Synthesis of covalently attached hexadecaanilines on carbon nanotubes: toward electronic nanocarbon preparation†
Long Y.
Chiang
*a,
Robinson
Anandakathir
a,
Tanya S.
Hauck
b,
Lawrence
Lee
b,
Taizoon
Canteenwala
a,
Prashant A.
Padmawar
a,
Kenneth
Pritzker
b,
Ferdinando F.
Bruno
c and
Lynne A.
Samuelson
c
aDepartment of Chemistry, University of Massachusetts Lowell, Lowell, MA 01854, USA. E-mail: Long_Chiang@uml.edu
bPathology and Laboratory Medicine, University of Toronto, Toronto, Canada
cU. S. Army RDECOM, Natick Soldier Center, Natick, MA 01760, USA
First published on 16th January 2010
Abstract
We describe the direct covalent-grafting synthesis of well-defined aniline oligomers, such as tetraaniline (A4) and hexadecaaniline (A16, major)/eicosaaniline (A20, minor), on the sidewalls of carbon nanotubes (CNTs), via dediazonization reaction, for achieving highly soluble nanomaterials suitable for printing purposes, with long-term physical stability. Chemically grafting a layer of electroactive hexadecaanilines on CNTs resembles semiconductive encapsulation of functionalized CNTs. The resulting covalent nanoconjugates SWNT–(A4)x, MWNT–(A4)x, SWNT–(A16/20)x, and MWNT–(A16/20)x were characterized by various spectroscopic and microscopic mapping methods. The combination of transmission electron microscopy (TEM) and electron energy loss spectroscopy (EELS) analyses provided direct evidence for A16/20 attachment to the CNTs, giving confirmation of the presence of heteroatoms surrounding the CNTs that was absent in the parent CNTs. Subsequent atom mapping in the vicinity of the tube structure allowed us to illustrate the 3D distribution of heteroatoms along the CNT surface.
Introduction
Research on supercapacitors1 and rechargeable batteries in a printable thin film form as the emerging alternative cost-effective energy storage system for portable electronic devices has become a recent focus of active study. The first few examples were demonstrated recently using formulated single-walled carbon nanotube (SWNT) solutions2 or polymer composites.3 In a similar aspect, simultaneous enhancement of the double-layer capacitance and Faradic pseudocapacitance can be achieved by the modification of carbon nanotubes (CNTs) with conductive polymers (CPs).4 This is due to the fact that conducting polymers often exhibit large specific capacitance,5 however, they also exhibit the disadvantage of a lower service-life during charge–discharge cycles as a result of possible degradation of redox sites in the polymer backbone. Among the variety of conducting polymers, polyanilines (PAni) exhibit better reversibility of electrochemical reduction–oxidation properties with higher ambient stability in air than other CPs. Polyanilines also show quick electrochemical switching and electrochromic effects with moderate conductivities, in various doped forms, and can be produced using facile preparation methods in aqueous media.6 Therefore, it is plausible to incorporate either conducting polymers7 or polyanilines8 onto CNTs, or related carbon materials, to produce one integrated nanocomposite electrode structure for realising the synergistic effect of nanomaterials exhibiting appreciable conductivity, long service-life cycle, and improved specific capacitance.For the preparation of CNT–PAni composites,9 a number of methods including a non-covalent solution coating processes, the wrapping of CNTs with conjugate polymers, or in situ polymerization of aniline in the presence of MWNTs10 have been applied, giving the electrical conductivity of the resulting composite materials in a range matching well with that of the parent CNTs.11 These non-covalent binding approaches may not prevent physical separation of CNT–PAni composite components under long-term or severe service conditions. Here, we report an alternative approach by direct covalent-grafting of well-defined aniline oligomers, such as tetraaniline (A4) and hexadecaaniline (A16, major)/eicosaaniline (A20, minor), on the sidewall of CNTs for achieving highly soluble nanomaterials suitable for printing purposes with long-term physical stability. Accordingly, the synthesis and characterization of soluble SWNT–(A4)x, MWNT–(A4)x, SWNT–(A16/20)x, and MWNT–(A16/20)x nanoconjugates, composed of oligoaniline moieties each being attached to a CNT by a covalent bond, are described. Chemical grafting a layer of electroactive hexadecaanilines on CNTs resembles the semiconductive encapsulation of functionalized carbon nanotubes. Hexadecaanilines in the emeraldine base (EB) form exhibit unique reversible redox potential waves and are susceptible to protonation and de-protonation cycles for conductivity switching at different oxidation states in close resemblance to those of a high-molecular-weight PAni, making them attractive for electroactive materials in modified CNT electrodes.
Results and discussion
Aryl diazonium salts are known to reacts with aromatic compounds typically giving rise to reactive aryl radicals or in some cases through an aryl cation as the intermediate. Electrochemical grafting of the aryl diazonium salt on a carbon surface i.e. pyrolitic graphite or a glassy carbon electrodes has been successfully demonstrated.12 In this case, reduction of aryl diazonium salts gives an aryl radical that covalently attaches to the carbon surface. Although electrochemically induced reaction is the most favorable process, thermal reaction is also efficacious. Derivatization of small-diameter nanotubes via electrochemical reduction of a variety of bulky small molecular aryl diazonium salts was reported leading to functionalized CNTs with significant improvement of solubility in organic solvents.13We produced the similar diazonium salt forms of tetraaniline (A4) and hexadecaaniline/eicosaaniline14 (A16/20) in their EB structures by reaction with isoamyl nitrite under sonication as a precursor step for the generation of the corresponding reactive intermediates in situ. Subsequent grafting of these diazonium intermediates onto the sidewall surface carbons of SWNTs and MWNTs in separate reactions was accomplished by repetitive cycles of sonication to afford a good yield of soluble SWNT–(A4)x, MWNT–(A4)x, SWNT–(A16/20)x, and MWNT–(A16/20)x products, respectively, as shown in Fig. 1. In this particular reaction, SWNTs exhibited a higher reactivity with diazonium–A16/20 than MWNTs, in terms of a relatively larger quantity in weight percent of soluble products obtained within a short reaction period at ambient temperature to 45 °C. It also required a smaller quantity of diazonium–A16/20 reagent, 8–10 fold in weight of the raw SWNTs, to complete the grafting conversion to the soluble products than that (10–15 fold in weight) for the similar reaction using MWNTs. Variation of the MWNT source tends to influence the degree of functionalization and the product yield. As a result, long nanotubes (purchased from Conyuan Biochemical Co., Taiwan), exhibiting good intrinsic electrical conductivity, and short nanotubes with a large number of defect sites, such as ball-milled pipes, gave less insoluble reaction residues with a highly soluble product yield. Interestingly, reacted MWNTs were found to possess a tendency to consume diazonium–A16/20 reagents faster than unreacted MWNTs which allowed clear separation of the DMF-soluble and -insoluble fractions as the highly functionalized and unreacted or partially functionalized MWNTs, respectively, if a smaller quantity of diazonium–A16/20 was used in a partial reaction. Accordingly, a negligible degree of A16/20 attachments on insoluble nanotube residues was substantiated by the IR spectra of the latter fraction showing a nearly identical profile to that of raw MWNTs with the absence of major hexadecaanilinyl optical absorption peaks. This phenomenon was also subsequently confirmed by subjecting the insoluble fractions to repeated reaction under the same conditions, resulting in the detection of more soluble MWNT–(A16/20)x materials. Both soluble SWNT–(A16/20)x and MWNT–(A16/20)x exhibit a dark blue color in DMF and DMSO solutions. A typical mass of blue DMF-soluble SWNT–(A16/20)x and MWNT–(A16/20)x solids was roughly 100 and 75 mg, respectively, obtained per 20 mg of the raw CNTs applied in a complete reaction. Remaining residual solids were roughly 30% by weight raw CNTs used, and were assumed to be a combination of non-CNT related carbon materials and partially functionalized CNTs. The higher product yield for SWNTs than MWNTs may be correlated to their higher total reactive surface area for the same weight. Either unreacted A4 or A16/20 was removed from the corresponding product fractions by re-precipitation and repeatedly washing of the solids with acetonitrile, THF, MeOH, and ether with occasional sonication until no significant color remained in the washings. Large solubility difference between CNT–(A16/20)x and A16/20 in THF allowed us to purify the products without difficulty.
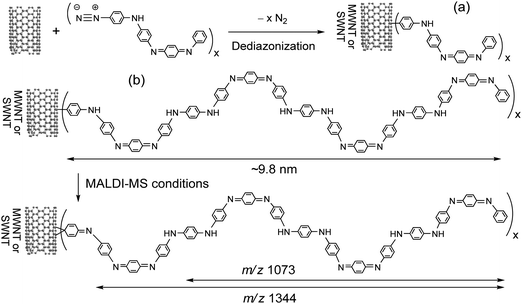 | ||
| Fig. 1 Synthesis and proposed fragmentations of (a) SWNT–(A4)x or MWNT–(A4)x and (b) SWNT–(A16)x or MWNT–(A16)x under experimental MALDI-MS conditions. | ||
The oligoanilines applied in this study are the major products synthesized from a sequence of procedures including controlled oligomerization of the well-defined starting aniline tetramer (A4), product separation, and repeated purification. Only an isolated aniline oligomer fraction showing a single chromatographic band in a liquid chromatographic (HPLC, Fig. S1 of the ESI†) experiment was used for the grafting reaction to CNTs. Its molecular weight was confirmed by matrix-assisted laser desorption ionization (MALDI-TOF) mass spectrometry (MS), using α-cyano-4-hydroxycinnamic acid as the matrix. The spectrum fits well with the mass of hexadecaaniline (A16) and eicosaaniline (A20) showing the corresponding group of protonated molecular ion mass (MH+) peaks centered at m/z = 1451 and 1813, respectively, as shown in Fig. 2(b). Based on the relative intensity of these two mass ion peaks and the separate end-group capping experiment of the amino group with an alkyl aldehyde giving the corresponding anilinic proton integration count in the 1H NMR spectrum, hexadecaanilines were elucidated to be the major oligomers with eicosaanilines as the minor components in the fraction. A distribution of molecular mass ion from m/z = 1447 to 1455 [inset of Fig. 2(b)] for the A16 peak was observed that revealed a slight variation of benzenoid vs. quinonoid moieties and protonated states in the structure of A16, with the latter being generated under MALDI-MS conditions. It was followed by a series of major fragmentation mass ion peaks at m/z = 1089, 727, and 366 corresponding to the mass of protonated dodecaanilines (A12H+), octaanilines (A8H+), and tetraanilines (A4H+), respectively, with several minor mass ion peaks of low peak intensity located between these major groups of peaks. The mass difference between minor mass ion peaks can be accounted for by the consecutive loss of one aniline unit of m/z = 90 or 91 for the moiety of quinonoid or benzenoid, respectively. The overall spectrum implied good stability of the A4 unit during the fragmentation processes, consistent with the MALDI mass spectrum of tetraanilne [Fig. 2(a)] showing only the protonated molecular mass ion (MH+) peaks at m/z = 365 and 366 with no obvious mass fragmentation at m/z = 260–350. Each A4 unit contains a quinonoid moiety in EB form that may contribute to the cause of the observed stability.
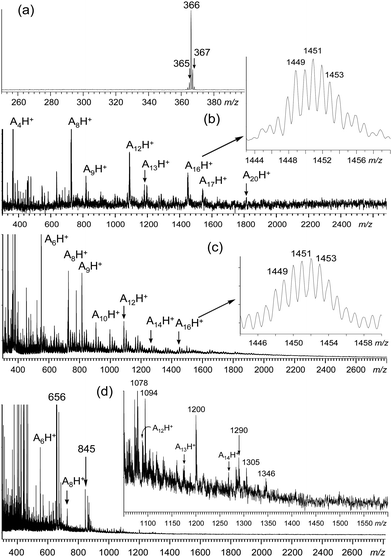 | ||
| Fig. 2 MALDI-TOF mass spectra of (a) tetraaniline (A4), (b) hexadecaaniline/eicosaaniline (A16/20), (c) DMF-soluble fraction of MWNTs and A16/20 mixture residue, and (d) oligoaniline-grafted MWNT products, MWNT–(A16/20)x. | ||
Facile detection of the A16-related molecular mass ion peaks allowed us to apply the same MS method to distinguish between physically absorbed vs. covalently bonded CNT–(A16)x samples. In the former case, the same quantity of A16/20 and carbon nanotubes, either SWNTs or MWNTs, as those applied to the chemical grafting reaction were subjected to sonication treatments in DMF, except in the absence of the isoamyl nitrite reagent. As a result, a nearly quantitative amount of A16/20 was recovered in the work-up by repeated THF washings of the mixture, which implies its low degree of incorporation or physical absorption on CNTs. This was also confirmed by the insoluble solid fraction showing nearly identical characteristic infrared absorption band profiles as those of the parent CNTs, revealing negligible presence of A16 that clearly ruled out the possibility of A16 physisorption on the isolated CNTs. Accordingly, MALDI-MS data of the DMF-soluble sample displayed a group of the highest mass ion peaks centered at m/z = 1452 [the inset of Fig. 2(c)] with a fragmentation pattern that closely resembles that of A16/20 in Fig. 2(b), indicating the presence of either free A16/20 or easy dissociation of A16/20 from CNTs if it was physically absorbed. One plausible explanation of low physisorption is given by the relative short molecular length of A16 of roughly 98 Å as estimated by a 3D molecular modeling technique (Fig. 1). Unlike high-molecular-weight polyanilines, the molecular length of A16/20 is apparently not long enough to wrap around a carbon nanotube.
In the case of covalently bonded MWNT–(A16/20)x samples, a dark blue solution of the DMF-soluble material fraction taken in DMSO was examined by the same MALDI-MS technique. As shown in the inset of Fig. 2(d), no obvious mass ion peaks in the region of m/z = 1445–1455 were detected indicating no free or physically absorbed A16 molecules in the sample. Interestingly, the highest mass ion peak located at m/z = 1346 agrees well with the mass of a protonated deaminated pentadecaaniline (A15H+–N) fragment. The absence of this mass ion peak in the mass spectrum of A16 [Fig. 2(b)] revealed no correlation of it to the fragmentation of free A16. In fact, it is plausible to expect the facile cleavage of a phenylquinonoid A14C6H4–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) bond generated under MALDI-MS conditions, as the partial structure shown in Fig. 1(b). Subsequent loss of three carbons, four carbons, and a phenyl group from m/z = 1346 led to a series of fragmented mass ion peaks at m/z = 1305, 1290, and 1268 (A14H+), respectively. These data provide clear evidence of the chemical attachment of A16 arms to MWNTs showing the corresponding fragmentation pattern in the spectrum. In the lower mass region, the related fragments at m/z = 1179, 1089, 727, and 550 were accounted for by the mass ions of A13H+, A12H+, A8H+, and A6H+, respectively. The lower relative peak intensity of A8H+ and A12H+ mass ions than those in the spectrum of A16 [Fig. 2(b)] may be indicative of the shift of quinonoid units along the oligoaniline chain once attached to a CNT. The phenomena should lead to a deviation of the oxidation state from its EB form. Further measurements are required to confirm this hypothesis.
bond generated under MALDI-MS conditions, as the partial structure shown in Fig. 1(b). Subsequent loss of three carbons, four carbons, and a phenyl group from m/z = 1346 led to a series of fragmented mass ion peaks at m/z = 1305, 1290, and 1268 (A14H+), respectively. These data provide clear evidence of the chemical attachment of A16 arms to MWNTs showing the corresponding fragmentation pattern in the spectrum. In the lower mass region, the related fragments at m/z = 1179, 1089, 727, and 550 were accounted for by the mass ions of A13H+, A12H+, A8H+, and A6H+, respectively. The lower relative peak intensity of A8H+ and A12H+ mass ions than those in the spectrum of A16 [Fig. 2(b)] may be indicative of the shift of quinonoid units along the oligoaniline chain once attached to a CNT. The phenomena should lead to a deviation of the oxidation state from its EB form. Further measurements are required to confirm this hypothesis.
Incorporation of A16 and A20 arms on CNTs was clearly revealed by the infrared spectra (Fig. 3) of DMF-soluble SWNT–(A16/20)x and MWNT–(A16/20)x fractions showing main absorption bands centered at 1592, 1508, 1309, 1176, 837, 757, and 696 cm−1 in close resemblance to those of A4 or A16/20. Similarly, as shown in Fig. 4, the UV–vis–NIR spectrum of soluble SWNT–(A16/20)x showed two main characteristic peaks of A16/20 moieties with a complete loss of near-IR features of the parent SWNTs, indicating disruption in the electronic structure of nanotubes upon covalent modification at its surface carbons.15 In the case of soluble MWNT–(A16/20)x, a slight blue-shift of two strong characteristic chromophore bands centered at 300–310 and 575–585 nm, corresponding to the absorption of benzenoid moieties (–NH–C6H4–NH–) and quinonoid moieties (–NC6H4N–), respectively, of MWNT–(A16/20)x was detected as compared with those of A16/20 (314 and 603 nm). The 1H NMR spectra of DMF-soluble MWNT–(A16/20)x in DMSO-d6 (Fig. S2 of the ESI†) showed no significant aliphatic proton peaks at δ 0.5–2.0 with the absence of aryloxylated carbon protons at roughly δ 3.2, indicating no incorporation of isoamyloxy groups on A16/20 moieties of MWNT–(A16/20)x. It showed broad aromatic proton peaks at δ 6.5–7.7 with the main peak centered at δ 7.1 that appeared to be identical to those of the parent hexadecaaniline–eicosaaniline. All spectroscopic characterizations substantiated that the observed good solubility of MWNT–(A16/20)x and SWNT–(A16/20)x in DMF and DMSO giving a dark-blue solution is attributed to the attachment of A16/20 chains to the CNTs.
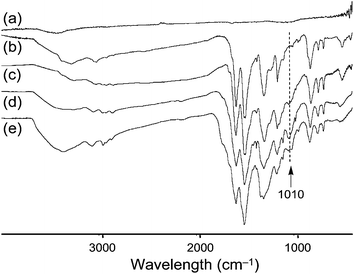 | ||
| Fig. 3 FTIR spectra of (a) MWNTs, (b) A16, and MWNT–A16 synthesized at (c) 25, (d) 60, and (e) 85 °C. | ||
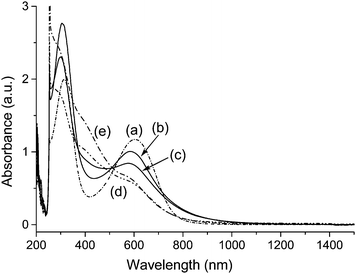 | ||
| Fig. 4 UV–vis–NIR spectra of (a) hexadecaaniline–eicosaaniline (A16/20, 3.0 × 10−4 M) and MWNT–(A16/20)x as DMF-soluble products isolated from the reaction carried out at (b) 25 °C, (c) 50 °C for 3.0 h, (d) 50 °C for 10 h, and (e) 85 °C in DMSO (2.0 mg mL−1). | ||
The degree or density of functionalization on MWNTs was estimated by elemental analyses of MWNT–(A16/20)x (DMF-soluble fraction) resulting in a N![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :C:H ratio of 20:132:73 (assuming A16 to be the major component with each A16 arm containing 73 Hs) on average. The calculated values were also based on the assumption that all residual solids remaining in the measurement vessel were composed of carbon, since incomplete combustion of the nanotube moiety during the analysis procedure is common for all samples. These ratios correlated to roughly the attachment of one A16 arm per 36 carbons of CNTs, indicating a relatively high degree of functionalization considering the bulky size of hexadacaanilines each in a calculated long axis length of roughly 9.8 nm. The slightly higher nitrogen element ratio measured than that of the individual A16 may be due to residual DMF trapped in the material. Results of these analyses agree approximately with the yield of soluble products isolated in roughly 4.0 fold of raw CNTs by weight after deduction of estimated amorphous carbon impurities.
:C:H ratio of 20:132:73 (assuming A16 to be the major component with each A16 arm containing 73 Hs) on average. The calculated values were also based on the assumption that all residual solids remaining in the measurement vessel were composed of carbon, since incomplete combustion of the nanotube moiety during the analysis procedure is common for all samples. These ratios correlated to roughly the attachment of one A16 arm per 36 carbons of CNTs, indicating a relatively high degree of functionalization considering the bulky size of hexadacaanilines each in a calculated long axis length of roughly 9.8 nm. The slightly higher nitrogen element ratio measured than that of the individual A16 may be due to residual DMF trapped in the material. Results of these analyses agree approximately with the yield of soluble products isolated in roughly 4.0 fold of raw CNTs by weight after deduction of estimated amorphous carbon impurities.
The combination of transmission electron microscopy (TEM) and electron energy loss spectroscopy (EELS) provides an important means for direct confirmation of oligoaniline grafting on the surface of the CNTs. In a control experiment, the absence of nitrogen in the blank (raw) SWNTs and MWNTs sample tubes, even after high contrast enhancement, facilitated the feasibility of applying EELS measurements to MWNT–(A16/20)x samples using the ionization edge of the nitrogen element in the region of 401 eV for the image mappingt. The covalent grafting of hexadecaaniline arms may be monolayer in nature. A high nitrogen-to-carbon element ratio (16 nitrogen atoms per A16) of hexadecaaniline improved the visibility of weak plasmon loss signals of the nitrogen in the monolayer. The image was further enhanced by magnification and contrast enrichment to overcome the background noise.
In the case of soluble MWNT–(A16/20)x samples, a bright-field TEM image of a selected nanotube was taken first, as shown in Fig. 5(a), as the reference of the tube shape and the width of external cross-sectional diameter. An EELS image of the same nanotube [Fig. 5(b)] and its contrast-enhanced image [Fig. 5(c)] were subsequently taken for comparison with the prior TEM image. It showed the location of nitrogen signals emitted from the A16/20 moieties of MWNT–(A16/20)x in the grafted region of the tube. Micrographs of both Fig. 5(b) and 5(c) were also arranged to illustrate the nitrogen signal intensity as a function of distance along the cross-section of the tube, with the result depicted in incremental pixels [Fig. 5(d)]. This intensity plot was then normalized by subtracting the value measured at the centre of the tube from each data point in a line plot into Fig. 5(e).
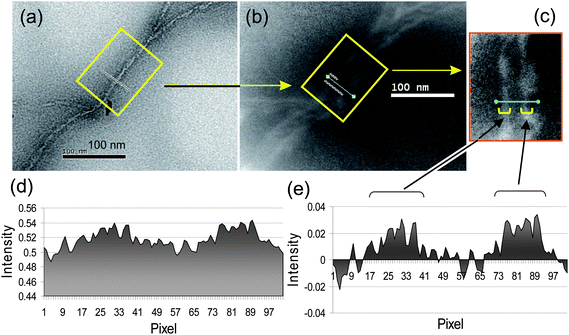 | ||
| Fig. 5 TEM micrographs of (a) the bright-field image of a MWNT–(A16/20)x tube, (b) EELS nitrogen mapping image of the same tube, (c) an enhanced EELS nitrogen image of the same tube segment marked by a rectangle in (b), (d) nitrogen signal intensity graph across the bar in the rectangle of (c), and (e) normalized nitrogen intensity data of (d) showing higher peak signal around the edges of the tube. | ||
Comparing Fig. 5(a) and 5(b), a nearly identical tube shape between TEM and EELS images of a MWNT–(A16/20)x sample tube segment was observed indicating that the location of nitrogen elements coincide within the tube region. No mismatching nitrogen signals outside the tube were detectable. Most significantly, the distribution of signal intensity was apparently fairly even along the long dimension of the tube revealing no large, differentiable disruption of hexadecaaniline functionalization on the carbons located at the flat surface region from the branched area, kink spots, and curve regions. As the nitrogen intensity [Fig. 5(d) and 5(e)] was mapped across a short tube segment perpendicular to the long tube dimension, two multiple peak regions were convoluted after intensity normalization. These two regions match well with the sidewall area of the nanotube indicating higher nitrogen content at both sides of the tubing image edge than the central tube area. That allowed us to visualize the actual A16/20-grafted domains along the long tube dimension and measure more accurately the edge-to-edge distance of the cross-section covered by A16/20 moieties. As a result, the linewidth shown in Fig. 5(c) corresponds to roughly 55–60 nm. As the average tube width of the blank MWNT control, measured separately, was in a range of 20–40 nm, the additional width of 20–35 nm should be attributed to the attachment of A16 at both image edges of the tube. This extra width agrees approximately with two calculated full molecular lengths of A16, each roughly 10 nm.
The electrochemical properties of MWNT–(A16/20)x were evaluated in DMF via cyclic voltammetry (CV). Prior to the measurement, the sample was deposited on a Pt-foil working electrode as a thin coating from DMSO solution. As shown in Fig. 6, the cyclic voltammograms of MWNT–(A16/20)x indicate the first (Eox1) and second oxidation (Eox2) potentials at −0.32 and 0.63 V, respectively, using Ag/AgCl as the reference electrode. They were accompanied by the corresponding first (Ered1) and second reduction (Ered2) potentials at −0.41 and 0.24 V, respectively. These reductive potentials were found to closely resemble those of the parent A16 (purified), being only 0.1 and 0.2 V, respectively, lower in value. The conductivity of doped MWNT–(A16/20)x was measured by the four-point probe method in sheet resistivity on a highly compressed pellet at ambient temperature. During the doping process, finely divided amorphous power of MWNT–(A16/20)x was subjected to exposure to gas-phase HCl in a closed container for a period of 12 h. As a result, a range of conductivities (σ) from 10−3 to 10−2 S cm−1 was measured for the DMF-soluble fraction of the products, as MWNT–(A16/20)x–(HCl)y, a similar value to that of HCl-doped A16/20 (neat). The conductivity is dependent on the dopant used and the doping level, with a higher σ value for HCl-saturated MWNT–(A16/20)x. The value falls in the similar range to that (σ = 0.01–0.1 S cm−1) of the parent MWNTs used. A gradual increase of the conjugative chain length above that of A16/20 is expected to significantly increase the conductivity above 10−2 S cm−1. Therefore, SWNT–(A16/20)x and MWNT–(A16/20)x become important precursor conjugates for the preparation of SWNT–(PAni)x and MWNT–(PAni)x, respectively, currently under investigation, with each arm having a significantly higher number of aniline repeating units than A16/20.
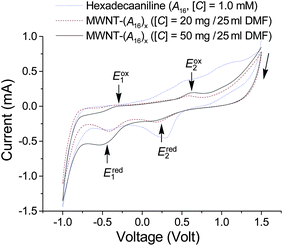 | ||
| Fig. 6 Cyclic voltammograms of hexadecaaniline (A16), and MWNT–(A16/20)x in DMF in two concentrations using Ag/AgCl as the reference electrode, a Pt-foil as the working electrode, a Pt-rod as the counter electrode, and HCl (1.0 M) as the electrolyte. | ||
Conclusion
We have described the direct covalent-grafting synthesis of well-defined aniline oligomers, such as tetraaniline (A4) and hexadecaaniline (A16, major)/eicosaaniline (A20, minor), on the sidewalls of carbon nanotubes (CNTs), via the dediazonization reaction of the corresponding diazonium salts, for achieving highly soluble nanomaterials suitable for printing purposes with long-term physical stability. Chemical grafting a layer of electroactive hexadecaanilines on CNTs resembles semiconductive encapsulation of functionalized CNTs. The resulting covalent nanoconjugates of SWNT–(A4)x, MWNT–(A4)x, SWNT–(A16/20)x, and MWNT–(A16/20)x were characterized by various spectroscopic and microscopic mapping methods. The combination of transmission electron microscopy (TEM) and electron energy loss spectroscopy (EELS) analyses provided direct evidence for the attachment of A16 to the CNTs confirming the presence of heteroatoms surrounding a CNT that were absent in the parent CNTs. Subsequent atom mapping along the vicinity of the tube structure allowed us to illustrate the 3D distribution of heteroatoms along the CNT surfaceCovalent bonding of hexadecaanilinyl arms to CNTs is expected to greatly improve their compatibility with conventional polymers for processing and high physical durability during their service life. Most importantly, preliminary results revealed that appreciable semiconductivity can be realized on HCl-doped CNT–(A16/20)x nanoconjugates while preserving their unique high mechanical strength, one of the intrinsic physical properties of CNTs.
Experimental methods
General
Both single-walled carbon nanotubes and multi-walled carbon nanotubes were purchased from Conyuan Biochemical Co., Taiwan. All other chemicals were purchased from Acros Ltd. 1H NMR and 13C NMR spectra were recorded on either a Bruker Spectrospin–500 or Bruker AC-250 spectrometer. Mass spectroscopic studies were performed by the use of positive ion matrix-assisted laser desorption ionization (MALDI-TOF) mass spectrometry (MS) using α-cyano-4-hydroxycinnamic acid as the matrix on a micromass M@LDI-LR mass spectrometer. Infrared spectra were recorded as KBr pellets on a Nicolet 750 series FTIR spectrometer. All transmission electron micrographs and electron energy loss spectroscopy (EELS) images were collected on a Philips Tecnai 20 TEM at an accelerating voltage of 200 keV.Synthesis of hexadecaaniline–eicosaaniline-grafted single-walled carbon nanotubes SWNT–(A16/20)x
A typical sample preparation procedure was carried out by taking SWNTs (∼2.0 nm in external diameter) or MWNTs (20–40 nm in external diameter) (20 mg) and freshly prepared hexadecaaniline–eicosaaniline (A16/20, ∼200 mg) in DMF (5.0 mL) under nitrogen. It was sonicated for 10 min and isoamyl nitrite (0.2 mL) in DMF (1.0 mL) was added. The mixture was further sonicated at 45 °C for 3.0 h. It was then centrifuged to collect the DMF-soluble fraction of the products. Remaining DMF-insoluble materials were washed with DMF (3 × 3.0 mL). The combined DMF-soluble extracts were passed through a celite bed to clear residual particles. The filtrate was added to diethyl ether (100 mL) to effect precipitation of dark solids. The solids were washed with diethyl ether (3 × 20 mL) and THF (3 × 20 mL) to remove unreacted A16/20. Further purification was achieved by dissolving the solids in DMF (3.0 mL) followed by re-precipitation of blackish solids upon the addition of THF (20 mL) with sonication. This procedure was repeated three times to afford relatively pure DMF-soluble products of the corresponding SWNT–(A16/20)x or MWNT–(A16/20)x in a yield of 100 or 75 mg, respectively.Acknowledgements
The authors at UML thank the support of Air Force Office of Scientific Research under the grant numbers FA9550-09-1-0380 and FA9550-09-1-0183 and U.S. Army Soldier Command under the contract number DAAD16-01-C-0011.References
- A. Nishino, J. Power Sources, 1996, 60, 137–147 CrossRef CAS For a review see: M. Jayalakshmi and K. Balasubramanian, Int. J. Electrochem. Sci., 2008, 3, 1196–1217 Search PubMed.
- M. Kaempgen, C. K. Chan, J. Ma, Y. Cui and G. Gruner, Nano Lett., 2009, 9, 1872–1976 CrossRef CAS; A. Kiebele and G. Gruner, Appl. Phys. Lett., 2007, 91, 144104–144107 CrossRef; M. Kaempgen, J. Ma, G. Gruner, G. Wee and S. G. Mhaisalkar, Appl. Phys. Lett., 2007, 90, 264104–264106 CrossRef; M. Shiraishi, T. Takenobu, T. Iwai, Y. Iwasa, H. Kataura and M. Ata, Chem. Phys. Lett., 2004, 394, 110–113 CrossRef CAS.
- For a review see: M. Moniruzzaman and K. I. Winey, Macromolecules, 2006, 39, 5194–5205 Search PubMed.
- S. R. Sivakkumar, W. Kim, J. A. Choi, D. R. Macfarlane, M. Forsyth and D. W. Kim, J. Power Sources, 2007, 171, 1062–1068 CrossRef CAS; C. F. Zhou, S. Kumar and C. D. Doyle, Chem. Mater., 2005, 17, 1997–2002 CrossRef CAS; Y.-K. Zhou, B.-L. He, W.-J. Zhou, J. Huang, X.-H. Li, B. Wu, H.-L. Li and et al, Electrochim. Acta, 2004, 49, 257–262 CrossRef CAS.
- A. Rudge, J. Davey, I. Raistrick, S. Gottesfeld and J. P. Ferraris, J. Power Sources, 1994, 47, 89–107 CrossRef CAS; B. C. Kim, J. M. Ko and G. G. Wallace, J. Power Sources, 2008, 177, 665–668 CrossRef CAS.
- A. G. Macdiarmid, J. C. Chiang and A. F. Richter, Synth. Met., 1987, 18, 285–290 CrossRef CAS; For reviews see: H. S. Nalwa, Handbook of Organic Conductive Molecules and Polymers,John Wiley & Sons, Chichester and New York, 1996, vol. 1–4 Search PubMed; S. Bhadra, D. Khastgir, N. K. Singha and J. H. Lee, Prog. Polym. Sci., 2009, 34, 783–810 Search PubMed; A. Malinauskas, J. Power Sources, 2004, 126, 214–220 CrossRef CAS.
- J. N. Coleman, S. Curran, A. B. Dalton, A. P. Davey, B. Mc Carthy, W. Blau and R. C. Barklie, Synth. Met., 1999, 102, 1174–1175 CrossRef CAS; H. S. Woo, R. Czerw, S. Webster, D. L. Carroll, J. W. Park and J. H. Lee, Synth. Met., 2001, 116, 369–372 CrossRef; C. Downs, J. Nugent, P. M. Ajayan, D. J. Duquette and K. S. V. Santhanam, Adv. Mater., 1999, 11, 1028–1031 CrossRef CAS; M. Cochet, W. K. Maser, A. M. Benito, M. A. Callejas, M. T. Martinez, J.-M. Benoit, J. Schreiber, S. Lefrant and O. Chauvet, Chem. Commun., 2001, 1450–1451 RSC; X. Zhang, J. Zhang, R. Wang and Z. Liu, Carbon, 2004, 42, 1455–1461 CrossRef CAS; M. Gao, S. Huang, L. Dai, G. Wallace, R. Gao and Z. Wang, Angew. Chem., Int. Ed., 2000, 39, 3664–3667 CrossRef CAS; W. Feng, X. D. Bai, Y. Q. Lian, J. Liang, X. G. Wang and K. Yoshino, Carbon, 2003, 41, 1551–1557 CrossRef CAS; S. R. C. Vivekchand, L. Sudheendra, M. Sandeep, A. Govindraj and C. N. R. Rao, J. Nanosci. Nanotechnol., 2002, 2, 631–635 CrossRef CAS; Z. Wei, M. Wan, T. Lin and L. Dai, Adv. Mater., 2003, 15, 136–139 CrossRef CAS.
- D.-W. Wang, F. Li, J. Zhao, W. Ren, Z.-G. Chen, J. Tan, Z.-S. Wu, I. Gentle, G. Q. Lu and H.-M. Cheng, ACS Nano, 2009, 3, 1745–1752 CrossRef CAS; P. Sivaraman, S. K. Rath, V. R. Hande, A. P. Thakur, M. Patri and A. B. Samui, Synth. Met., 2006, 156, 1057–1064 CrossRef CAS; K. S. Ryu, Y. Lee, K. S. Han, Y. J. Park, M. G. Kang, N. G. Park and S. H. Chang, Solid State Ionics, 2004, 175, 765–768 CrossRef CAS; W. C. Chen and T. C. Wen, J. Power Sources, 2003, 117, 273–282 CrossRef CAS; Y.-R. Lin and H. Teng, Carbon, 2003, 41, 2865–2871 CrossRef CAS; E. Frackowiak and F. Beguin, Carbon, 2001, 39, 937–950 CrossRef CAS.
- M. Baibarac, I. Baltog, S. Lefrant, J. Y. Mevellec and O. Chauvet, Chem. Mater., 2003, 15, 4149–4156 CrossRef CAS; M. Lefenfeld, G. Blanchet and J. A. Rogers, Adv. Mater., 2003, 15, 1188–1191 CrossRef CAS; G. M. Spinks, V. Mottaghitalab, M. Bahrami-Samani, P. G. Whitten and G. G. Wallace, Adv. Mater., 2006, 18, 637–642 CrossRef CAS.
- H. Zengin, W. Zhou, J. Jin, R. Czerw, D. W. Smith Jr., L. Echegoyen, D. L. Carroll, S. H. Foulger and J. Ballato, Adv. Mater., 2002, 14, 1480–1483 CrossRef CAS.
- P. C. Ramamurthy, A. M. Malshe, W. R. Harrell, R. V. Gregory, K. McGuire and A. M. Rao, Solid-State Electron., 2004, 48, 2019–2024 CrossRef CAS.
- M. Delamar, R. Hitmi, J. Pinson and J. M. Saveant, J. Am. Chem. Soc., 1992, 114, 5883–5884 CrossRef CAS; P. Allongue, M. Delamar, B. Desbat, O. Fagebaume, R. Hitmi, J. Pinson and J.-M. Saveant, J. Am. Chem. Soc., 1997, 119, 201–207 CrossRef CAS; C. Saby, B. Ortiz, G. Y. Champagne and D. Belanger, Langmuir, 1997, 13, 6805–6813 CrossRef CAS.
- J. L. Bahr, J. Yang, D. V. Kosynkin, M. J. Bronikowski, R. E. Smalley and J. M. Tour, J. Am. Chem. Soc., 2001, 123, 6536–6542 CrossRef CAS; J. L. Bahr and J. M. Tour, Chem. Mater., 2001, 13, 3823–3824 CrossRef CAS; C. A. Dyke and J. M. Tour, J. Am. Chem. Soc., 2003, 125, 1156–1157 CrossRef CAS.
- W. J. Zhang, J. Feng, A. G. MacDiarmid and A. J. Epstein, Synth. Met., 1997, 84, 119–120 CrossRef CAS; V. Anantharaj, L. Y. Wang, T. Canteenwala and L. Y. Chiang, Chem. Soc., Perkin Trans., 1999, 1, 3357–3366 Search PubMed; T. Canteenwala, S. Patil, M. Halder and L. Y. Chiang, Synth. Met., 2003, 135–136, 795–797 CrossRef CAS.
- P. Kim, T. W. Odom, J. L. Huang and C. M. Lieber, Phys. Rev. Lett., 1999, 82, 1225–1228 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: HPLC chromatogram of oligoaniline fractions and 1H NMR spectra of A16/20 and MWNT–A16/20. See DOI: 10.1039/b9nr00255c |
| This journal is © The Royal Society of Chemistry 2010 |