Quantum dot-based theranostics
Yi-Ping
Ho
and
Kam W.
Leong
*
Department of Biomedical Engineering, Duke University, CIEMAS 1395, PO Box 90281, Durham, NC 27708, USA. E-mail: kam.leong@duke.edu; Fax: +1 919 660 0031; Tel: +1 919 660 8466
First published on 6th October 2009
Abstract
Luminescent semiconductor nanocrystals, also known as quantum dots (QDs), have advanced the fields of molecular diagnostics and nanotherapeutics. Much of the initial progress for QDs in biology and medicine has focused on developing new biosensing formats to push the limit of detection sensitivity. Nevertheless, QDs can be more than passive bio-probes or labels for biological imaging and cellular studies. The high surface-to-volume ratio of QDs enables the construction of a “smart” multifunctional nanoplatform, where the QDs serve not only as an imaging agent but also a nanoscaffold catering for therapeutic and diagnostic (theranostic) modalities. This mini review highlights the emerging applications of functionalized QDs as fluorescence contrast agents for imaging or as nanoscale vehicles for delivery of therapeutics, with special attention paid to the promise and challenges towards QD-based theranostics.
Introduction
Emergence of novel nanoparticles, namely colloidal particles of 5–50 nm in diameter, such as semiconductor nanocrystal and metallic nanoparticles, has fundamentally changed the bioanalytical measurement landscape.1–4 Luminescent semiconductor nanocrystals, colloquially known as quantum dots (QDs), stand among the research tools in chemistry, physics, and biology as one of the most exciting developments. These inorganic fluorescent nanocrystals typically comprise periodic groups of II–VI (e.g. CdSe and CdTe) or III–V (e.g. InP and InAs) semiconductor materials. As a photon of proper energy impinges a semiconductor, exciting an electron from the valence band into the conduction band, it generates an electron–hole pair (or exciton) that is weakly bound by Coulomb forces. For semiconductor nanocrystals with all three dimensions less than the Bohr exciton radius (typically a few nanometres), their energy levels are quantized (due to quantum-confinement effect, henceforth named quantum dots), and the spacing of which can be controlled by the crystal sizes.5,6 This effect leads to the superior optical properties of QDs, such as narrow, symmetric and size-tunable emission spectra, and broad excitation spectra, rendering them particularly valuable for multicolor fluorescent applications. Other commonly discussed benefits of QDs over organic fluorophores or fluorescent proteins include stronger fluorescence (∼10–100 times brighter) and higher fluorescence stability against photobleaching (∼100–1000 times more stable),7,8 which facilitate the long-term monitoring of intermolecular and intramolecular interactions in live cells and organisms. Consequently, since the first demonstration of colloidal quantum dots for biological labeling in 1998,9,10 subsequent innovations have centered on the exploration of QDs for biomedical applications.Synthesis and surface modification of quantum dots
Among the array of synthetic routes devised for the preparation of quantum dots (reviewed in refs. 8,11–16), the predominant approach is to coat a CdSe core with a ZnS layer to obtain the best crystalline quality and monodispersity. Passivation by the ZnS layer protects the core from oxidation, reduces toxicity by preventing the CdSe from leaching out to the surrounding solutions, and also enhances the photoluminescence yield. However, the ZnS-coated QDs are only soluble in nonpolar organic solvents. Due to the aqueous nature of the biological environment, altering the QD surface properties from hydrophobic to hydrophilic becomes an essential step for QDs to be useful in biological applications. Although the synthesis of QDs has been performed directly in aqueous solution, the products of the aqueous schemes are largely polydispersed and rarely match the quality of those synthesized through high-temperature routes with hydrophobic organic solvent/ligand mixtures.7,17,18 Furthermore, inorganic materials such as QDs have little to no innate biological specificity. They must rely on conjugation with biological molecules such as aptamers,19antibodies,20oligonucleotides,21peptides,22–25folates,26 and small molecule ligands to gain biological affinity.27 After much effort to alter the properties of QDs, such as stability, monodispersity, crystallinity, solubility, and biocompatibility,9,20,28–30QDs have evolved from an interesting curiosity to a widely used research tool for diagnostics, cell and molecular biology studies, and in vivo bioimaging.11,12,31QD-based multiplexed biosensing and FRET
Quantum dots have become popular fluorescent cellular probes for light microscopy (LM), again because of their unique optical and physical properties. Notably, their electron-dense semiconductor cores can be directly imaged by electron microscopy (EM) even without any contrasting treatment.32 Collectively, their distinct size, shape and elemental fingerprint facilitate multilabeling for correlative microscopy with LM and EM.33 A widely adopted mechanism of QD-based fluorescence biosensing is through QD-mediated Förster Resonance Energy Transfer (FRET ). Since the first theoretical predication and experimental demonstration,34,35 respectable progress has been made in the past few years on the use of QD-FRET based biosensors ,36,37 particularly on bioanalysis (nucleic acids, proteins, and immunoassays ) and intracellular sensing. QDs possess several unique optical properties over organic fluorophores that can benefit FRET configurations, including broad absorption, size-dependent narrow emission and strong resistance to photobleaching. Their emission spectra are usually fairly symmetric and narrow (typically 10–20 FWHM), tunable across a wide range by changing the size and composition of the QD core, once again due to the quantum confinement effect. When paired with an organic fluorophore, crosstalk, the spectrum overlap between the donor and acceptor emissions, can be effectively minimized. Meanwhile, the QD absorption has an increased probability at higher energies, resulting in a broader absorption over the entire spectral range extending from the characteristic emission band to the UV band. This feature enables the use of UV-range excitation, lessening the possibility of direct-acceptor excitation. Perhaps more importantly, excitation at a single wavelength can excite multiple QDs to emit in non-overlapping, narrow spectral ranges that can still be discriminated. This renders QDs well suited towards multicolor applications38,39 and even mulitiplexed FRET , which would be particularly valuable for cellular interactions involving multiple entities or events.Nanoparticle-mediated cellular responses
Despite the tremendous therapeutic potential of nanoparticles in medicine, the fundamental information regarding the physicochemical interaction between nanoparticles and cells (i.e. membrane surfaces, endosomal compartments, cytoplasm, and other organelles) is relatively limited (recently reviewed in ref. 40). Cellular uptake of nanoparticles is modulated by size,41 shape and angle of curvature,42–44 effective surface charge (zeta-potential) and surface functionalization.45 For example, gold nanoparticles45 and QD conjugates46 exhibit different cell-membrane penetration and cytotoxicity characteristics depending on the types of ligand on their surfaces. It would be valuable to investigate whether there exists an intrinsic generalized correlation between the physicochemical properties of nanoparticles and cellular responses. Otherwise, findings for a particular nanomaterial, such as gold nanoparticles (currently the most studied because of their ease of synthesis and characterization), may be irrelevant for other types of nanomaterials (such as QDs), or even invalid for the same nanomaterial that is produced through a different synthetic route. For instance, why does the cytotoxicity of gold nanoparticles differ with size and surface decoration?42,47 A better understanding of nanoparticle-mediated cellular responses, therefore, would assist nanoparticle design by providing insights on the uptake kinetics and intracellular behavior to complement the readout of therapeutic efficacy or marker gene expression levels.Scope of this mini review
Theranostics, a term coined for combining diagnostics and therapeutics, integrates real-time evaluation with delivery of a medication. QDs excel in imaging applications; they may also serve as particulate delivery vehicles if the biocompatibility issue can be managed. Such multifunctional nanoparticles offer synergistic advantages over any single-modal nanoparticle alone.48–51 Through surface immobilization of ligands and conjugation of “drugs” on QD one can construct an “all-in-one” multifunctional nanoplatform that features targeting, therapeuctic and imaging modalities. Multifunctional QDs therefore may have the potential to meet the requirements of a theranostic system, which ideally, should possess a number of the following characteristics (Fig. 1): (a) accumulate in the pathological zone, targeting specific cell types, (b) penetrate the cells efficiently, with minimal cytotoxicity, (c) overcome the intracellular delivery barriers, allowing efficient intracellular trafficking, (d) respond to local stimuli, releasing the therapeutic agents, and (e) bear a diagnostic agent (optical or magnetic), allowing for real-time monitoring of the treatment. Current success of QD-based theranostics remains at the stage of ex vivo, due to the challenges of navigating the biological barriers in vivo and imaging deep tissues. With respect to multifunctional nanoparticles constructed on metallic and magnetic nanoplatforms constructed from various types of nanoparticles, the readers are referred to two recent reviews.52,53 In this mini review, we explore specifically whether multifunctional QDs would be able to tackle the challenges in theranostics. We will begin with coverage of QDs carrying single-function modalities, paying special attention to the emerging application of QDs for drug and gene delivery. We will then describe the integration of multiple-function modalities in highlighted examples to illustrate the potential and challenges of QD-based theranostics for future nanomedicine.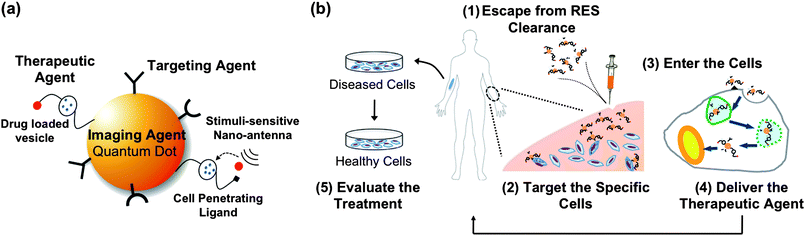 | ||
| Fig. 1 (a) Schematic representation of a multimodal QD, where the QD serves as both a diagnostic agent (imaging) and a nanoscaffold to incorporate multiple functional modalities, such as a targeting ligand (peptide, antibody, or protein) and a therapeutic agent. Upon interacting with the target cell, the cell-penetrating ligand can then be exposed, allowing the multifunctional QD to enter the cell. Stimuli-sensitive antennae may be triggered by local stimuli (pH, temperature, or enzyme), allowing subsequent intracellular release of the drug from the drug-loaded vesicle. (b) Requirements of an ideal theranostic process in the human body may include: (1) escape from the clearance of reticuloendothelial system (RES, mainly liver, spleen, and bone marrow), allowing longer blood circulation time, (2) accumulate in the pathological zone, targeting specific cell types, (3) penetrate the cell efficiently, leaving minimum damage to the cell, (4) overcome the delivery barriers, leading to efficient intracellular release, and (5) bear a diagnostic agent (imaging, optical or magnetic), allowing for real-time monitoring of the treatment, while maintaining minimum toxicity to the healthy cells. | ||
Quantum dots in diagnostics and therapeutics
Cell-specific targeting and subsequent uptake
Amidst substantial progress in in vitro diagnostics,54QDbioconjugates have emerged as imaging probes for recognition of specific cell types, tissues and organelles11,13,14,55,56 that are poised for or being exploited in clinical translation,57 especially for cancer research and therapy. The interaction of drug-carrying vehicles with the cell membrane and subsequent penetration is the primary requisite for a successful diagnostic and therapeutic process, where the diseased cells are firstly located, following by subsequent cellular uptake and release of therapeutic agent to the cytosol or nucleus of cells.58,59 Static immunostaining of cellular targets with QDs was firstly demonstrated by Wu in 2003 and shown to be both brighter and more photostable than comparable organic fluorophores without non-specific labeling.28 Akerman et al. reported the use of QD–peptide conjugates to specifically target the tumor tissues in live cells.22 Although the QD signal was not detected in vivo, in vitro histological results revealed that QD–peptide targeted tumor vasculatures in mice and PEG-coating reduced the reticuloendothelial clearance. Inspired by the in vitro success, Gao et al.20 developed an ABC triblock copolymer -coated QD probe to target and image prostate cancer in vivo. The tumor site could be actively probed by the antibody-conjugated QDs and imaged in living animals, after proper signal post-processing. Because the large size and immunogenicity of antibodies may affect their pharmacological behavior, small ligands, such as peptides23 and aptamers,19 were engaged to conjugate QDs for similar purposes. It is worthwhile noting that the capability of acquiring multiple biomarkers on the surface renders QD conjugates advantageous with improved binding affinities for the receptors on the cells, due to the polyvalency effect.22,60,61Cellular uptake of exogenous material generally occurs through internalization mechanisms including phagocytosis, macropinocytosis, receptor-mediated endocytosis, and clathrin- and caveolin-independent endocytosis.62 In Parak et al.'s study, undecorated QDs were adopted as an alternative marker over gold nanoparticles for phagokinetic tracking to monitor cell motility as a potential assay for cancer metastasis,63 where QDs were passively uptaken via non-specific endocytosis along the migratory pathway of human mammary epithelial tumor cells. Delivery assisted by transfection agent (liposome, micelle or polymer) or ligand-modification on the QDs is usually more specific and efficient than non-specific endocytosis alone.64,65 A general observation is that the endocytosed QDs are often trapped in endosomes and lysosomes, visualized as punctate fluorescence staining, hence limiting their delivery to the cytosol, as shown in Fig. 2(a) and (b). The aggregation, more problematically, prevents their entry into the nucleus. It also generates undesirable background noise and complicates subsequent analysis or single-molecule studies.
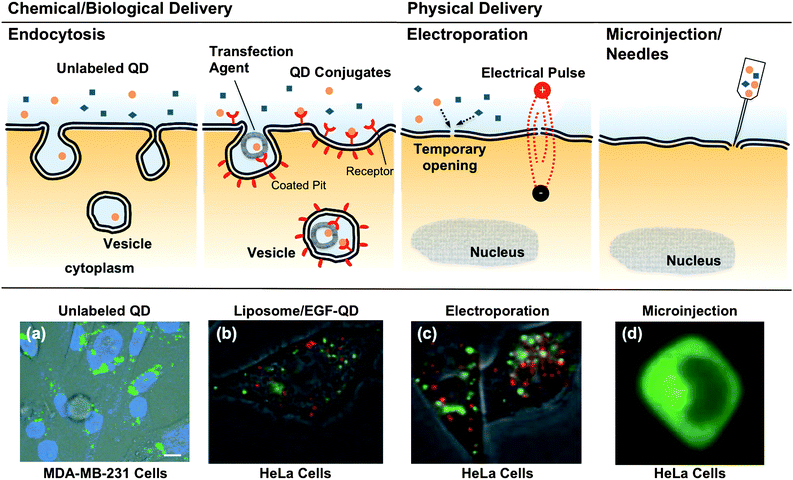 | ||
| Fig. 2 Cellular internalization of QD conjugates by chemical/biological and physical approaches. (a) Unconjugated QDs typically are uptaken via non-specific endocytosis, resulting in aggregation in the cytosol, shown as punctate fluorescence staining.63 (b) Delivery assisted by transfection agent (liposome, micelle or polymer) or ligand-modification on the QDs is usually more specific and efficient than non-specific endocytosis alone.64 (c) The electrical field in electroporation temporarily permeabilizes the cell membrane, allowing the QD conjugates to be delivered directly into the cells to achieve high gene expression. However, aggregations are still observed in the endolysosomes and cytosol for both transfection agent and electroporation assisted deliveries.64 (d) Microinjection or needle penetration, allows QDs to be delivered directly into the cytoplasm or even nucleus, and bypass the endosome/lysosome, thus avoiding enzymatic degradation.64QD conjugates delivered through different approaches result in contrasting patterns in cell labeling. Figures reprinted with permission from Wiley-VCH Verlag GmbH & Co. | ||
Physical approaches, such as microinjection,30electroporation,64,66 and via the recently developed “membrane-penetrating needles”67 have been applied to deliver QDs into cells. Electroporation, applying a short and intense electric pulse to reversibly permeabilize the cell membrane, allows extracellular molecules to enter cells.68 Aggregation of QDs was observed in the cytosol,64 presumably due to the electric field inducing complexation between cell membrane and the QD conjugates, the aggregated complexes then internalized subsequently after membrane resealing. Empirical optimization of the electrical pulse and field strength parameters is generally required for optimal delivery to assorted cell types. Microinjection allows QDs to be directly delivered to the cytoplasm or even nucleus of individual cells, and to bypass the endosome/lysosome, thus avoiding enzymatic degradation.30 In a recent report, Yum and co-workers developed a membrane-penetrating nanoneedle, capable of delivering monodispersed QDs into specific cells, as shown in Fig. 3(a).67 Monodispersed QDs can be identified at the single-molecule level, having single QDs distinguished from the aggregates through the blinking phenomenon (intermittent fluorescence).10 The ability to deliver controlled amounts of QDs with spatial and temporal precision is particularly useful for single-molecule studies. However, in addition to the low efficiency and labor-intensive procedures, as in the case of microinjection, physical delivery typically requires delicate instrumentation and optimization for different cell lines , and possibly induces irreversible damage to the cells.
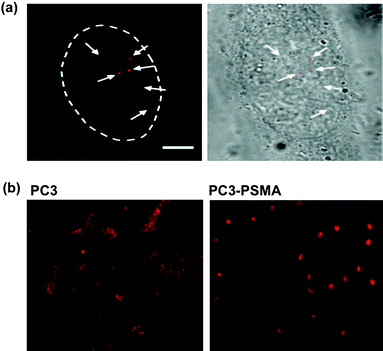 | ||
| Fig. 3 Cellular delivery of QDs. (a) Monodispered QDs can be delivered by microinjection. The single nature of QDs can be validated by the blinking phenomenon.67 The ability to deliver controlled amounts of QDs with spatial and temporal precision is particularly useful for single-molecule studies. (b) The labeling pattern of QDs is phenotype -dependent. Unlabeled QDs are found aggregated throughout the cytoplasm in the PC3 cells, as previously observed along the endosomal pathway. Strikingly, a single clump of QDs is localized around the perinuclear region of PC3-PSMA cells.69 As a result, cancer phenotypes can easily be identified by the contrasting labeling pattern of QDs. Figures reprinted with permission from the American Chemical Society and Wiley-VCH Verlag GmbH & Co. | ||
QD conjugates internalized through different mechanisms (endocytosis, receptor-mediated endocytosis, electroporation or microinjection) usually lead to concomitant variation in the resulting patterns of cellular labeling (Fig. 2).65 Recently, QDs trafficking, even without bioconjugation, were found to be phenotype -dependent in the model of PC3 cells.69 As shown in Fig. 3(b), unlabeled QDs were found aggregated throughout the cytoplasm, as previously observed along the endosomal pathway (Fig. 2).63 Surprisingly, a single clump of QDs was localized around the perinuclear region of PC3-PSMA cells. From a diagnostics point of view, cancer-cell phenotype can easily be pinpointed by this contrasting pattern of QDlabeling, although investigations on different cancer cell types would be necessary to establish the robustness and generality of this phenomenon prior to clinical translation. Perhaps one of the most important tasks regarding cellular delivery of QDs is a better understanding of the internalization mechanisms.46 It would be of particular interest to correlate the internalization and delivery mechanisms with the chemical (e.g. surface coating, length of spacer and density of coating) and physical (e.g. size, surface charge) properties of the QD conjugates against assorted cell-types. A clearer mechanistic understanding of intracellular delivery would facilitate the rational design of “ideal” functionalized QDs, where one may optimize the number of conjugates per QD, the arrangement of different functional modalities, and their binding strength to the QD nanoscaffold, towards controlled theranostics.
Intracellular drug/gene trafficking
Synthetic gene or drug delivery systems with increasing effectiveness are constantly being reported.70,71 However, one major remaining challenge is the real-time and long-term imaging of the payload transport and release, which may reveal critical delivery barriers. Organic fluorophores are commonly used for this purpose. However, they are susceptible to photobleaching and cannot be effectively applied to time-lapse studies of intracellulardrug/gene trafficking.8,72 To this end, functionalized QDs were employed as intracellulartracers of plasmidDNA (pDNA) delivery.73 Ho et al. developed a QD-FRET system to investigate the structural composition and dynamic behavior of polymeric DNA nanocomplexes intracellularly.74 As shown in Fig. 4(a), the FRET system is constructed upon complexation between pDNA and cationic polymers; the complex coacervation brings the donor (605QD, labeled on the pDNA) and acceptor (Cy5, functionalized on the polymer) into a close proximity, resulting in efficient FRET .21 The high signal-to-noise ratio offered by QD-FRET allows characterization of single nanocomplexes, and also provides a convenient method to intracellularly track the polymeric DNA nanocomplexes over time and digitally monitor their unpacking behavior with conventional or confocal microscopy. From quantitative image-based analysis, distributions of released plasmid within the endo/lysosomal, cytosolic, and nuclear compartments form the basis for constructing a three-compartment first-order kinetics model. The unpacking kinetics for the chitosan, polyethyleneimine, and polyphosphoramidate DNAnanoparticles (polyplexes) correlates well with transfection efficiencies. The study illustrates that QD-FRET -enabled detection of polyplex stability combined with image-based quantification is a valuable method for studying mechanisms involved in polyplex unpacking and trafficking within live cells.75 Further incorporation of two-step FRET , shown in Fig. 4(b), where the QD is paired with an additional nuclear dye (ND) on the pDNA, serves as a novel approach to study both polyplex dissociation and DNA degradation in a simultaneous and non-invasive manner.76 The integration of single-particle FRET provides valuable insight to the heterogeneity of nanocomplexes, enabling elucidation of structure-function relationship, which would facilitate the optimization of gene carrier characteristics such as molecular weight, charge density, and chemical composition. Although the labeled polyplexes were found to be similar to their unlabeled counterparts in physical properties, their transfection efficiency was relatively low compared to the reported value, presumably due to the large size of the DNA nanocomplexes (typically∼100–200 nm)42 and the aggregation observed around the perinuclear region.74,75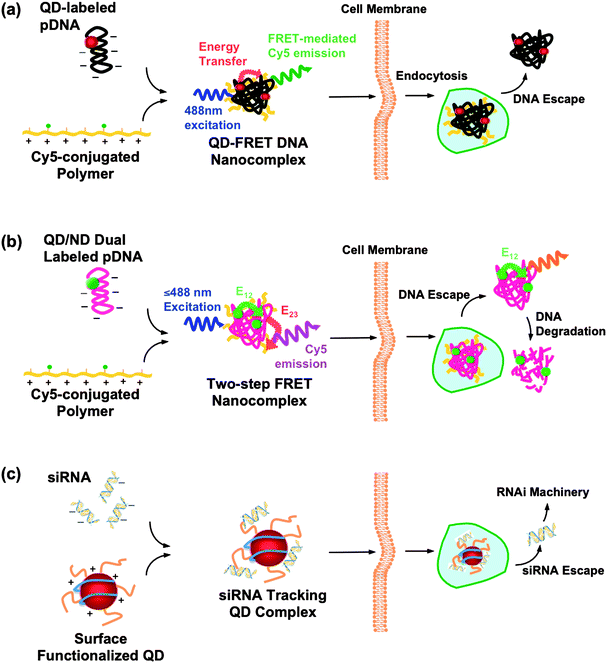 | ||
| Fig. 4 QD conjugates/complexes for intracellulargene trafficking. (a) QD-FRET system: QDs, as an energy donor, are conjugated onto the plasmidDNA (pDNA), whereas Cy5, the energy acceptor, is functionalized on the cationic polymer. The FRET -mediated Cy5 emission upon complex coacervation provides a digital indicator of the interaction between pDNA and the polymer. Consequently, the FRET signal is abrogated when DNA is released into the cytosol.74,75 (b) Two-step FRET system: the two-step energy transfer is constructed from the QD donor to the first acceptor, nuclear dye (ND, energy transfer E12), as a relay donor to the second acceptor, Cy5 (energy transfer E23). Similar to the one step QD-FRET system, the “OFF” signal from Cy5 (from E23) signifies the DNA escape. Moreover, dual-labeled pDNA provides an additional dimension after the DNA unpacks from the complexes, allowing simultaneous detection of DNA release and degradation, during gene delivery.76 (c) siRNA tracking system: siRNA/QDs complexes are generated with surface modified QDs (proton-sponge coating79 or Amphipol80) or transfection agent (polymers55 or Lipofatamine23) encapsulated QDs, to trace siRNA delivery. | ||
RNAi-based gene silencing harnesses an endogenous cellular regulatory mechanism in which small interfering RNAs (siRNAs < 30 bp) bind to and mediate the destruction of specific mRNA molecules. The gene silencing thus greatly relies on a well-designed siRNAs, with high accessibility and affinity to the complementary site of the target mRNA. It is thus necessary to visualize the transport and release of siRNA from the carrier for optimization. In this regard, QDs were introduced as a traceable marker to shed light on the siRNA delivery, shown in Fig. 4(c).31 The report described the co-transfection of QD/siRNA with the well-developed liposome system (Lipofectamine 2000).31 However, chloroquine, an anti-malarial drug is required as a lysosomotropic agent to facilitate the endosomal escape,77 limiting its in vivo applications. Subsequently, Tan and co-workers ameliorated the transfection system with a non-viral vector, chitosan polyplexes, doped with QDs as a siRNA tracking system.78 Recently, Gao's group developed both the proton-sponge-coated and Amphipol-modified QDs80 to further enhance the delivery efficiency. Amphipols, comprised of a strongly hydrophilic backbone, are able to graft to the hydrophobic chains of transmembraneproteins, making them soluble in their native form.81 Thus, Amphipol-mediated delivery permits better integrity of the cell membrane when compared to micelle-based delivery. Similarly, siRNA/QDs were ferried into the cytoplasm through Amphipol-mediated delivery, where Amphipol not only serves a gene carrier but also protects siRNA from enzymatic degradation. Single QDs could be observed when complexed with Amphipols and siRNAin vitro, presumably due to the shorter strand of siRNA when compared with pDNA.74,75 The individually distinct QD is of particular interest for single-molecule studies, whereas the compactness of the siRNA/QD complex is of greater importance towards efficient delivery. However, QD aggregates were observed after endosomal escape, probably due to replacement of siRNA by other anionic biomolecules, resulting in non-specific interactions among QDs. Although the proton-sponge coating was found to enhance the gene silencing activity by 10–20 fold,79 the mechanism for this improvement is unclear. Current siRNA/QD delivery systems only allow monitoring of the siRNA delivery when the siRNA and QD are not separated. An additional imaging modality would better elucidate the delivery barriers of siRNA.
To aid pharmacokinetic and pharmacodynamic evaluations, Manabe et al. have proposed a QD–drugtracer system,82 where QDs were conjugated with Catrophil, an anti-hypertensive drug. The concentration of QD–captrophil in plasma decreased exponentially, yielding a half-life comparable to that of unconjugated captrophil. However, the QD–captrophil conjugates showed weaker activity than the unconjugated captrophil in reducing the blood pressure of hypertensive rats. The decrease in efficacy was attributed to the non-specific uptake by macrophages and endothelia cells. Further investigation would be required to ascertain that the therapeutic effect did not stem from the detached captrophil.
QD-based theranostics?
Multimodal quantum dots for simultaneous diagnostics and therapeutics
Perhaps the most successful demonstration towards developing QD-based theranostics, to our knowledge, is the QD–aptamer(Apt)–doxorubicin(Dox) conjugate, shortened as QD–Apt(Dox), presented by Bagalkot et. al. for synchronous cancer imaging and traceable drug delivery.83 The targeting modality, RNA aptamer, was functionalized onto the diagnostic modality (QDs) to pinpoint the prostate-specific membrane antigen (PSMA) expressed in LNCaP cells. The therapeutic modality, doxorubicin (Dox), a widely used anthracyclinedrug, was intercalated into the aptamer. The sensing of drug loading and release relies on the bi-FRET (dual donor–quencher ) design, as illustrated in Fig. 5(a): (1) in the drug-loading state: both QD and Dox fluorescence were turned “OFF”, because the QD fluorescence was quenched by the Dox, and the Dox fluorescence was in turned quenched by the aptamer, (2) in the drug-release state: the Dox was released from the QD–Apt complex, thus turning both QD and Dox fluorescence back “ON”, and (3) during drug transport: the Dox fluorescence was traced. This multifunctional QD was demonstrated in vitro to enhance the therapeutic specificity against the targeted LNCaP cells compared to non-specific PC3 cells. The release and transport of the drug can be followed by the Dox fluorescence, but it remains unclear whether the retained Dox fluorescence is due to physical dissociation from the conjugate or the enzymatic degradation of the aptamer. Although it may require further optimization prior to in vivo application, this work presents an exciting advance in the field. It is also worthy to note that the QD–liposome (QD–L) system may constitute another multifunctional platform for imaging and therapy, exemplified in Fig. 5(b). QDs are typically incorporated into the bilayer membrane,84–86 or functionalized onto a liposome,87 forming a QD–lipid vesicle. Liposomes have long been used as a nonviral drug and gene carrier. The combination of liposomes and QDs reduces the cytotoxicity of the QDsin vivo,85,86 while the labeling specificity remains comparable to other approaches.85–88 We envisage that liposomes and other well-developed polymeric gene carriers may also assist multifunctional QDs towards the ultimate goal of theranostics.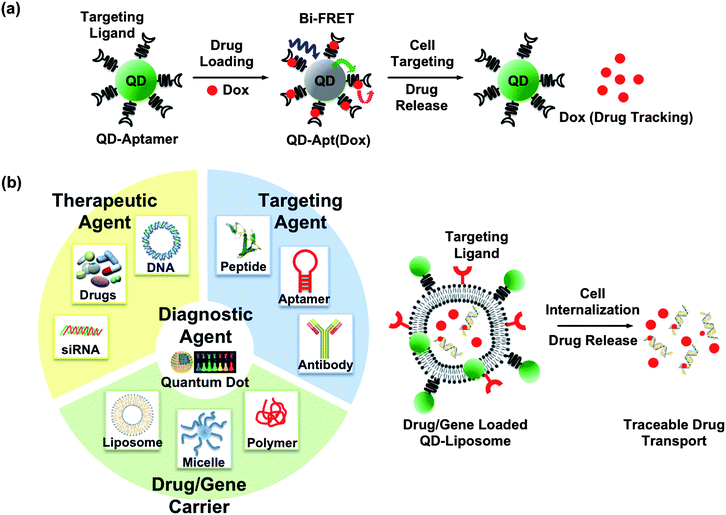 | ||
| Fig. 5 Possible constructs of multifunctional QDs for theranostics: (a) QD–aptamer(Apt)–doxorubicin(Dox) conjugate, shortened as QD–Apt(Dox), is presented for synchronous cancer imaging and traceable drug delivery towards QD-based theranostics. The targeting modality, RNA aptamer, is functionalized onto the diagnostic modality (QDs) to probe the cancer cells. The therapeutic modality, doxorubicin (Dox), is intercalated into the aptamer. The sensing of drug loading and release relies on the bi-FRET (dual donor–quencher ) design. In the drug-loading state: both QD and Dox fluorescence are turned “OFF”, since the QD fluorescence is quenched by the Dox and the Dox fluorescence is in turn quenched by the aptamer. In the drug release state: the Dox is released from the QD–Apt complex, turning both QD and Dox fluorescence back “ON”. During drug transport: the Dox fluorescence was used as a tracable dye. (Figure adapted from ref. 83). (b) Representation of an idealized nanoplatform of an “all-in-one” workstation. Multiply functionalized QDs may constitute an integrated nanoplatform, for example, able to target the tumor, transport/release the drug payload, and image the therapeutic response simultaneously. The QD–liposome (QD–L) system, although not experimentally demonstrated yet, is envisaged as a potential candidate towards QD-based therapeutics. In the current QD–L system, QDs are typically incorporated into the bilayer membrane, or functionalized onto a liposome, forming a QD–lipid vesicle. Liposomes have proved to be excellent drug and gene carriers. Integration of QD–L with other targeting ligands and therapeutic agents may achieve the goal of theranostics. | ||
Summary and challenges
In summary, quantum dots (QDs) have proven themselves as a powerful imaging agent for in vivo and in vitro applications. As applications of QDs in delivery of therapeutics emerge, the potential of multifunctional QDs serving as theranostic agents moves closer towards reality. Progress so far has paved the way towards the lofty objective of building an “all-in-one” personalized medical workstation. However, significant challenges abound. The major and lingering concern for QDs remains their inherent cytotoxicity. Surface oxidation and leaching-out of heavy metal ions from the core remains a threat.89,90 Short term in vitro cytotoxicity assays are inadequate to assess their toxicity because the coating might erode only after prolonged biological exposure. The toxicity has a strong dependency on the physicochemical properties of QDs, such as size, surface charge and surface coating materials, in addition to the dosage of QDs and the duration of exposure.91In vivo studies are paramount to define the biocompatibility of QDs and their conjugates.92–94 One also has to be aware that modifications to reduce cytotoxicity may also compromise the functionality of the QDs. For example, PEG-ylation may improve aqueous dispersion, prevent aggregation, and in some cases, enhance the optical properties of QDs, but it will also significantly reduce cellular uptake.95 We must correlate the QD-mediated cellular responses with the physicochemical properties of QDs to advance the field. While much effort has centered on optimization of the suface modifications of CdSe QDs, alternative approaches to synthesize more biological- and environmentally-friendly QDs, such as silicon-based QDs,96 should be pursued. Nontoxic QDs may hold the key to the future of QD-based theranostics. The main challenge is whether their photophysical properties, solubilities and biocompatibilities can match those of group II–VI or III–VQDs in biological fluids.The utility of QDs as a tag for imaging, while indisputably superior in the optical realm, does raise question on relevance and potential interference.97 The additional mass of QDs added to a biomolecule may alter their diffusivity. The QDs might also perturb the native conformation of a protein. Efforts to synthesize QD conjugates with a thinner coating, and hence reducing the overall size, would somewhat alleviate the concern. Smaller QDs with comparable photophysical properties would also benefit the QD-FRET construct by enhancing the energy-transfer efficiency.98 Another fertile research direction that might advance QD-based theranostics, is microfluidics-based synthesis. High-quality QDs with respect to uniform composition and size control is of critical importance. Microfluidic microreactors offer advantages in enhanced heat/mass transfer, low power/sample consumption, low production cost, high throughput synthesis and screening, and parallel sample processing. Previous studies have shown that microfluidics is capable of generating uniform microenvironments (microreactors, microcapillary, continuous or segmented microfluidics)99–102 for monodispersed and customizable nanoparticle synthesis.103–105 When collectively advances in making well-controlled, multifunctional, relatively non-toxic QD conjugates become a reality, so will QD-based theranostics. The rewards of nanomedicine call for such effort.
Acknowledgements
Support by NIH (EB002849 and HL89764) is acknowledged.Notes and references
- C. M. Niemeyer, Angew. Chem., Int. Ed., 2001, 40, 4128–4158 CrossRef CAS.
- S. G. Penn, L. He and M. J. Natan, Curr. Opin. Chem. Biol., 2003, 7, 609–615 CrossRef CAS.
- E. Katz and I. Willner, Angew. Chem., Int. Ed., 2004, 43, 6042–6108 CrossRef CAS.
- M. De, P. S. Ghosh and V. M. Rotello, Adv. Mater., 2008, 20, 4225–4241 CrossRef CAS.
- M. Nirmal and L. Brus, Acc. Chem. Res., 1999, 32, 407–414 CrossRef CAS.
- D. Norris and M. Bawendi, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 53, 16338–16346 CrossRef CAS.
- M. A. Correa-Duarte, M. Giersig and L. M. Liz-Marzan, Chem. Phys. Lett., 1998, 286, 497–501 CrossRef CAS.
- U. Resch-Genger, M. Grabolle, S. Cavaliere-Jaricot, R. Nitschke and T. Nann, Nat. Methods, 2008, 5, 763–775 CrossRef CAS.
- M. Bruchez Jr., M. Moronne, P. Gin, S. Weiss and A. P. Alivisatos, Science, 1998, 281, 2013–2016 CrossRef CAS.
- W. C. W. Chan and S. M. Nie, Science, 1998, 281, 2016–2018 CrossRef CAS.
- X. Michalet, F. F. Pinaud, L. A. Bentolila, J. M. Tsay, S. Doose, J. J. Li, G. Sundaresan, A. M. Wu, S. S. Gambhir and S. Weiss, Science, 2005, 307, 538–544 CrossRef CAS.
- A. M. Smith, H. Duan, A. M. Mohs and S. Nie, Adv. Drug Delivery Rev., 2008, 60, 1226–1240 CrossRef CAS.
- A. P. Alivisatos, W. Gu and C. Larabell, Annu. Rev. Biomed. Eng., 2005, 7, 55–76 CrossRef CAS.
- I. L. Medintz, H. T. Uyeda, E. R. Goldman and H. Mattoussi, Nat. Mater., 2005, 4, 435–446 CrossRef CAS.
- C. Murray, C. Kagan and M. Bawendi, Annu. Rev. Mater. Sci., 2000, 30, 545–610 CrossRef CAS.
- D. J. Norris, A. L. Efros and S. C. Erwin, Science, 2008, 319, 1776–1779 CrossRef CAS.
- A. L. Rogach, D. Nagesha, J. W. Ostrander, M. Giersig and N. A. Kotov, Chem. Mater., 2000, 12, 2676–2685 CrossRef CAS.
- N. Gaponik, D. V. Talapin, A. L. Rogach, K. Hoppe, E. V. Shevchenko, A. Kornowski, A. Eychmuller and H. Weller, J. Phys. Chem. B, 2002, 106, 7177–7185 CrossRef CAS.
- T. C. Chu, F. Shieh, L. A. Lavery, M. Levy, R. Richards-Kortum, B. A. Korgel and A. D. Ellington, Biosens. Bioelectron., 2006, 21, 1859–1866 CrossRef CAS.
- X. Gao, Y. Cui, R. M. Levenson, L. W. Chung and S. Nie, Nat. Biotechnol., 2004, 22, 969–976 CrossRef CAS.
- C. Y. Zhang, H. C. Yeh, M. T. Kuroki and T. H. Wang, Nat. Mater., 2005, 4, 826–831 CAS.
- M. E. Akerman, W. C. Chan, P. Laakkonen, S. N. Bhatia and E. Ruoslahti, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 12617–12621 CrossRef CAS.
- W. B. Cai, D. W. Shin, K. Chen, O. Gheysens, Q. Z. Cao, S. X. Wang, S. S. Gambhir and X. Y. Chen, Nano Lett., 2006, 6, 669–676 CrossRef CAS.
- F. Pinaud, D. King, H. P. Moore and S. Weiss, J. Am. Chem. Soc., 2004, 126, 6115–6123 CrossRef CAS.
- M. Stroh, J. P. Zimmer, D. G. Duda, T. S. Levchenko, K. S. Cohen, E. B. Brown, D. T. Scadden, V. P. Torchilin, M. G. Bawendi, D. Fukumura and R. K. Jain, Nat. Med., 2005, 11, 678–682 CrossRef CAS.
- D. J. Bharali, D. W. Lucey, H. Jayakumar, H. E. Pudavar and P. N. Prasad, J. Am. Chem. Soc., 2005, 127, 11364–11371 CrossRef CAS.
- D. S. Lidke, P. Nagy, R. Heintzmann, D. J. Arndt-Jovin, J. N. Post, H. E. Grecco, E. A. Jares-Erijman and T. M. Jovin, Nat. Biotechnol., 2004, 22, 198–203 CrossRef CAS.
- X. Wu, H. Liu, J. Liu, K. N. Haley, J. A. Treadway, J. P. Larson, N. Ge, F. Peale and M. P. Bruchez, Nat. Biotechnol., 2003, 21, 41–46 CrossRef CAS.
- D. Gerion, F. Pinaud, S. C. Williams, W. J. Parak, D. Zanchet, S. Weiss and A. P. Alivisatos, J. Phys. Chem. B, 2001, 105, 8861–8871 CrossRef CAS.
- B. Dubertret, P. Skourides, D. J. Norris, V. Noireaux, A. H. Brivanlou and A. Libchaber, Science, 2002, 298, 1759–1762 CrossRef CAS.
- A. A. Chen, A. M. Derfus, S. R. Khetani and S. N. Bhatia, Nucleic Acids Res., 2005, 33, e190 CrossRef.
- B. N. G. Giepmans, T. J. Deerinck, B. L. Smarr, Y. Z. Jones and M. H. Ellisman, Nat. Methods, 2005, 2, 743–749 CrossRef CAS.
- B. N. G. Giepmans, S. R. Adams, M. H. Ellisman and R. Y. Tsien, Science, 2006, 312, 217–224 CrossRef CAS.
- D. M. Basko, V. M. Agranovich, F. Bassani and G. C. La Rocca, Phys. Status Solidi A, 2000, 178, 69–72 CrossRef CAS.
- D. M. Willard, L. L. Carillo, J. Jung and A. Van Orden, Nano Lett., 2001, 1, 469–474 CrossRef CAS.
- I. L. Medintz and H. Mattoussi, Phys. Chem. Chem. Phys., 2009, 11, 17–45 RSC.
- W. R. Algar and U. J. Krull, Anal. Bioanal. Chem., 2008, 391, 1609–1618 CrossRef CAS.
- H. X. Xu, M. Y. Sha, E. Y. Wong, J. Uphoff, Y. H. Xu, J. A. Treadway, A. Truong, E. O'Brien, S. Asquith, M. Stubbins, N. K. Spurr, E. H. Lai and W. Mahoney, Nucleic Acids Res., 2003, 31, e43 CrossRef.
- Y. P. Ho, M. C. Kung, S. Yang and T. H. Wang, Nano Lett., 2005, 5, 1693–1697 CrossRef CAS.
- A. E. Nel, L. Mädler, D. Velegol, T. Xia, E. M. V. Hoek, P. Somasundaran, F. Klaessig, V. Castranova and M. Thompson, Nat. Mater., 2009, 8, 543–557 CrossRef CAS.
- J. Rejman, V. Oberle, I. S. Zuhorn and D. Hoekstra, Biochem. J., 2004, 377, 159–169 CrossRef CAS.
- W. Jiang, B. Y. Kim, J. T. Rutka and W. C. Chan, Nat. Nanotechnol., 2008, 3, 145–150 CrossRef CAS.
- B. D. Chithrani, A. A. Ghazani and W. C. W. Chan, Nano Lett., 2006, 6, 662–668 CrossRef CAS.
- Y. Geng, P. Dalhaimer, S. Cai, R. Tsai, M. Tewari, T. Minko and D. E. Discher, Nat. Nanotechnol., 2007, 2, 249–255 CrossRef CAS.
- A. Verma, O. Uzun, Y. Hu, Y. Hu, H.-S. Han, N. Watson, S. Chen, D. J. Irvine and F. Stellacci, Nat. Mater., 2008, 7, 588–595 CrossRef CAS.
- L. W. Zhang and N. A. Monteiro-Riviere, Toxicol Sci., 2009, 110, 138–155 CrossRef CAS.
- Y. Pan, S. Neuss, A. Leifert, M. Fischler, F. Wen, U. Simon, G. Schmid, W. Brandau and W. Jahnen-Dechent, Small, 2007, 3, 1941–1949 CrossRef CAS.
- V. P. Torchilin, Adv. Drug Delivery Rev., 2006, 58, 1532–1555 CrossRef CAS.
- K. Park, S. Lee, E. Kang, K. Kim, K. Choi and I. C. Kwon, Adv. Funct. Mater., 2009, 19, 1553–1566 CrossRef CAS.
- A. K. Salem, P. C. Searson and K. W. Leong, Nat. Mater., 2003, 2, 668–671 CrossRef CAS.
- S. Santra, C. Kaittanis, J. Grimm and J. M. Perez, Small, 2009, 5, 1862–1868 CrossRef CAS.
- J. Gao, H. Gu and B. Xu, Acc. Chem. Res., 2009, 42, 1097–1107 CrossRef CAS.
- M. E. Gindy and R. K. Prud'homme, Expert Opin. Drug Delivery, 2009, 6, 865–878 Search PubMed.
- K. E. Sapsford, T. Pons, I. L. Medintz and H. Mattoussi, Sensors, 2006, 6, 925–953 CrossRef CAS.
- X. Gao, L. Yang, J. A. Petros, F. F. Marshall, J. W. Simons and S. Nie, Curr. Opin. Biotechnol., 2005, 16, 63–72 CrossRef CAS.
- X. Michalet, F. Pinaud, T. D. Lacoste, M. Dahan, M. P. Bruchez, A. P. Alivisatos and S. Weiss, Single Mol., 2001, 2, 261–276 CAS.
- I. L. Medintz, H. Mattoussi and A. R. Clapp, Int. J. Nanomed., 2008, 3, 151–167 Search PubMed.
- R. C. Mulligan, Science, 1993, 260, 926–932 CrossRef CAS.
- P. D. Robbins and S. C. Ghivizzani, Pharmacol. Ther., 1998, 80, 35–47 CrossRef CAS.
- M. Mammen, S. K. Choi and G. M. Whitesides, Angew. Chem., Int. Ed., 1998, 37, 2754–2794 CrossRef.
- J. J. Storhoff, A. A. Lazarides, R. C. Mucic, C. A. Mirkin, R. L. Letsinger and G. C. Schatz, J. Am. Chem. Soc., 2000, 122, 4640–4650 CrossRef CAS.
- T. Kirchhausen, Nat. Rev. Mol. Cell Biol., 2000, 1, 187–198 CrossRef CAS.
- W. J. Parak, R. Boudreau, M. Le Gros, D. Gerion, D. Zanchet, C. M. Micheel, S. C. Williams, A. P. Alivisatos and C. Larabell, Adv. Mater., 2002, 14, 882–885 CrossRef CAS.
- A. M. Derfus, W. C. W. Chan and S. N. Bhatia, Adv. Mater., 2004, 16, 961–966 CrossRef CAS.
- J. B. Delehanty, H. Mattoussi and I. L. Medintz, Anal. Bioanal. Chem., 2009, 393, 1091–1105 CrossRef CAS.
- F. Q. Chen and D. Gerion, Nano Lett., 2004, 4, 1827–1832 CrossRef CAS.
- K. Yum, S. Na, Y. Xiang, N. Wang and M. Yu, Nano Lett., 2009, 9, 2193–2198 CrossRef CAS.
- S. Somiari, J. Glasspool-Malone, J. J. Drabick, R. A. Gilbert, R. Heller, M. J. Jaroszeski and R. W. Malone, Mol. Ther., 2000, 2, 178–187 CrossRef CAS.
- S. Barua and K. Rege, Small, 2009, 5, 370–376 CrossRef CAS.
- Y.-C. Tseng, S. Mozumdar and L. Huang, Adv. Drug Delivery Rev., 2009, 61, 721–731 CrossRef CAS.
- D. Putnam, Nat. Mater., 2006, 5, 439–451 CrossRef CAS.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Kluwer Academic Plenum, New York, 1999 Search PubMed.
- C. Srinivasan, J. Lee, F. Papadimitrakopoulos, L. K. Silbart, M. Zhao and D. J. Burgess, Mol. Ther., 2006, 14, 192–201 CrossRef CAS.
- Y. P. Ho, H. H. Chen, K. W. Leong and T. H. Wang, J. Controlled Release, 2006, 116, 83–89 CrossRef CAS.
- H. H. Chen, Y. P. Ho, X. Jiang, H. Q. Mao, T. H. Wang and K. W. Leong, Mol. Ther., 2008, 16, 324–332 CrossRef CAS.
- H. H. Chen, Y. P. Ho, X. Jiang, H. Q. Mao, T. H. Wang and K. W. Leong, Nano Today, 2009, 4, 125–134 CrossRef.
- A. M. Derfus, A. A. Chen, D. H. Min, E. Ruoslahti and S. N. Bhatia, Bioconjugate Chem., 2007, 18, 1391–1396 CrossRef CAS.
- W. B. Tan, S. Jiang and Y. Zhang, Biomaterials, 2007, 28, 1565–1571 CrossRef CAS.
- M. V. Yezhelyev, L. Qi, R. M. O'Regan, S. Nie and X. Gao, J. Am. Chem. Soc., 2008, 130, 9006–9012 CrossRef CAS.
- L. Qi and X. Gao, ACS Nano, 2008, 2, 1403–1410 CrossRef CAS.
- Y. Gohon, T. Dahmane, R. W. H. Ruigrok, P. Schuck, D. Charvolin, F. Rappaport, P. Timmins, D. M. Engelman, C. Tribet, J. Popot and C. Ebel, Biophys. J., 2008, 94, 3523–3537 CrossRef CAS.
- N. Manabe, A. Hoshino, Y. Liang, T. Goto, N. Kato and K. Yamamoto, IEEE Trans. NanoBiosci., 2006, 5, 263–267 CrossRef.
- V. Bagalkot, L. Zhang, E. Levy-Nissenbaum, S. Jon, P. W. Kantoff, R. Langer and O. C. Farokhzad, Nano Lett., 2007, 7, 3065–3070 CrossRef CAS.
- G. Gopalakrishnan, C. Danelon, P. Izewska, M. Prummer, P. Y. Bolinger, I. Geissbühler, D. Demurtas, J. Dubochet and H. Vogel, Angew. Chem., Int. Ed., 2006, 45, 5478–5483 CrossRef CAS.
- W. T. Al-Jamal, K. T. Al-Jamal, P. H. Bomans, P. M. Frederik and K. Kostarelos, Small, 2008, 4, 1406–1415 CrossRef CAS.
- W. T. Al-Jamal, K. T. Al-Jamal, B. Tian, A. Cakebread, J. M. Halket and K. Kostarelos, Mol. Pharmaceutics, 2009, 6, 520–530 CrossRef CAS.
- K. C. Weng, C. O. Noble, B. Papahadjopoulos-Sternberg, F. F. Chen, D. C. Drummond, D. B. Kirpotin, D. Wang, Y. K. Hom, B. Hann and J. W. Park, Nano Lett., 2008, 8, 2851–2857 CrossRef CAS.
- G. D. Bothun, A. E. Rabideau and M. A. Stoner, J. Phys. Chem. B, 2009, 113, 7725–7728 CrossRef CAS.
- E. Chang, N. Thekkek, W. W. Yu, V. L. Colvin and R. Drezek, Small, 2006, 2, 1412–1417 CrossRef CAS.
- A. M. Derfus, W. C. W. Chan and S. N. Bhatia, Nano Lett., 2004, 4, 11–18 CrossRef CAS.
- A. Hoshino, N. Manabe, K. Fujioka, K. Suzuki, M. Yasuhara and K. Yamamoto, J. Artif. Organs, 2007, 10, 149–157 Search PubMed.
- M. L. Schipper, G. Iyer, A. L. Koh, Z. Cheng, Y. Ebenstein, A. Aharoni, S. Keren, L. A. Bentolila, J. Li, J. Rao, X. Chen, U. Banin, A. M. Wu, R. Sinclair, S. Weiss and S. S. Gambhir, Small, 2009, 5, 126–134 CrossRef CAS.
- T. J. Daou, L. Li, P. Reiss, V. Josserand and I. Texier, Langmuir, 2009, 25, 3040–3044 CrossRef CAS.
- M. L. Schipper, Z. Cheng, S. Lee, L. A. Bentolila, G. Iyer, J. Rao, X. Chen, A. M. Wu, S. Weiss and S. S. Gambhir, J. Nucl. Med., 2007, 48, 1511–1518 CrossRef CAS.
- H. Mok, K. H. Bae, C. H. Ahn and T. G. Park, Langmuir, 2009, 25, 1645–1650 CrossRef CAS.
- J. H. Warner, A. Hoshino, K. Yamamoto and R. D. Tilley, Angew. Chem., Int. Ed., 2005, 44, 4550–4554 CrossRef CAS.
- S. Wennmalm and S. M. Simon, Annu. Rev. Biochem., 2007, 76, 419–446 CrossRef CAS.
- D. Zhou, L. Ying, X. Hong, E. A. Hall, C. Abell and D. Klenerman, Langmuir, 2008, 24, 1659–1664 CrossRef CAS.
- E. M. Chan, R. A. Mathies and A. P. Alivisatos, Nano Lett., 2003, 3, 199–201 CrossRef CAS.
- J. Wagner and J. M. Kohler, Nano Lett., 2005, 5, 685–691 CrossRef CAS.
- H. Song, D. L. Chen and R. F. Ismagilov, Angew. Chem., Int. Ed., 2006, 45, 7336–7356 CrossRef CAS.
- J. B. Edel, R. Fortt, J. C. DeMello and A. J. DeMello, Chem. Commun., 2002, 1136–1137 RSC.
- A. Jahn, W. N. Vreeland, M. Gaitan and L. E. Locascio, J. Am. Chem. Soc., 2004, 126, 2674–2675 CrossRef CAS.
- Y. J. Song, J. Hormes and C. Kumar, Small, 2008, 4, 698–711 CrossRef CAS.
- A. T.-H. Hsieh, N. Hori, R. Massoudi, P. J.-H. Pan, H. Sasaki, Y. A. Lin and A. P. Lee, Lab Chip, 2009, 9, 2638–2643 RSC.
| This journal is © The Royal Society of Chemistry 2010 |