DOI:
10.1039/B9NR00140A
(Paper)
Nanoscale, 2010,
2, 282-286
Microwave-assisted synthesis of Pt/CNT nanocomposite electrocatalysts for PEM fuel cells
Received
17th June 2009
, Accepted 5th September 2009
First published on 12th October 2009
Abstract
Microwave-assisted heating of functionalized, single-wall carbon nanotubes (FCNTs) in ethylene glycol solution containing H2PtCl6, led to the reductive deposition of Pt nanoparticles (2.5–4 nm) over the FCNTs, yielding an active catalyst for proton-exchange membrane fuel cells (PEMFCs). In single-cell testing, the Pt/FCNT composites displayed a catalytic performance that was superior to Pt nanoparticles supported by raw (unfunctionalized) CNTs (RCNTs) or by carbon black (C), prepared under identical conditions. The supporting single-wall carbon nanotubes (SWNTs), functionalized with carboxyl groups, were studied by thermogravimetric analysis (TGA), cyclic voltammetry (CV), and Raman spectroscopy. The loading level, morphology, and crystallinity of the Pt/SWNTcatalysts were determined using TGA, SEM, and XRD. The electrochemically active catalytic surface area of the Pt/FCNT catalysts was 72.9 m2/g-Pt.
1. Introduction
Because of their extraordinary electrical, mechanical, and structural properties, carbon nanotubes (CNTs) have been widely studied as supports for Pt catalysts in fuel cell applications.1–5 Pt nanoparticles generally display superior electrocatalytic performance when supported on carbon nanotubes compared to carbon black .6,7 However, to ensure efficient electrocatalysis, the deposited Pt must have a small particle size, narrow size distribution, and excellent dispersion on the support.8 Over the last few decades, material synthesis techniques based on microwave chemistry have enjoyed significant attention due, in no small measure, to their ability to better control particle shape, size, and size distribution, amongst others.9–11 Microwave irradiation has many advantages over conventional heating, including more rapid heating, selective materials coupling, and enhanced reaction kinetics; these make the process an attractive alternative route for materials synthesis.12–19 An important aspect of forming Pt nanoparticles on CNTs is the method of activating the substrate surface. CNTs are chemically inert and generally do not provide enough surface groups for Pt deposition. Sonochemical treatment using extremely aggressive reagents is probably the most common method of surface activation since a mixture of strong acids will typically not only remove most impurities on CNTs, but also allow the tubes to be functionalized with groups such as carboxyl (–COOH), hydroxyl (–OH) and carbonyl (>C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) that are suitable for anchoring Pt metal ions.4,19–22
O) that are suitable for anchoring Pt metal ions.4,19–22
In this work, we report a microwave-assisted method for preparing nanostructured Pt catalysts supported on sonochemically-treated SWNTs for a proton-exchange membrane electrolyte fuel cell (PEMFC). We show that the microwave-assisted process reduces the Pt salt precursor, leading to the formation of 2.5–4 nm Pt nanoparticles on the CNT substrates. Electrochemical measurements indicate that the Pt nanocatalysts exhibit enhanced electrochemical active surface areas and improved fuel cell performance on the sonochemically-treated CNTs than on raw (unfunctionalized) CNTs or on carbon black .
2. Experimental
2.1 Materials
Single-wall CNTs were purchased from Unidym, Inc., USA. Carbon black (Vulcan XC-72R) was supplied by Cabot Corp. Nafion® 117 solution (5 wt%), Ethylene glycol, H2PtCl6·xH2O, sulfuric acid, nitric acid and other solvents were purchased from Sigma-Aldrich.
2.2 Sonochemical treatment of CNTs
Functionalized SWNTs (FCNTs) were prepared using a conventional sonochemical treatment method. The raw CNTs (RCNTs) were dispersed in a mixture of concentrated H2SO4 and 70% HNO3 (1 : 3 molar ratio) and sonicated for 2 h. These resulting functionalized SWNTs were washed with Milli-Q water several times until the pH value was close to 7. The solid FCNTs powder was isolated viacentrifugation and dried in a vacuum oven at 85 °C for 24 h.
2.3 Preparation of supported Pt nanoparticles
Pt nanoparticles were synthesised on the FCNTs or RCNTs by microwave heating a solution of H2PtCl6 dissolved in ethylene glycol (EG) according to the following procedure. 40 mg of FCNTs were dispersed in 25 mL EG solvent. Then 1.0 mL 0.05 M H2PtCl6 in EG was added and the resulting solution adjusted to pH ca. 12 by adding 0.5 mL of 0.05 M NaOH aqueous solution. The solution was thereafter heated using a household microwave oven (National 700 W, Japan) for two 75 s intervals at 700 W. The solution was then stirred overnight to allow for complete deposition of the particles over the CNT supports. The solid Pt/FCNTs catalyst was recovered viacentrifugation and washed with acetone four times, then dried in a vacuum oven at 85 °C overnight. For comparative purposes, a sample of Pt-loaded carbon black was also prepared under identical conditions.
2.4 Characterization of Pt/FCNTs, Pt/RCNTs and Pt/C
The effect of the sonochemical treatment on the surface functionality of CNTs was examined using a JOBIN YVON HR800 confocal Raman spectrometer (HORIBA, Ltd., France) with 632.8 nm diode laser excitation on a 900 lines mm−1 grating at room temperature. Thermogravimetric analysis (TGA) was performed using a Q500 TGA analyzer(TA Instruments, UK) to evaluate the residual level of impurity in the CNTs before and after the sonochemical treatment. We also employ this technique to calculate the loading level of platinum metal on the different CNT supports. The crystallinity of the platinum nanoparticles were examined by X-ray powder diffraction (XRPD) analysis, performed using GBC MMA diffraction equipment (GBC Scientific Equipment Pty Ltd, Australia) with monochromatic Cu Kα radiation. The spectra were recorded at a low scan rate of 1 deg min−1 at a range of 2θ from 15° to 90° in order to ensure fine crystalline structures were obtained for the Pt catalysts. Bright-field transmission electron microscopy images were taken using a transmitted electron detector (TED on image) in a JEOL JSM7500FA cold-field-gun scanning electron microscope (CFG-SEM) (JEOL Ltd., Japan) operating at 30.0 kV.
2.5 Electrochemical measurements
All electrochemical measurements were carried out in a standard three-electrode cell using a CHI 720C electrochemical Biopotentiostat (CH Instruments, USA) at ambient temperature. A Pt mesh and a Ag/AgCl electrode (3 M NaCl) served as the counter and reference electrodes, respectively.
2.6 Fuel cell performance
Membrane electrode assemblies (MEAs) were prepared through two steps; (1) by brushing the catalyst suspension over the carbon paper of the gas diffusion membrane and (2) pressing the anode and cathode on either side of the Nafion membrane at 130 °C. For the comparative studies, a commercially-available Pt/Vulcan XC 72R (Johnson Matthey, U.K.) was used as the anode catalyst, and the microwave-prepared Pt catalyst was used as the cathode catalyst. The prepared MEAs were tested using a 850e fuel cell test system (Scribner Associates, Inc., USA) in a single fuel cell hardware assembly (Fuel Cell Technologies, Inc., USA) fed with humidified pure H2 and O2, respectively. The dimensions of the MEA electrode used in the testing single-cell was 5 cm2 with Pt loading of 0.4 mg cm−2 on both anode and cathode catalyst layers. The gas flow rates were fixed at 0.8 L min−1. The fuel cell measurements were carried out at 80 °C under 100 kPa back-pressure. The polarization curves were recorded using a scan current method by holding the cell at each current-density value for 60 s in order to obtain the steady-state voltage value.
3. Results and discussion
3.1 Characterization of SWNTs
The TGAspectra for the SWNTs before and after ultrasonication were obtained (Fig. 1). The initial mass loss for the FCNTs below 100 °C is attributed to the removal of water and residual solvent. The mass loss between 200 °C and 400 °C was due to burning of the amorphous carbon and the removal of functionalities. As can be seen in Fig. 1, the mass loss was substantially less for the RCNTs in this temperature range. Final residual weights of 3.29% and 9.28% were obtained for the FCNTs and RCNTs, respectively, indicating that an improved purity of 96.7% SWNTs was obtained after ultrasonication.
 |
| | Fig. 1
TGA curves of FCNTs and RCNTs under air atmosphere at a heating rate of 10 °C per min. | |
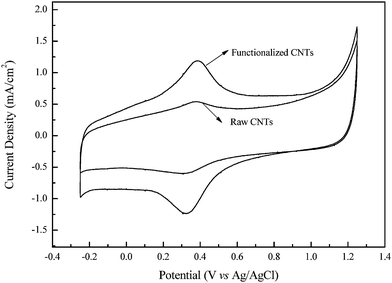
Fig. 2 shows the cyclic voltammograms in 0.5 M H2SO4 solution of glassy carbon electrodes upon which fixed weights of FCNTs or RCNTs were cast. As can be seen, the redox peaks corresponding to the reduction and oxidation of the FCNT carboxyl groups (0.3–0.4 V) were significantly enlarged after the sonochemical treatment. This confirms that additional carboxyl groups were successfully introduced on the surface of the SWNTs through the sonochemical treatment.24
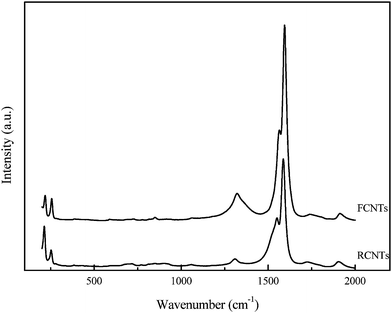
Raman spectroscopy was used to study defects and the extent of functionalization on the surface of the CNTs before and after functionalization (Fig. 3). The Raman spectra of both the RCNTs and the FCNTs display a strong peak at about 1592 cm−1 (G-band) and a weak peak at 1330 cm−1 (D-band). These correspond to the sp3- and sp2- hybridized carbons present, signifying graphitic disorder and the ordered state on the CNT surface, respectively. The intensity of the D-band describes the degree of disorder present along the tube, indicating the presence of pentagons, heptagonal defects, the pentagon–heptagon pairs, or line defects. The extent of the modification and associated defects on the CNTs can be evaluated by the ratio of the intensities of the D-band and G-bands. For the FCNTs and RCNTs, intensity ratios ID/IG of 0.133 and 0.068 were observed, respectively. The strong acid treatment used to produce carboxylic acid sites on the surface of the CNTs, therefore also leads to the formation of significant structural defects suitable for the subsequent deposition of Pt.3,25,26
3.2 Characterization of Pt nanoparticles loaded on CNT supports
After the Pt loading using microwave-assisted heating, the TGAspectra of Pt/FCNTs, Pt/RCNTs and the control Pt/C in an air atmosphere were obtained (Fig. 4) to monitor the loading level of Pt nanoparticles. As can be seen, the highest residual mass was observed from Pt/RCNTs due to the fact that they contained more metal impurities (catalyst used for CNT growth) than Pt/FCNTs. The mass loadings of Pt on the different substrates were determined as 20.31 wt%, 17.20 wt% and 19.51 wt% for Pt/FCNTs, Pt/RCNTs and Pt/C, respectively, by subtracting the mass percentage of impurities in the FCNTs and the RCNTs before Pt loading. Thus, the Pt-loading level on the FCNTs (20.31 wt%) is higher than on the RCNTs (17.20 wt%). This might be due to fewer functional groups and surface defects on the RCNTs. The S-TEM image depicted in Fig. 5 indicates that the Pt nanoparticles were uniformly distributed over the FCNTs with an average size of 2.5–4 nm. Such an arrangement is crucial for efficient electrochemical operation.
 |
| | Fig. 4
TGA curves of Pt/FCNTs, Pt/RCNTs and Pt/C under air atmosphere at a heating rate of 10 °C per minute. | |
The crystalline nature of the supported Pt nanoparticles was examined by X-ray diffraction (XRD) (Fig. 6). The XRD patterns show similar crystalline properties for the Pt catalysts deposited on all three substrates. The diffraction peaks at Bragg angles of about 39°, 46°, 67°, and 81° correspond to the (111), (200), (220) and (311) Pt facets. These patterns are in good agreement with those of the reference pattern (JCPDS 75-1621) for Pt.7
3.3 Electrochemical activity of the supported Pt catalysts
The electrocatalytic activity of the Pt nanoparticles is generally closely related to the electrochemically active surface area of the Pt on the supporting FCNTs, RCNTs, and carbon black . Cyclic voltammograms were recorded (Fig. 7) in 0.5 M H2SO4 to determine the atom H adsorption on each of the Pt/FCNT, Pt/RCNT, and Pt/C electrodes, deposited by casting in equal weights on a glassy carbon electrode. The electrochemically active catalytic surface area (Sact) for the deposited Pt could be estimated using the formula below, from the average charge transfer (QH) based on integrated values of hydrogen adsorption–desorption in the range −0.2–0.0 V.20,26
Assuming the charge per real cm2 of Pt with monolayer adsorption of hydrogen is Q0H = 210 µC cm−1, then according to the CV curves in Fig. 7, the estimated Sact values are: 72.9 m2/g-Pt (Pt/FCNT electrode), 48.2 m2/g-Pt (Pt/RCNT electrode), and 10.7 m2/g-Pt (Pt/C electrode). This suggests that the Pt/FCNTs have the highest value of Sact, which is consistent with improved adhesion, accessibility, and dispersion of the Pt on the FCNT support. The Pt-loaded FCNTs would then also be expected to display the best performance in a PEM fuel cell.
3.4 Fuel cell performance
A comparison of the PEM fuel cell performance as a cathode catalyst, using Pt loaded onto the three different supports, FCNTs, RCNTs and C, is shown in Fig. 8. The depicted polarization curves were recorded under the same working conditions (0.4 mg-Pt/cm2) and using the same type of anode catalyst, Pt/Vulcan XC 72R. As can be seen, the best fuel cell performance, with the highest current and power density output was obtained using the Pt/FCNTs as the cathode catalyst. The single-cell PEMFC test rig with Pt/FCNTs as the cathode catalyst, displays a current density of ca. 560 mA cm−2 at a potential of 0.6 V. The power density is then ca. 340 mW cm−2. Under the same operating conditions, the PEMFC with Pt/RCNTs displays a current density of around 480 mA cm−2 at a potential of 0.6 V, with an accompanying power density of <280 mW cm−2, which is 15% lower compared with Pt/FCNTs. This trend is consistent with the observed electrochemically catalytic surface areas of the Pt/FCNTs and Pt/RCNTs. Sonochemical treatment of the SWNTs also results in enhanced surface functionalization, which likely provides for better adherence of the Pt nanoparticles on the support surface. This would also be expected to give a better performance at higher current densities. The weakest fuel cell performance was obtained using Pt/C. This can be attributed to the lower surface area and poorer electrical properties compared with SWNTs.
 |
| | Fig. 8 Polarization curves of a testing single PEM fuel cell using Pt/FCNTs, Pt/RCNTs, and Pt/C as the cathode catalyst, respectively. | |
The experimentally obtained polarization data were analysed using the semiempirical equation.27
where
V and
I are the experimentally measured cell voltage and current, and
b and
R are the Tafel slope and the total uncompensated dc resistance. The curve-fitting results for
Fig. 8 are listed in
Table 1.
Table 1 Kinetic parameters derived from a regression analysis of the polarization curves in Fig. 8
|
Catalyst
|
V
0
|
b/V decade−1 |
R/Ω cm2 |
| Pt/FCNTs |
0.896 |
0.059 |
0.262 |
| Pt/RCNTs |
0.871 |
0.067 |
0.433 |
| Pt/C |
0.856 |
0.059 |
0.501 |
As can be seen, the PEMFC employing Pt/FCNTs has the lowest dc resistance (0.262 Ω cm2), compared to Pt/RCNTs (0.433 Ω cm2) and Pt/C (0.501 Ω cm2). The three supported catalysts display similar Tafel slopes for oxygen reduction (50–70 mV decade−1). This is in accord with the previous reports for Pt catalysts in fuel cell.3,9,28
4. Conclusion
We have shown that the microwave-assisted technique is an effective method for preparing supported nanostructured Pt catalysts for PEM fuel cells. Sonochemical treatment readily produces functionality on the surface of the CNTs to which Pt ions can anchor during the microwave treatment. Pt nanoparticles supported on functionalized SWNTs (Pt/FCNTs) by these means exhibit a significantly enhanced electrochemically active catalytic surface area and PEM fuel cell performance when compared to controls: Pt/RCNTs or Pt/C. This is undoubtedly due to better accessibility and dispersion in the Pt nanoparticles. As such, these results demonstrate that the microwave-assisted process offers a very promising method for loading Pt nanoparticles onto CNTs. It also highlights the potential utility of functionalized SWNTs as nanostructured support materials in PEM fuel cell applications.
Acknowledgements
The authors gratefully thank the following for financial support; the Australia Research Council and the Department of Education, Science and Technology (Australia) (International Linkage Project).
References
-
M. Meyyappan, Carbon Nanotubes: Science and Applications, CRC Press, Boca Raton, 2005 Search PubMed.
- Y. Mu, H. Liang, J. Hu, L. Jiang and L. Wan, J. Phys. Chem. B, 2005, 109, 22212 CrossRef CAS.
- A. Leela Mohana Reddy and S. Ramaprabhu, J. Phys. Chem. C, 2007, 111, 16138 CrossRef.
- N. Rajalakshmi, H. Ryu, M. M. Shaijumon and S. Ramaprabhu, J. Power Sources, 2005, 140, 250 CrossRef CAS.
- J. J. Niu and J. N. Wang, Electrochim. Acta, 2008, 53, 8058 CrossRef CAS.
- W. Li, C. Liang, J. Qiu, W. Zhou, H. Han, Z. Wei, G. Sun and Q. Xin, Carbon, 2002, 40, 791 CrossRef CAS.
- W. Li, C. Liang, W. Zhou, J. Qiu, Z. Zhou, G. Sun and Q. Xin, J. Phys. Chem. B, 2003, 107, 6292 CrossRef CAS.
- T. S. Ahmadi, Z. L. Wang, T. C. Green, A. Henglein and M. A. El-Sayed, Science, 1996, 272, 1924 CrossRef CAS.
- Z. Liu, L. M. Gan, L. Hong, W. Chen and J. Y. Lee, J. Power Sources, 2005, 139, 73 CrossRef CAS.
- W. X. Chen, J. Y. Lee and Z. Liu, Chem. Commun., 2002, 2588 RSC.
- W. Yu, W. Tu and H. Liu, Langmuir, 1999, 15, 6 CrossRef CAS.
- S. A. Galema, Chem. Soc. Rev., 1997, 26, 233 RSC.
-
D. Bogdal, A. Prociak, Microwave-Enhanced Polymer Chemistry and Technology, Blackwell Publishing Ltd, UK, 2007 Search PubMed.
-
D. Bogdal, Microwave-assisted Organic Synthesis – One Hundred Reaction Procedures, Amsterdam, Elsevier, 2005 Search PubMed.
- D. L. Boxall and C. M. Lukehart, Chem. Mater., 2001, 13, 806 CrossRef CAS.
- W. Tu and H. Liu, Chem. Mater., 2000, 12, 564 CrossRef CAS.
- S. Komarneni, D. Li, B. Newalkar, H. Katsukl and A. S. Bhalla, Langmuir, 2002, 18, 5959 CrossRef CAS.
- X.-H. Liao, J.-M. Zhu, J.-J. Zhu, J.-Z. Xu and H.-Y. Chen, Chem. Commun., 2001, 937 RSC.
- A. L. Washington II and G. F. Strouse, J. Am. Chem. Soc., 2008, 130, 8916 CrossRef CAS.
- Yangchuan Xing, J. Phys. Chem. B, 2004, 108, 19255 CrossRef CAS.
- M. Holzinger, O. Vostrovsky, A. Hirsch, F. Hennrich, M. Kappes, R. Weiss and F. Jellen, Angew. Chem., Int. Ed., 2001, 40, 4002 CrossRef CAS.
- V. Georgakilas, K. Kordators, M. Prato, D. M. Guldi, M. Holzinger and A. Hirsch, J. Am. Chem. Soc., 2002, 124, 760 CrossRef CAS.
- Z. D. Wei, C. Yan, Y. Tan, L. Li, C. X. Sun, Z. G. Shao, P. K. Shen and H. W. Dong, J. Phys. Chem. C, 2008, 112, 2671 CrossRef CAS.
- S. Wang, X. Wang and S. P. Jiang, Langmuir, 2008, 24, 10505 CrossRef CAS.
- R. V. Hull, L. Li, Y. Xin and C. C. Chusuei, Chem. Mater., 2006, 18, 1780 CrossRef CAS.
- R. Woods, J. Electroanal. Chem., 1976, 9, 1 CAS.
- E. A. Ticianelli, C. R. Derouin, A. Redondo and S. Srinivasan, J. Electrochem. Soc., 1988, 135, 2209 CAS.
- A. Parthasarathy, S. Srinivasan, A. J. Appleby and C. R. Martin, J. Electroanal. Chem., 1992, 339, 101 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2010 |
Click here to see how this site uses Cookies. View our privacy policy here.