: Ion channels and bacterial outer membrane proteins, in Advances in Protein Chemistry, ed. F. M. Richards, D. S. Eisenberg, and J. Kuriyan, 2003, Academic Press, vol. 66, pp. 159–193, Search PubMed.
- T. Allen, O. Andersen and B. Roux, Energetics of ion conduction through the gramicidin channel, Proc. Natl. Acad. Sci. U. S. A., 2003, 101, 117–122 CrossRef.
- L. Monticelli, K. Robertson, J. MacCallum and D. Tieleman, Computer simulation of the KvAP voltage-gated potassium channel: steered molecular dynamics of the voltage sensor, FEBS Lett., 2004, 564, 325–332 CrossRef CAS.
- S. Noskov, S. Berneche and B. Roux, Control of ion selectivity in potassium channels by electrostatic and dynamic properties of carbonyl ligands, Nature, 2004, 431, 830–834 CrossRef CAS.
- K. M. Robertson and D. P. Tieleman, Orientation and interactions of dipolar molecules during transport through OmpF porin, FEBS Lett., 2002, 528, 53–57 CrossRef CAS.
- M. Ceccarelli, C. Danelon, A. Laio and M. Parrinello, Microscopic mechanism of antibiotics translocation through porin, Biophys. J., 2004, 87, 58–64 CrossRef CAS.
- A. Aksimentiev et al., Computer Modeling in Biotechnology: a Partner in Development, Nanostructure Design: Methods and Protocols, vol. 474 of Methods in Molecular Biology, 2008, Humana Press, Totowa, NJ, pp. 181–234 Search PubMed.
- J. Comer, V. Dimitrov, Q. Zhao, G. Timp and A. Aksimentiev, Microscopic mechanics of hairpin DNA translocation through synthetic nanopores, Biophys. J., 2009, 96, 593–608 CrossRef CAS.
- A. Milchev, K. Binder and A. Bhattacharya, Polymer translocation through a nanopore induced by adsorption: Monte Carlo simulations of a coarse-grained model, J. Chem. Phys., 2004, 121, 6042–6051 CrossRef CAS.
- S. Matysiak, A. Montesi, M. Pasquali, A. Kolomeisky and C. Clementi, Dynamics of polymer translocation through nanopore: Theory meets experiment, Phys. Rev. Lett., 2006, 96, 118103 CrossRef.
- S. J. Marrink, A. H. de Vries and A. E. Mark, Coarse grained model for semiquantitative lipid simulations, J. Phys. Chem. B, 2004, 108, 750–760 CrossRef CAS.
- J. Chuang, Y. Kantor and M. Kardar, Anomalous dynamics of translocation, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2001, 65, 011802 CrossRef.
- C. Y. Kong and M. Muthukumar, Modeling of polynucleotide translocation through protein pores and nanotubes, Electrophoresis, 2002, 23, 2697–2703 CrossRef CAS.
- M. Muthukumar and C. Y. Kong, Simulation of polymer translocation through protein channels, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 5273–5278 CrossRef CAS.
- C. Forrey and M. Muthukumar, Langevin dynamics simulations of ds-DNA translocation through synthetic nanopores, J. Chem. Phys., 2007, 127, 015102 CrossRef.
- M. Muthukumar, Translocation of a confined polymer through a hole, Phys. Rev. Lett., 2001, 86, 3188–3191 CrossRef CAS.
- S. S. Chern, A. E. Cárdenas and R. D. Coalson, Three-dimensional dynamic Monte Carlo simulations of driven polymer transport through a hole in a wall, J. Chem. Phys., 2001, 115, 7772–7782 CrossRef CAS.
- C. Chen and E. Peng, Nanopore sequencing of polynucleotides assisted by a rotating electric field, Appl. Phys. Lett., 2003, 82, 1308–1310 CrossRef CAS.
- H. C. Loebl, R. Randel, S. P. Goodwin and C. C. Matthai, Simulation studied of polymer translocation through a channel, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2003, 67, 041913 CrossRef CAS.
- M. G. Fyta, S. Melchionna, E. Kaxiras and S. Succi, Multiscale coupling of molecular dynamics and hydrodynamics: Application to DNA translocation through a nanopore, Multiscale Model. Simul., 2006, 5, 1156–1173 CrossRef.
- M. Fyta, S. Melchionna, S. Succi and E. Kaxiras, Hydrodynamic correlations in the translocation of a biopolymer through a nanopore: Theory and multiscale simulations, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2008, 78, 036704 CrossRef.
- A. J. Storm, C. Storm, J. Chen, H. Zandbergen, J.-F. Joanny and C. Dekker, Fast DNA translocation through a solid-state nanopore, Nano Lett., 2005, 5, 1193–1197 CrossRef CAS.
- S. Reboux, F. Capuani, N. Gonzalez-Segredo and D. Frenkel, Lattice-Boltzmann simulations of ionic current modulation by DNA translocation, J. Chem. Theory Comput., 2006, 2, 495–503 CrossRef CAS.
- M. Gracheva, A. Xiong, A. Aksimentiev, K. Schulten, G. Timp and J.-P. Leburton, Simulation of the electric response of DNA translocation through a semiconductor nanopore-capacitor, Nanotechnology, 2006, 17, 622–633 CrossRef CAS.
- D. E. Shaw et al., Anton, a special-purpose machine for molecular dynamics simulation. International Symposium on Computer Architecture, 2007, ACM Press, New York, pp. 1–12, Search PubMed.
- F. Khalili-Araghi, E. Tajkhorshid and K. Schulten, Dynamics of K+ ion conduction through Kv1.2, Biophys. J., 2006, 91, L72–L74 CrossRef CAS.
- P. W. Atkins, Physical Chemistry, 1998, Oxford University Press Search PubMed.
- S. Pezeshki, C. Chimerel, A. N. Bessonov, M. Winterhalter and U. Kleinekathofer, Understanding ion conductance on a molecular level: An all-atom modeling of the bacterial porin OmpF, Biophys. J., 2009, 97, 1898–1906 CrossRef CAS.
- C. Chimerel, L. Movileanu, S. Pezeshki, M. Winterhalter and U. Kleinekathofer, Transport at the nanoscale: temperature dependence of ion conductance, Eur. Biophys. J., 2008, 38, 121–125 CrossRef CAS.
- P. F. Batcho, D. A. Case and T. Schlick, Optimized particle-mesh Ewald/multiple-time step integration for molecular dynamics simulations, J. Chem. Phys., 2001, 115, 4003–4018 CrossRef CAS.
- R. A. Böckmann, B. L. de Groot, S. Kakorin, E. Neumann and H. Grubmüller, Kinetics, statistics, and energetics of lipid membrane electroporation studied by molecular dynamics simulations, Biophys. J., 2008, 95, 1837–1850 CrossRef.
- P. S. Crozier, D. Henderson, R. L. Rowley and D. D. Busath, Model channel ion currents in NaCl-extended simple point charge water solution with applied-field molecular dynamics, Biophys. J., 2001, 81, 3077–3089 CrossRef CAS.
- W. Nonner, A. Peyser, D. Gillespie and B. Eisenberg, Relating microscopic charge movement to macroscopic currents: The ramo-shockley theorem applied to ion channels, Biophys. J., 2004, 87, 3716–3722 CrossRef CAS.
- S. M. Bezrukov and J. J. Kasianowicz, Current noise reveals protonation kinetics and number of ionizable sites in an open protein ion channel, Phys. Rev. Lett., 1993, 70, 2352–2355 CrossRef CAS.
- J. J. Kasianowicz and S. M. Bezrukov, Protonation dynamics of the α-toxin ion channel from spectral analysis of ph dependent current fluctuations, Biophys. J., 1995, 69, 94–105 CrossRef CAS.
- M. Sotomayor, V. Vasquez, E. Perozo and K. Schulten, Ion conduction through MscS as determined by electrophysiology and simulation, Biophys. J., 2007, 92, 886–902 CrossRef CAS.
- E. R. Cruz-Chu, A. Aksimentiev and K. Schulten, Ionic current rectification through silica nanopores, J. Phys. Chem. C, 2009, 113, 1850–1862 CrossRef CAS.
- J. C. Phillips, R. Braun, W. Wang, J. Gumbart, E. Tajkhorshid, E. Villa, C. Chipot, R. D. Skeel, L. Kale and K. Schulten, Scalable molecular dynamics with NAMD, J. Comput. Chem., 2005, 26, 1781–1802 CrossRef CAS.
- M. Wanunu, J. Sutin, B.M., A. Chow and A. Meller, DNA translocation governed by interactions with solid-state nanopores, Biophys. J., 2008, 95, 4716–4725 CrossRef CAS.
- U. Mirsaidov, D. Wang, J. Comer, W. Timp, V. Dimitrov, J. W. ShimA. Aksimentiev, and G. Timp, Trapping a double-stranded DNA molecule in a nanopore to discriminate between base-pairs, 2009, submitted.
- I. C. Yeh and G. Hummer, Diffusion and electrophoretic mobility of single-stranded RNA from molecular dynamics simulations, Biophys. J., 2004, 86, 681–689 CrossRef CAS.
- Q. Zhao, J. Comer, V. Dimitrov, A. Aksimentiev and G. Timp, Stretching and unzipping nucleic acid hairpins using a synthetic nanopore, Nucleic Acids Res., 2008, 36, 1532–1541 CrossRef CAS.
- B. Dorvel, G. Sigalov, Q. Zhao, J. Comer, V. Dimitrov, U. Mirsaidov, A. Aksimentiev and G. Timp, Analyzing the forces binding a restriction endonuclease to DNA using a synthetic nanopore, Nucleic Acids Res., 2009, 37, 4170–4179 CrossRef CAS.
- A. Meller, L. Nivon and D. Branton, Voltage-driven dna translocations through a nanopore, Phys. Rev. Lett., 2001, 86, 3435–3438 CrossRef CAS.
- D. B. Wells, V. Abramkina and A. Aksimentiev, Exploring transmembrane transport through alpha-hemolysin with grid-based steered molecular dynamics, J. Chem. Phys., 2007, 127, 125101 CrossRef.
- B. Isralewitz, S. Izrailev and K. Schulten, Binding pathway of retinal to bacterio-opsin: A prediction by molecular dynamics simulations, Biophys. J., 1997, 73, 2972–2979 CrossRef CAS.
- H. Grubmüller, B. Heymann and P. Tavan, Ligand binding and molecular mechanics calculation of the streptavidin-biotin rupture force, Science, 1996, 271, 997–999 CrossRef CAS.
- S.-J. Marrink, O. Berger, P. Tieleman and F. Jähnig, Adhesion forces of lipids in a phospholipid membrane studied by molecular dynamics simulations, Biophys. J., 1998, 74, 931–943 CrossRef CAS.
- C. Jarzynski, Nonequilibrium equality for free energy differences, Phys. Rev. Lett., 1997, 78, 2690–2693 CrossRef CAS.
- S. Park and K. Schulten, Calculating potentials of mean force from steered molecular dynamics simulations, J. Chem. Phys., 2004, 120, 5946–5961 CrossRef CAS.
- Y. Wang, K. Schulten and E. Tajkhorshid, What makes an aquaporin a glycerol channel: A comparative study of AqpZ and GlpF, Structure, 2005, 13, 1107–1118 CrossRef CAS.
- J. Gumbart and K. Schulten, Molecular dynamics studies of the archaeal translocon, Biophys. J., 2006, 90, 2356–2367 CrossRef CAS.
- H. Chang, F. Kosari, G. Andreadakis, M. A. Alam, G. Vasmatzis and R. Bashir, DNA-mediated fluctuations in ionic current through silicon oxide nanopore channels, Nano Lett., 2004, 4, 1551–1556 CrossRef CAS.
- R. M. M. Smeets, U. F. Keyser, D. Krapf, M. Y. Wu, N. H. Dekker and C. Dekker, Salt dependence of ion transport and DNA translocation through solid-state nanopores, Nano Lett., 2006, 6, 89–95 CrossRef CAS.
- E. R. Cruz-Chu, A. Aksimentiev and K. Schulten, Water-silica force field for simulating nanodevices, J. Phys. Chem. B, 2006, 110, 21497–21508 CrossRef CAS.
- M. Bates, M. Burns and A. Meller, Dynamics of DNA molecules in a membrane channel probed by active control techniques, Biophys. J., 2003, 84, 2366–2372 CrossRef CAS.
- J. Nakane, M. Wiggin and A. Marziali, A nanosensor for transmembrane capture and identification of single nucleic acid molecules, Biophys. J., 2004, 87, 615–621 CrossRef CAS.
- B. McNally, M. Wanunu and A. Meller, Electromechanical unzipping of individual DNA molecules using synthetic sub-2 nm pores, Nano Lett., 2008, 8, 3418–3422 CrossRef CAS.
- G. Manning, Limiting laws and counterion condensation in polyelectrolyte solutions, J. Phys. Chem., 1981, 85, 1506 CrossRef CAS.
- D. Long, J. L. Viovy and A. Ajdari, Simultaneous action of electric fields and nonelectric forces on a polyelectrolyte: Motion and deformation, Phys. Rev. Lett., 1996, 76, 3858 CrossRef CAS.
- B. Luan and A. Aksimentiev, Electro-osmotic screening of the dna charge in a nanopore, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2008, 78, 021912 CrossRef.
- S. Ghosal, Electrokinetic-flow-induced viscous drag on a tethered dna inside a nanopore, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2007, 76, 061916 CrossRef.
- M. Wiggin, C. Tropini, V. Tabard-Cossa, N. Jetha and A. Marziali, Nonexponential kinetics of dna escape from alpha-hemolysin nanopores, Biophys. J., 2008, 95, 5317–5323 CrossRef CAS.
- T. Chou, Enhancement of charged macromolecule capture by nanopores in a salt gradient, J. Chem. Phys., 2009, 131, 034703 CrossRef.
- M. Wanunu, W. Morrison, Y. Rabin, A. Y. Grosberg and A. Meller, Electrostatic focusing of unlabelled DNA into nanoscale pores using a salt gradient, Nat. Nanotechnol., 2009 DOI:10.1038/nnano.2009.379.
| This journal is © The Royal Society of Chemistry 2010 |