By what means should nanoscaled materials be constructed: molecule, medium, or human?
Katsuhiko
Ariga
*,
Xianluo
Hu
,
Saikat
Mandal
and
Jonathan P.
Hill
World Premier International (WPI) Research Center for Materials Nanoarchitectonics (MANA), National Institute for Materials Science (NIMS), 1-1 Namiki, Tsukuba, Ibaraki 305-0044, Japan. E-mail: ARIGA.Katsuhiko@nims.go.jp; Fax: +81 29 860 4832; Tel: +81 29 860 4957
First published on 8th October 2009
Abstract
There is great potential in nanoscale science and technology, and construction of macrosized materials and systems possessing nanoscale structural features is a crucial factor in the everyday application of nanoscience and nanotechnology. Because nanoscale substances are often constructed through self-assembly of unit molecules and nanomaterials, control of the self-assembly process is required. In order to establish general guidelines for the fabrication of materials with nanoscale structural characteristics, i.e., nanoscaled materials, we introduce here examples of recent research in related fields categorised as: (i) self-assembled structures with forms generally determined by intrinsic interactions between molecules and/or unit nanomaterials, (ii) self-assemblies influenced by their surrounding media, especially interfacial environments, (iii) modulation of self-assembly by artificial operation or external stimuli. Examples are not limited to organic molecules, which are often regarded as the archetypal species in self-assembly chemistry, and many examples of inorganic assemblies and hybrid structures are included in this review.
 Katsuhiko Ariga | Katsuhiko Ariga is principal investigator of World Premier International (WPI) Research Center for Materials Nanoarchitectonics (MANA), National Institute for Materials Science (NIMS). He also works as a director of the Supermolecules group at NIMS and professor of Tokyo University of Science. His major interests are the fabrication of novel nanostructures based on molecular recognition and self-assembly, including Langmuir–Blodgett films, layer-by-layer films, and mesoporous materials. |
 Xianluo Hu | Xianluo Hu is currently a JSPS postdoctoral fellow under the supervision of Dr Katsuhiko Ariga at the National Institute for Materials Science (NIMS). He received his Ph.D. in Environmental Science from The Chinese University of Hong Kong in 2007, and then worked as a Research Associate in Department of Chemistry, The Chinese University of Hong Kong. His most recent research interests include fabrication and assembly of nanostructured materials for environmental and energy transfer applications. |
 Saikat Mandal | Saikat Mandal is a postdoctoral researcher (JSPS fellow) in the Supermolecules Group at the National Institute for Materials Science. His work is concerned with the synthesis of metal and semiconductor nanoparticles in thin film form, core–shell nanostructures and assembly of metal nanoparticles using different scaffolds. |
 Jonathan P. Hill | Jonathan P. Hill is a senior researcher in the Supermolecules Group at the National Institute for Materials Science (NIMS). Prior to working in NIMS, he occupied postdoctoral positions in Tokyo, University of Karlsruhe (Germany), and Osaka. His research interests include supramolecular science, chemistry of the tetrapyrroles, molecular self-assembly in the bulk state and at interfaces, and the sensing, optical and electronic properties of molecules. |
Introduction
Rapid developments in nanoscience have led to the discovery of unexpected phenomena at the nanoscale1 as well as novel techniques for construction of nanoscale objects.2 From the point of view of the public, nanotechnology appears to entail the fabrication of miniature machines which, for example, might be able to move within a human body and repair damaged tissues or compose supercomputers small enough to fit in a pocket. However, to exploit nanoscience and nanotechnology, construction of macro-sized materials and systems containing nanoscale structural features is important. To realise practical devices from nanomaterials utilizing their unique properties, individual nanostructures must be assembled into a predesignated form, within which individual components can interact in a fashion appropriate to the required function. Nanotechnology provides the tools for humans to ‘play’ with the ultimate toy box which contains all atoms and molecules, i.e. Nature, and the possibilities for creating new entities are limitless. In scientific terms, nanotechnology deals directly with the manipulation of individual atoms and thereby the programmed formation of superstructures. There has already been considerable success in the use of microstructures over the past decade. Various fabrication techniques categorized as top-down technology can be used to construct integrated microstructures. Appearance of cellular phones and mobile devices is a reflection of the great success of these methods.However, there exists a question of whether the successful application of microfabrication can be extended to bulk materials and systems having nanoscale structural features. This extension would be of great utility but can not be easily achieved for materials requiring nanoscale structural precision. Two major approaches, “top-down” (engineering down) and “bottom-up” (engineering up) methods, are being used for the generation of organized nanoscale assemblies. Top down approaches involve generation of patterned structures by suitable lithographic or ion implantation techniques and these mark the beginning of silicon-integrated chip technology. However it is known that the current rate of miniaturization (governed by so-called Moore's law) in silicon memory technology will be affected very soon by the physical limits of device dimensions imposed by ultra-violet, electron/ion beam and soft X-ray lithographic techniques. This is the reason why the bottom-up approaches are gradually gaining in importance relative to the top-down approaches. Concurrently therefore, material fabrication based on bottom-up-type self-assembly has become more important. Formation and preparation of nanoscale objects rely strongly on self-assembly of unit molecules and nanomaterials.3 If self-assembly processes are determined solely by molecules and unit materials structures, formation of the final products must be performed by designing appropriate molecular structures, and has been attractively termed “molecular programming”.4 However, this is disadvantageous for development of practical applications. Modulation of self-assembly processes by human intervention and/or external stimuli is required.
From this point of view, we classify examples of recent research on self-assembled materials having nanoscale structural features into three categories: (i) self-assembled structures with forms generally determined by intrinsic interactions between molecules and/or unit nanomaterials, (ii) self-assemblies influenced by their surrounding medium, especially an interfacial environment; (iii) modulation of self-assembly by artificial operation or external stimuli. Although the examples described represent only a small portion of the huge volume of research in this hot area, we hope that this review will provide some useful general guidelines for the fabrication of materials with nanoscale features.
By molecules or materials: spontaneous assembly
Molecules often self-assemble into objects of nanoscale structural precision through various molecular interactions such as electrostatic interaction and hydrogen bonding (Fig. 1). This process occurs spontaneously and requires no human intervention. Information contained in a molecular structure often decides the final stationary status of its assemblies. This kind of spontaneous assembly has been mostly investigated for organic materials over a wide range of molecular weight: small molecules to large polymeric compounds.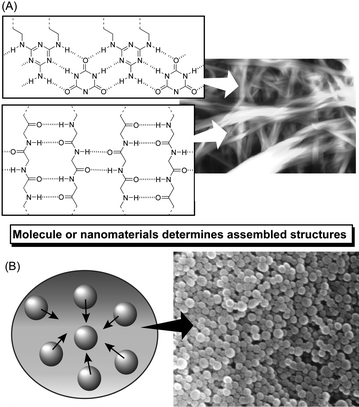 | ||
| Fig. 1 Schematic illustration of the formation of self-assembled materials based on molecular interaction (A) and interaction between nanomaterials (B). | ||
(i) Organic molecules
Spontaneous assembly often results in objects composed of many units by continuous extension of molecular interactions. Yagai, Ajayaghosh, and coauthors showed self-organization of hydrogen-bonded rosette assemblies of oligo(p-phenyleneethynylene) attached melamine derivatives and a cyanurate derivative in aliphatic solvents, leading to the formation of toroidal objects of nanometer dimensions.5 In the assembly, π-electronic segments are spatially arranged in closed circular structures reminiscent of the light-harvesting systems of purple photosynthetic bacteria. Shape-defined assemblies have also received attention. In particular, cell-like vesicular assemblies remain a central target because of their potential application in drug delivery and their fundamental importance in biomimetics .6 Araki and coworkers showed that molecular design to sandwich two-dimensional hydrogen-bonding networks between nonpolar protective layers results in fabrication of nano- and microcapsules with high stability even in aqueous media.7 Vesicular structures were prepared through injection of a small amount of THF solution of guanosine derivatives into deionized water followed by sonication. Cell-like high stability, which is ascribed to the two-dimensional hydrogen-bonding networks of the vesicular membrane, was demonstrated. The strategy of protecting the two-dimensional hydrogen-bonded networks with nonpolar protective layers should be effective in the design of highly stable supramolecular materials. Lee and coworkers used two-dimensional self-assembly of dumbbell-shaped rod amphiphiles to form a capsule structure with lateral nanopores.8 Interestingly, the lateral nanopores undergo a transition from the open state to the closed state upon heating, which is capable of blocking material transport through the membrane. Capsule structures with reversibly gated lateral pores can be considered as synthetic analogues to viral capsids.Large molecules such as polymers can act as building units of spontaneously assembled objects. Schappacher and Deffieux reported an efficient route to design large macrocyclic polymers of controlled molar mass using block copolymers.9 The macrocyclic brushes obtained can be self-assembled into supramolecular tubes. Hanson et al. prepared water-in-oil-in-water double emulsions through a simple process that can be stabilized over many months using single-component, synthetic amphiphilic diblock copolypeptide surfactants, poly(L-lysine HBr)x-b-poly(racemic-leucine)y where x ranged from 20 to 100, and y ranged from 5 to 30 residues.10 These surfactants even stabilize droplets subjected to extreme flow, leading to direct, mass production of robust double nanoemulsions that are amenable to nanostructured encapsulation applications in foods, cosmetics and drug delivery. Winnik, Manners, and coworkers demonstrated that living, crystallization-driven polymerizations offer exceptional potential for the controlled generation of complex and hierarchical micelle architectures.11Self-assembly of polyisoprene-block-poly(ferrocenyldimethylsilane) (PI-b-PFS) diblock copolymers in a selective solvent for polyisoprene yields micelles with a crystalline poly(ferrocenyldimethylsilane) core and a polyisoprenecorona. The morphology formed depends on the ratio of the degrees of polymerization of the two blocks. The polymer with a block ratio of 6:1 forms cylindrical micelles, while the polymer with a block ratio of 1:1, gives tape-like platelet structures.
The specific functions of the objects are often dependent on their shape and design. In this context dissociable molecular assembly provides reversible shape-switching of molecular complexes. The combination of reversible bonds and a morphologically controlled unit in a single complex system would make it possible to switch reversibly between a macrocycle and a polymer. Based on this concept, Ulrich and Lehn reported a system that displays combined constitutional/morphological/motional dynamics in which the morphological change triggered by a metal ion results in a reversible constitutional switch between macrocycle and polymer states.12 Templating by metal ions was used to pre-organize unit molecules to increase the efficiency of macrocycle formation, leading to the stabilization of a given macrocycle among a virtual library. The proposed process is based on the ability to switch a tridentate terpyridine-type ligand molecule reversibly between an “extended” W-shaped state and a “compact” U-shaped state, resulting in reversible switching between macrocycle and polymer states upon addition of a metal ion or its removal by the addition of a competitive binder. An appropriately assembled system permits movement of the component units even in assembled structures. Akutagawa, Nakamura, and coworkers demonstrated ferroelectricity and polarity control in solid-state flip-flop supramolecular rotators using m-fluoroanilinium as a rotor within a hydrogen-bonded assembly of dibenzo[18]crown-6 with Ni(2-thioxo-1,3-dithiole-4,5-dithiolate)2− anion.13 The rotor unit m-fluoroanilinium exhibited dipole rotation by the application of an electric field, and the crystal showed a ferroelectric transition at 348 K. This example suggests a new strategy for chemical design of dipole units enabling control of the nature of the ferroelectric transition temperature.
Non-covalent interactions such as hydrogen bonding, dipole–dipole, electrostatic, van der Waals, and hydrophobic interactions dictate self assembly between molecules and can result in superstructures or supramolecules. Among the non-covalent interactions, hydrogen bonding often plays a critical role in supramolecular self-assembly. Materials assembled through reversible non-covalent interactions lead to self-healing properties. Leibler and coworkers synthesized molecules that associate together to form both chains and cross-links through hydrogen bonds.14 Multitopic molecules with attach functional groups such as amidoethyl imidazolidone, di(amidoethyl)urea and diamido tetraethyl triurea were used to form multiple hydrogen bonds. Unlike conventional cross-linked or thermoreversible rubbers made of macromolecules, these hydrogen-bonded materials can be repaired simply by bringing together fractured surfaces resulting in self-healing at room temperature even after mechanical breaking or cutting. Self-healing functions can be also constructed using properly designed catalytic systems. Braun proposed a generalized approach to self-healing polymer-coating systems.15 In that case, the self-healing coating contained microencapsulated catalyst and a phase-separated healing agent as droplets in a matrix formed on a metallic substrate. When the coating layer was damaged catalyst and healing agent were released and mixed only in the damaged region. Self-healing occurred by the subsequent cross-linking of the polymer (polydimethylsiloxane), thus protecting the substrate from the environment.
Pisignano synthesized photoswitchable nanofibers of 1′,3′-dihydro-1′,3′,3′-trimethyl-6-nitrospiro[2H-1-benzopyran-2,2′-(2H)-indole] based on reversible transformation between two molecular photochemical states, which exhibit different chemicophysical characteristics including absorption, emission, dipole moment, surface energy.16 The resulting photochromism relies on the transformation of a non-polar, colorless closed form, spiropyran, into a highly polar, isomeric, colored open form (trans-merocyanine) with extended π-electron delocalization by the photochemical cleavage of the C–O bond in the molecular ring, which occurs upon ultraviolet exposure. The good stability and cycling performance might lead to application of this system in sensors, lab-on-a-chip technologies, and optical interconnects. Aizenberg and coworkers reported the induction of capillarity-driven self-organization of a nanobristle into helical clusters and demonstrated the fabrication of nontrivial, hierarchically assembled, coiled mesostructures over large areas, in which neither the assembling elements nor the environment are chiral.17 The structures obtained may serve as the seed for the spontaneous breaking of symmetry on large scales.
In nature, self-assembling and disassembling complexes of proteins and nucleic acids bound to a variety of ligands perform intricate and diverse dynamic functions. In particular, DNA fragments are programmable and undergo specific complexation with particular substrates. Therefore, preparation of nanoobjects through self-assembly of DNA strands with designer sequences has been investigated.18 For example, recently Pierce and coworkers demonstrated growth of branching nanofibers using programmed DNA strands.19 Ballister, Zuckermann, and coworkers demonstrated a method for generating discretely structured proteinnanotubes from the simple ring-shaped building block, homohexameric Hcp1 from Pseudomonas aeruginosa.20 The ends and interior of the tubes could be independently and specifically functionalized to generate nanocapsules using a reactive dendrimer as a plug.
Functional molecules such as fullerene and porphyrins can be assembled into materials with nanoscale structural characteristics.21 Nakanishi et al. proposed a novel approach to fabricate robust and durable artificial nanocarbon superhydrophobic surfaces, exhibiting fractal morphology on both nano- and micrometer length scales.22 A fullerene derivative bearing three eicosyloxy chains self-assembled into globular objects of macroscopic dimensions with a wrinkled, flake-like sub-micrometer structure in 1,4-dioxane. Surfaces covered with a thin film prepared through casting of these objects exhibited superhydrophobicity and a surprising durability. Hierarchical fractal structures are believed to promote low adhesion, low friction, and non-wetting characteristics useful in micro/nano-electromechanical systems, microfluidics, or self-cleaning surfaces. Hupp, Nguyen, and coworkers reported a hierarchical strategy for producing mesoscopic objects from discrete amphiphilic porphyrins.23 Macroscopic prisms were self-assembled in the presence of a simple surfactant or selectively partitioned into hydrophobic patterns on glass surfaces.
(ii) Inorganic materials
Spontaneous assemblies are not only limited to organic substances but inorganic nanoscale substances such as nanoparticles, nanorods, and nanowires can be also self-assembled into organized structures through several interactions including capillary force and surface reaction.24 The study of collective properties of assembled inorganicnanostructures has become an important aspect of nanotechnology research in recent years. Recently, Yang and coworkers reported spontaneous self-assembly of shaped silver nanoparticles into three-dimensional plasmonic crystals that display a frequency-selective response in the visible wavelengths.25 Extensive long-range order mediated by exceptional colloid monodispersity gives rise to optical pass bands that can be tuned by particle volume fraction. Because this colloid system has the capability for batch fabrication in a massively parallel manner, both fabricating micrometer-sized devices and large paint-on coatings become possible. Fang and coworkers prepared high-quality cubic PbTe nanocrystals that could be assembled into square arrays, two-dimensional patterns and three-dimensional simple cubic super crystals.26 The unsaturated base oleylamine plays a very important role in the self-assembly process. Combination of structured cubic building blocks and long-range ordered assembly should lead to applications in electronic and photonic devices. Hu, Yu, and coworkers developed a rapid microwave-polyol process that offers an efficient pathway to monodisperse ZnO colloidal clusters in large quantities.27 The size of the clusters composed of small primary nanocrystals can be tuned precisely from about 57 to 274 nm by simply varying the amount of Zn complex precursors. Sensors made of ZnO assembled clusters exhibit high sensitivity for humidity measurement at room temperature.Recently Li and coworkers proposed a versatile approach to assemble nanocrystals to colloid spheres.28 Their method is based on a designed oil-in-water microemulsion system where nanocrystals assemble and aggregate spontaneously by the hydrophobic van der Waals interaction of the surfactant ligands absorbed to the nanocrystals. During controlled evaporation of a low-boiling oil solvent, micrometer-sized three-dimensional spaces were provided by the microemulsion droplets. Zerrouki et al. used a colloidal system to explore a simple forcing route to chiral structures.29 They showed that magnetic colloids can rapidly self-assemble into a variety of isomeric forms. Depending on the shape of the colloidal blocks relative to their spontaneous direction of magnetization, these colloids assemble into structures that must fulfill both steric and magnetic constraints. This induces a fixed repeated isomeric configuration or tacticity. If the size ratio between the spheres is large enough, a single helicity is adopted, either right- or left-handed. The realization of chiral colloidal clusters opens up a new link between colloidal science and chemistry. Very recently, Ji et al. reported the controlled assembly of monodispersed gold nanoparticles into multi-dimensional (one-, two-, and three-dimensional) structures by using ionic and nonionic surfactants.30 A ligand-driven self-assembly strategy, in which charged ligands interact with and partially or fully replace the initial stabilizer molecules, led to assembly of the gold nanoparticles. The optical absorption spectra for differently dimensional assemblies were compared. The monodispersed gold nanoparticles exhibited the well-known localized surface plasmon resonance bands that were shifted depending on particle dimensionality. The shift in the localized surface plasmon resonance band has important connotations for the preparation of waveguides in the visible frequency range and might offer many excellent opportunities for photonics, sensing, and imaging applications.
In another approach, Somorjai and coworkers reported the design of a model catalytic system that consists of a Pt metal core coated with a mesoporous silica shell (Pt@mSiO2), which is stable at elevated temperatures.31 The Pt@mSiO2nanoparticles maintained their core–shell configurations up to 750 °C and exhibited high catalytic activity for ethylenehydrogenation and COoxidation. The chemistry involved in coating mesoporous silica onto nanoparticles' surfaces is straightforward. Li and coworkers reported a simple controlled preparation of hierarchical hollow microspheres and microcubes of MnO2nanosheets through self-assembly with an intermediate crystal-templating process.32 Hierarchical hollow manganese dioxide can be synthesized by a shape-preserving KMnO4oxidation of micrometer-scale manganese carbonate structures at room temperature, followed by the selective removal of MnCO3 with HCl. The resulting MnO2 nanomaterials exhibited activity in the removal of organic pollutants from waste water and are expected to be useful in many other applications. Mandal et al. proposed the novel concept of gold cold fusion for gold nanoparticle assembly and extension of their nanostructures (Fig. 2).33 Using this concept, fabrication of two-dimensional web-like nanonetworks of gold became possible. Molecular assemblies stabilized by hydrogen bonding provided excellent templates leading to highly integrated structures of gold through room-temperature (i.e., cold) fusion. Since gold is a powerful medium for organization of functional organic molecules by the self-assembled monolayer technique, the methodology proposed here should find widespread use for two-dimensional integration of organic functional units, possibly leading to nanonetwork media for brain-like information processing.
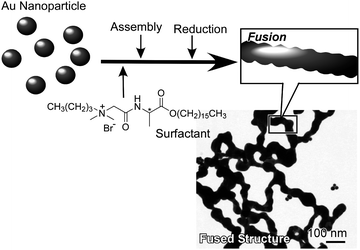 | ||
| Fig. 2 Gold cold fusion process to fabricate two-dimensional network nanostructures from gold nanoparticles. | ||
Acharya et al. successfully synthesized very narrow free quantum rods of 1.7 nm in diameter, which introduced the opportunity to test a variety of properties in the strong confinement regime.34 The strong confinement effect can be found in absorption, photoluminescence, and Raman spectroscopy. Because these rods fluoresce strongly and display discrete narrow optical spectra with robust stability, the nanorods should be of great use for biological labeling, in fluorescence resonance energy transfer, and in optoelectronics applications. In addition, the nanorods formed vortex patterns consisting of micron-sized domains originating from a center of declination on a variety of solid substrates upon spin casting. This unique pattern formation probably is a result of a delicate balance of the radially outward centrifugal force, frictional forces acting at the interface of the surfactant-coated nanorods and the substrates, and rod dipole-surface charge interaction. Li, Cao, and coworkers synthesized cylindrical supercystalline superparticle structures from CdSe/CdS nanorods.35 Anisotropic interparticle interactions between nanorods, as well as solvophobic interactions between the superparticles and the surrounding solvent, play major roles in controlling the shape of the superparticles. According to their sizes (or volumes), the structures obtained exhibit linearly polarized emission, and so these materials might be useful as functional components in devices such as polarized light-emitting diodes and electrooptical modulators. Liu, Stoyanov, and coworkers reported the production of superstable foams with a bimodal size distribution stabilized by modified CaCO3 rods.36 Because of their superstability and stiffness, bubbles stabilized by CaCO3 rods are packed into two-dimensional binary colloidal crystals, which show pronounced effects of bimodality. Kong and coworkers proposed preparation of self-assembled, free-standing paper-like structures of cryptomelane-type manganese oxide nanowires from a self-assembly process.37 The membrane was composed of three-dimensional porous nanostructures, exhibited a superhydrophilic character but became superhydrophobic upon casting with a thin layer of hydrophobic molecules. These two extreme wetting characteristics were completely switchable upon coating with or removal of the hydrophobic molecules at elevated temperatures.
(iii) Hybrids and composites
Spontaneous assemblies from mixtures of organic and inorganic components sometimes provide unique hybrid objects with nanoscale structural characteristics which differ from their one component counterparts. Using hierarchic construction, broader functionality can be introduced by incorporating hard inorganic nanomaterials into soft organic/biological systems. DNA is among the most commonly used biological templates for assembly of nanoparticles. Sharma, Chhabra, and coworkers investigated control of DNA tubes through integration of gold nanoparticles.38DNA tubes are known to form through either self-association of multi-helix DNA bundle structures or closing up of two-dimensional DNA tile lattices. By the attachment of single-stranded DNA to gold nanoparticles, nanotubes of various three-dimensional architectures could form, ranging in shape from stacked rings to single spirals, double spirals, and nested spirals. Chemical appendages sometimes assist the self-assembly of the main components into unique structures. This kind of assembly can be termed ‘guided-assembly’. Several examples are available in the spontaneous assemblies of DNA-conjugated nanomaterials. Maye et al. proposed a novel high-throughput method for designing and fabricating clusters using DNA-encoded nanoparticles assembled on a solid support in a stepwise manner.39 This method efficiently generates remarkably high yields of well-defined dimer clusters and Janus (two-faced) nanoparticles. Willner and coworkers developed a unique DNA scaffold for the one-step self-assembly of hierarchical nanostructures onto which multiple proteins or nanoparticles are positioned on a single template with precise relative spatial orientation.40 The architecture is a topologically complex ladder-shaped polycatenane in which the “rungs” of the ladder are used to bring together the individual rings of the mechanically interlocked structure, and the “rails” are available for hierarchical assembly. The effectiveness of this approach was demonstrated using proteins, complementary DNA, and gold nanoparticles. This DNA template strategy can lead to multiple nanostructures with precise spatial control that could be useful in catalysis, biosensing, and nanomaterials design.There also exist several non-DNA-based approaches. For example, Kumar and coworkers demonstrate that spherical nanoparticles uniformly grafted with macromolecules (‘nanoparticle amphiphiles’) self-assemble robustly into a variety of anisotropic superstructures when they are dispersed in the corresponding homopolymer matrix.41 Their approach to nanoparticleself-assembly enables considerable control for the creation of polymer nanocomposites with enhanced mechanical properties. Chang, Xu and coworkers studied TiO2 nanoleaves as the building blocks to form self-assembled nanostructuresvia metal ligand interaction.42 The TiO2 nanoleaves were modified with a Zn(II) porphyrin derivative through a general ligand exchange method which used trans-2,2′-ethylene-4,4′-bipyridyl to interconnect the zinc metal centers of the porphyrin moiety on the [101] face of the TiO2 nanoleaves.
Shimizu and coworkers prepared antimicrobialnanotubes consisting of Ag-embedded peptidic lipid bilayer membranes.43 The synthetic peptidic lipid, 2-(2-tetradecanamidoacetamido)acetic acid can self-assemble into hollow cylindrical structures. Ag ions coordinate to the negatively charged surface carboxylate groups and the resultant chiral coordination motif eventually causes the nanosheets to twist or coil, leading to Ag+-complexed lipidnanotubes. Upon exposure to UV radiation, the Ag cations are reduced to Ag atoms, which form Ag nanoparticles. The obtained hybrids possess antimicrobial activity against E. coli, contributing to the release of Ag ions into the aqueous culture. Acharya et al. demonstrated that dispersion of ultranarrow ZnS nanorods encapsulated by a fluid-like soft organic layer in the nematic liquid crystal ZLI-4792 results in a novel soft-matter-type blend.44 Local ordering affects significantly the global ordering of the blend, allowing a more rapid response of the electro-optic properties (Fig. 3). An external field affects the orientational order of the blend within which the orthogonal state of polarization persists, allowing the electro-optic properties to be tailored between vertical and grazing view angles. This approach of blending ultrasmall ZnS nanorods with nematic ZLI-4792 LC could be used to design blends consisting of other elongated nanomaterials and different types of liquid crystals, and could also be used as the basic component of displays of enhanced contrast and low driving voltage operation.
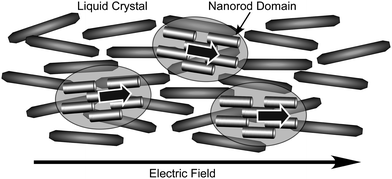 | ||
| Fig. 3 Control of liquid crystal orientation by external electric fields with the aid of blended nanorods. | ||
Inorganic components are very useful for formation of rigid framework structures, while organic units play important roles in flexibly connecting framework components yielding varieties of nano-organized structures. Typical examples can be seen in rigid porous frameworks constructed through a coordination-type self-assembly process. Some of these are known as metal organic frameworks (MOFs) and/or coordination polymers.45 Appropriate selection of organic ligands can lead to regular porous structures with various sizes and geometries. Yaghi and coworkers extensively developed MOF families using oxo-bridged coordination complexes and organic ligands.46 Recently, they developed novel framework materials, ZIF (zeolitic imidazolate frameworks), that are assembled from metal ions such as zinc and cobalt ions and imidazole-type ligands.47 Porous ZIFs have huge surface areas exhibiting selective adsorption of CO2 from a CO/CO2 mixed flow under particular conditions. Adsorption quantities of CO2 gas could be as high as ca. 83 L per 1 L ZIF. Such materials have great potential in ameliorating some of the causes of global warming.
Transcription of organic assemblies into inorganic structures is also a powerful method for fabricating inorganic materials containing nanoscale structural features and can be used for synthesis of mesoporous materials including mesoporous silica,48 mesoporous carbon,49 amongst others50 (Fig. 4). A similar strategy has been used for structure transcription from gel fibers to inorganic substances such as nanotubes51 (Fig. 4). The concept of structure transcription has also been developed. Bein and coworkers proposed the “brick and mortar” approach for formation of mesoporous crystalline materials.52 Their approach is based on the fusion of preformed titania nanocrystalline “bricks” with surfactant-templated sol -gel titania “mortar”, which acts as a structure-directing matrix and as a chemical glue. The “mortar” was added to colloidal solution of “bricks” at ratios ranging from 0 to 100 wt% for the preparation of the nanocomposite films. The dip-coated titania films were calcined in order to induce further crystal growth and to remove the copolymer used for pore formation. Introduction of crystalline titaniananoparticles into surfactant-templated titania precursor sols has a dramatic influence on the porous structure and crystallinity of the calcined films. Brezesinski et al. reported the synthesis and characterization of polymer-templated sol -gel and nanocrystal-based anatase TiO2thin films .53 Both materials have a highly crystalline pore-solid architecture and a large surface area, resulting in considerable enhancement of the electrochemical properties. The materials obtained are expected to show high levels of capacitive charge storage and high insertion capacities. Jin, Raines and coworkers demonstrated that synthetic peptides related to collagen retain their ability for self-assembly into micrometer-long fibers.54 Modification with colloidal gold nanoparticles led to gold-decorated collagen-like fibers that could be further processed into metal nanowires by metallization through electroless silver plating under mild conditions. Wiesner and coworkers presented results from the self-assembly of block copolymers with ligand-stabilized platinum nanoparticles, leading to lamellar and inverse hexagonal hybrid mesostructures with high nanoparticle loadings.55Pyrolysis of the latter hybrid produced an ordered mesoporous platinum-carbon nanocomposite with open and large pores (larger than 10 nm). Removal of the carbon leads to ordered porous platinum mesostructures. The platinum-carbon nanocomposite has very high electrical conductivity (400 siemens per cm) for an ordered mesoporous material fabricated from block copolymerself-assembly. Iyoda and coworkers demonstrated preparation of a highly ordered array of mesoporous silica nanorods with tunable aspect ratios, through an integrated strategy of block copolymerlithography and evaporation-induced self-assembly.56Block copolymerthin films with normal, cylindrical domains were used as external scaffolds to define the morphology of SiO2nanorods. Internal mesoporous structures rely on structure-directing by surfactant cetyltrimethylammonium bromide. Highly ordered tunable SiO2nanorod arrays have tremendous potential for sensory, optoelectronic, and on-chip separation applications. Kyotani, Akagi and coworkers synthesized helical carbon and graphitic films from helical polyacetylene film, where iodine doping is quite effective in increasing carbon yield and in preserving both the helical nanofibril structure and the spiral morphology of the helical polyacetylene films.57 The carbon film obtained by carbonization at 800 °C can be graphitized by heat treatment at 2600 °C while retaining their original nanofibril-fabricated structures.
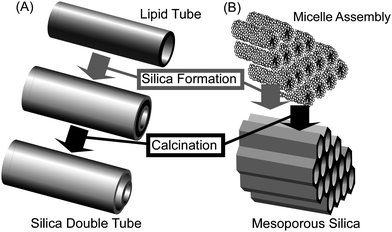 | ||
| Fig. 4 Example of structural transcription: (A) formation of silica nanotube; (B) preparation of mesoporous silica. | ||
As descibed above, coordination assembly is one of the most powerful methods to provide nanoscaled porous materials. Recently, Kitagawa and coworkers report the porous coordination polymer, {[Cd(bpndc)(bpy)]}n (bpndc = benzophenone-4,4′-dicarboxylate, bpy = 4,4′-bipyridyl), which possessed a kinetic gate-opening function.58 The formation of an intermediate, which can be described as a gate-opening process, governs the gate-opening pressure and enhances the difference between gases. The gate-opening process could be associated with the condensation of adsorbate on a crystal surface. Modification of a crystal surface could provide new mechanisms for adsorption phenomena. The same research team reported selective (stepwise) sorption properties of two isomorphous frameworks, synthesized from a tripodal symmetrical ligand with highly flexible arms and secondary functional groups.59 This assembled material shows sponge-like dynamic behavior, directly visualized by single crystal to single crystal transformations, accompanied with size- and affinity-based selective sorption. Mirkin and coworkers developed a new class of micrometer-sized amorphous infinite coordination polymer particles from the coordination chemistry of a metallo-salen building block and Zn ions.60 The spherical infinite coordination polymer particles were stable up to more than 300 °C under high vacuum and showed moderately high H2 uptake with almost no N2 adsorption.
Spontaneous assembly between organic and inorganic components provides various types of supramolecular structures. Lee et al. synthesized organic-inorganic hybrid rotaxanes with inorganic and organic structural units linked together mechanically at the molecular level.61 Structural units (dialkylammonium groups) in dumb-bell-shaped organic molecules templated the assembly of essentially inorganic heterometallic (Co(II), Cr(III) and/or Cu(II)) rings about organic ammonium axles to form rotaxanes consisting of various numbers of rings and axles. One of the rotaxanes behaved as a molecular shuttle where the ring moves between two binding sites on the axle in a large-amplitude motion typical of some synthetic molecular machine systems. Tsuda, Aida, and coworkers demonstrated the formation of an inorganic/organic polypseudorotaxane by the template-assisted cofacial assembly of a ring-shaped molybdenum cluster with a rigid-rod molecule having a high affinity toward the molybdenum cluster surface.62 Tanaka et al. successfully aligned Cu and Hg ions within an artificial duplex structure using different metal base pairs.63 By varying the number and the sequence of the various metal-complexing units it was possible to control the number and the position of the metal ions inside the duplex. This programmed assembly allows one to increase significantly the complexity and diversity of potential metal arrays under precise control of the sequence and the distance between the individual ions. Müller and coworkers synthesized hybrid nanowires with a silsesquioxane core and a shell made up from oligo(ethyleneglycol) methacrylate units.64 The nanowires obtained are soluble in water and many organic solvents and show lyotropic liquid-crystalline structures that could be pyrolysed to silica nanowires. Their approach provides a route to the controlled fabrication of inorganic or hybrid silica nanostructures by living polymerization techniques.
By medium: medium-induced assembly, especially at interfaces
Materials' assemblies at surfaces and interfaces often show different characteristics from those in bulk media. Often highly aligned structures can be obtained based on surface morphology (Fig. 5). Therefore, these assemblies can be recognized as interface-induced assembly in which the interface directs the materials' assemblies. In addition, self-assembly processes at the surfaces are important for surface coating that is important in various applications including anti-corrosion, sensing device preparation, and biomedical applications.65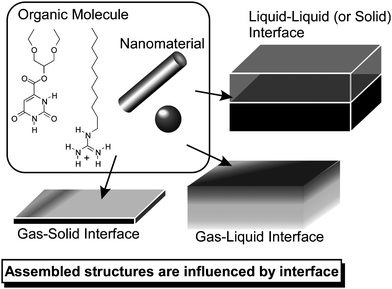 | ||
| Fig. 5 Formation of self-assembled structures at various interfaces. | ||
(i) Solid interfaces
Interface-induced assembly is effective for alignment of bulk-materials. Sibener and coworkers proposed a new methodology for aligning nanoscale cylindrical diblock copolymer (polystyrene-block-poly(ethylene-alt-propylene)) domains over macroscopic length scales.66 Alignment was initiated by a wetting layer of polystyrene domains on the vertical sidewalls of lithographic channels and extended throughout, and even beyond, the confined volume by coarsening of these edge-aligned domains. Cylinders guided to align in this manner could accommodate lithographic imperfections without introducing structural defects. Ryu and Park reported a solid-phase growth of vertically aligned peptidenanowires having a high aspect ratio (at least 100) and a long persistence length (more than 10 mm) by a high temperature aniline vapor aging process, starting from an amorphous peptidethin film .67 Fabrication of a micro-pattern of the peptidenanowires became possible by combining a simple soft-lithographic technique and the high-temperature aniline vapor-aging process. Sofos, Goldberger and coworkers prepared nanoscale lamellar photoconductive hybrids.68 The obtained hybrid electronically active materials consisting of nanoscale alternating layers of inorganic and organic components were attained in essentially one step by integrating molecular self-assembly and electrodeposition. The presence of conjugated moieties in the organic not only added an optoelectronic component to the hybrid, but also synergistically stabilized the lamellar architecture on thermal conversion of the insulator Zn(OH)2 to the semiconductor ZnO. Their method offers a promising approach towards the synthesis and device integration of previously inaccessible bifunctional p-type/n-type hybrid materials with lamellar architecture.Zhou and coworkers reported fabrication of a perpendicular nanopin film fabricated using a bottom-up process through chemical bath deposition.69 Solutions for the chemical bath deposition process were prepared by dissolving CoCl2·6H2O and NH2CO. Following the deposition of brucite-type cobalt hydroxide on a glass substrate, the films were coated with lauric acid. The resulting film showed superhydrophobicity with a water contact angle of 178°. Wu and Coffer investigated gold-induced self-assembly of germanium square micropatterns through annealing of Au-coated n-type Ge (100) wafer.70 As a demonstration of their utility in possible patterning of biomaterials, the Ge micropatterns were used as templates to fabricate 1D calcium carbonatenanowires by an electrochemical deposition method using simulated body fluid or aqueous solutions of CaCl2·2H2O and NaHCO3 as electrolytes. Jin and coworkers demonstrated a strategy for the assembly of large area arrays of single semiconductor nanocrystals directly onto a polymer surface from low-temperature aqueous solutions using self-assembled block co-polymer, poly(styrene)-block-poly(acrylic acid), nanostructured templates.71 The hydrophilic –COOH surface groups of the poly(acrylic acid) domains interact favorably with the metal ions creating localized regions of higher metal-ion concentration, causing preferential heterogeneous nucleation at these sites.
Atomically flat interfaces are an excellent medium for alignment of functional molecules such as that seen in highly organized molecular patters of porphyrins72 and fullerenes73 (Fig. 6). Kumpf and coworkers reported on two molecular adsorbate systems of Sn- and Cu-phthalocyanine on Ag(111) which exhibit fundamentally different behaviors. On varying coverage, the structure of the molecular adsorbate layer changes continuously, and no discrete phase transitions occur.74 This is caused by a dominant substrate-mediated repulsive intermolecular interaction between the molecules (exceeding van der Waals and other attractive forces). Despite the long-range order in so-called “point-on-line” structures, there is still an effective intermolecular repulsion that minimizes the local molecular density in the layer. Puigmartí-Luis et al. prepared supramolecular wires using organic molecules which are self-assembled at a surface under ambient conditions in a liquid.75Tetrathiafulvalene (TTF) units stack flat on the graphite surface in the absence of strong intermolecular interactions. Introduction of amide functional groups in the structure generated hydrogen bonded chains to modify the molecular orientation and create one-dimensional assemblies. Lei et al. investigated the assembly of a TTF derivative, with only one side-chain containing an amido group that adsorbs in a head-to-head and tail-to-tail manner.76 With the aid of intermolecular hydrogen bonds, the TTF moieties were also tilted at a large angle from the surface forming π-stacks. Polymorphous multilayer adsorption is observed and high density cross junctions of the supramolecular fibres are formed reproducibly. Lehn, Samor and coworkers presented STM visualization at the liquid-solid interface at room temperature of the formation of supramolecular hydrogen-bonded polymers with either a linear or a zigzag geometry on highly oriented pyrolytic graphite (HOPG) surfaces.77 The ditopic molecular components bearing complementary hydrogen-bonding recognition groups, either a Janus-type cyanuric wedge or a barbituric wedge, and a corresponding receptor unit were used for formation of a two-dimensional network. Gambardella et al. showed that the supramolecular assembly of Fe and 1,4-benzenedicarboxylic acid molecules on a Cu surface results in ordered arrays of high-spin mononuclear Fe centres on a grid.78 Lateral coordination with the molecular ligands yields unsaturated yet stable coordination bonds, which enable chemical modification of the electronic and magnetic properties of the Fe atoms independently from the substrate. The easy magnetization direction of the Fe centres can be switched by oxygen adsorption, thus opening a way to control the magnetic anisotropy in supramolecular layers akin to that used in metallic thin films .
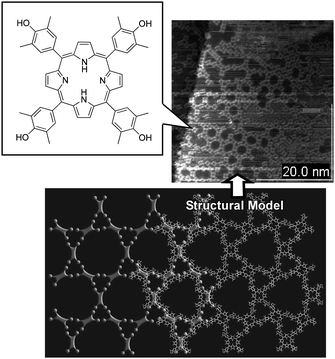 | ||
| Fig. 6 Hydrogen bonded array of porphyrin derivative molecules as an example of molecular assembly on a solid surface. | ||
Regular assemblies from two or more components at solid interfaces have also been investigated. Zimmt and coworkers demonstrated that weak interactions, designed to introduce recognition and selectivity, can be combined with molecular shape (chain length) to direct self-assembly of patterned cocrystal monolayers.79 The formation of two distinct, molecular structure-specific morphologies demonstrates that recognition and selection based on the (12-methoxydodecyloxymethyl)/(2-(10-ethoxydecyloxy)ethyl) side-chain pair can be used to design patterned monolayer cocrystals with feature sizes as small as one molecule and unit cells that extend over many molecules. Yoshimoto, Itaya and coworkers proposed a guide for designing two-dimensional alternate arrays consisting of bicomponents and for producing patterns of fullerene nanodots on surface-supported supramolecular nanostructures from the solution phase.80 The formation of a uniquely nanostructured, bimolecular chessboard consisting of zinc phthalocyanine and zinc octaethylporphyrin molecules and a nanohexagon consisting of zinc tetraphenylporphyrin and zinc octaethylporphyrin molecules were demonstrated. The supramolecular assembly of C60 on the bimolecular chessboard on Au(111) yielded a regularly arranged fullerene nanodot pattern. The results obtained provide a path toward the epitaxial growth of crystals on surface-supported supramolecular nanostructures for creation of a hybrid bilayer of supramolecularly assembled fullerenes. Wee and coworkers presented a new strategy for fabrication of surface nanotemplates featuring well-ordered nanocavity arrays composed of C60 molecules, referred to as a C60 nanomesh, which was formed by precisely adjusting the binary molecular phases of C60 and pentacene on Ag(111).81 This C60 nanomesh can serve as an effective template to selectively accommodate guest C60 molecules at the nanocavities, thereby leading to the formation of an ordered two-dimensional C60 array with a large intermolecular distance. de Oteyza et al. investigated mixtures of diindenoperylene and fluorinated copper-phthalocyanines, two molecules used as donor and acceptor semiconductors.82 The growth of binary layers resulted in films with strongly enhanced intermolecular interactions and consequently reduced molecule-substrate interactions. This new insight into the interplay among the aforementioned interactions provides a novel strategy for balancing the critical interactions in the assembly processes by the appropriate choice of molecular species in binary supramolecular assemblies, and thereby controls the self-assembly of functional organic nanostructures. Madueno et al. showed that the two strategies can indeed be combined to create integrated hybrids between network from 2,4,6-triamino-1,3,5-triazine and perylene-3,4,9,10-tetracarboxylic diimide and self-assembled monolayer (SAM) systems that are sufficiently robust for further processing.83 The honeycomb network involving three hydrogen bonds per synthon possessed sufficient stability to act as a template in subsequent processes. The combination of the network with SAMs offers considerable design flexibility, with the network providing an exact definition of structures in the substrate plane and the SAM permitting separate surface modification.
(ii) Liquid interfaces
Unlike solid interfaces, liquid interfaces provide dynamic media for molecular assemblies where dynamic control of orientation and alignment are highly expected. The most studied system is the air-water interface which has long been recognized as an excellent medium for the assembly of inorganic ions, biomacromolecules and inorganic nanoparticles. The well-known Langmuir–Blodgett (LB) technique has been used to prepare two-dimensional molecular assemblies at an air-water interface. In particular, the air-water interface can promote molecular interactions between water-insoluble membrane components and water soluble substances,84 resulting in regular patterns of organic molecules.85 In addition, molecules confined at the air-water interface must experience modulation of their conformational changes sometimes leading to unique arrangements of molecules. Zhang et al. reported a controllable fabrication of supramolecular nanocoils and straight nanoribbons of an anthracene derivative, N-(2-(2-(9-anthrylmethylene)hydrazino)-2-oxoethyl)dodecanamide, and their morphologically dependent photoswitching.86 When the Langmuir film of N-(2-(2-(9-anthrylmethylene)hydrazino)-2-oxoethyl)dodecanamide at the air-water interface is compressed to different surface pressures, nanostructures with different morphologies can be obtained in the deposited LB films. Nanocoils formed at a lower surface pressure, whereas the coiled structures were transformed to straight nanoribbons when the surface pressure was increased. While the nanocoils did not show any photocurrent response, the nanoribbons exhibited a switchable photocurrent. Stupp and coworkers reported the self-assembly of macroscopic sacs and membranes at the interface between two aqueous solutions, one containing a megadalton polymer (polysaccharidehyaluronic acid) and the other, small self-assembling molecules (peptide amphiphile) bearing opposite charge.87The air-water interface provides the opportunity to assemble and align inorganic substances such as nanoparticles, nanocrystals, nanowires, and nanosheets.88 Rogach, Chi and coworkers presented a patterning technique that allows the alignment of luminescent CdSe and core/shell CdSe/ZnS nanoscrystals into large area periodic lateral structures.89 Selective deposition of nanocrystals onto organic channel-like templates fabricated by the LB technique was achieved by anisotropic dewetting of the nanocrystal carrier solvent (1-phenyloctane). The filling fraction of nanocrystals within the channels was controlled by their concentration in solution and the deposition time. Yang and coworkers demonstrated the bottom-up fabrication of large-area, two-dimensional plasmonic materials composed of polyhedral Ag nanocrystals, where plasmon resonances can be explicitly tuned by means of nanocrystal shape and arrangement.90 In the LB technique, a colloidal solution of nanocrystals is spread at an air-water interface, forming an isotropic monolayer of floating nanocrystals whose density can be controlled by surface pressure through isothermal compression or expansion. The capability of controlling interparticle spacing, density and packing symmetry allows for tunability of the optical response over the entire visible range. This assembly strategy offers a new, practical approach to making novel plasmonic materials for application in spectroscopic sensors, subwavelength optics and integrated devices that utilize field-enhancement effects.
Lieber and coworkers reported a solution-based approach for hierarchically organizing nanowire building blocks en masse into integrated arrays tiled over large areas.91Nanowires were aligned with controlled nanometer to micrometer scale pitch using the LB technique, transferred to planar substrates in a layer-by-layer process to form parallel and crossed nanowire structures over centimeter length scales, and then efficiently patterned into repeating arrays of controlled dimensions and pitch using photolithography. Further efforts focused on increasing the structural complexity provided combinations of logic and memory arrays which would be necessary for nanocomputing or even integrated sensing and processing functions. Li and Wu investigated the coassembly behavior of graphene oxide nanosheets and Na0.44MnO2nanowires.92 Upon addition of graphene oxide nanosheets to an aqueous nanowire suspension, concentration enrichment and orientation alignment of nanowires at the air-water interface was observed. At some critical surface concentration, nanowire self-alignment occurred. Their method was extended to alignment of other oxidenanowires such as sodium titanate and TiO2nanowires. Yang and coworkers proposed a facile self-assembly approach to prepare macroscopic graphene oxide membranes at a liquid/air interface by evaporating a hydrosol of graphene oxide.93 This method is easy to scale up, and the resulting free-standing membranes showed excellent mechanical and optical properties. Studart and coworkers showed that layered hybrid films combining high tensile strength and ductile behaviors could be obtained through bottom-up colloidal assembly of strong submicrometer-thick ceramic platelets within a ductile polymer matrix.94 In the first step two-dimensional assemblies of colloidal platelets were prepared at the air-water interface. Adsorption of platelets at the air-water interface was favored through the attachment of slightly hydrophobic amine-terminated silane species on the platelet surface. Then, the two-dimensional assembled platelets were transferred to a glass substrate by dip-coating and subsequently spin-coated with an organic layer of chitosan solution. Repetition of these steps in a sequential manner leads to multi-layered inorganic-organic films. Free-standing high-strength films could be obtained by peeling off from the substrate with a razor blade.
The LB technique is versatile in the sense that it can be extended to the preparation of hybrid assemblies.95 Melosh and coworkers developed a hybrid lipid/inorganic controlled-release system by creating spanning lipid bilayers to seal chip-based chemical reservoirs.96 It is possible to produce a system with high spatial and temporal resolution, which also provides zero leakage in the “off” state, protein adsorption resistance, no toxic byproducts, and a simple external optical trigger. Gleiche et al. presented a fast and simple method to generate extended patterned surfaces with controlled wetting properties on the nanometre scale, without using any lithographic processes.97 The technique utilizes wetting instabilities that occur when monomolecular layers of phospholipids are transferred onto a solid substrate by the LB method. Modified surfaces were used as templates for patterning a wide variety of molecules and aggregates, including dyes and gold nanoclusters, into approximately parallel channels, with a spatial density of up to 20000 cm−1.
In addition to the above-mentioned examples, unusual assemblies can be obtained by using a liquid-liquid interface. For example, Xia and Wang demonstrated a new and facile way of creating macroscopic, dried, freestanding films of close-packed metallic nanoparticle monolayers by means of interfacial self-assembly.98 The gold nanoparticles were found to self-assemble spontaneously into a monolayer film at the pentanol/water interface, a process which was accelerated by adding a small amount of ethanol or gently shaking. After 3 h heating, these islands eventually merged into a large film on the aqueous phase. After drying in ambient conditions for 1 day, the resulting films were fished from the surface of the aqueous phase using copper wire loops; they exhibited a strong golden luster. Sathish et al. demonstrated controlled formation of two-dimensional objects such as hexagons and rhombi through self-assembly and their selective shape-shifting into one-dimensional rods through solvent-dependent changes of crystal lattice, all from pure C60 using a liquid-liquid interface (Fig. 7).99 Uniformly shaped rhombi and hexagons were obtained at t-butyl alcohol/toluene and i-propyl alcohol/CCl4 interfaces, respectively. Nanorhombi were converted into short nanorods upon exposure to water.
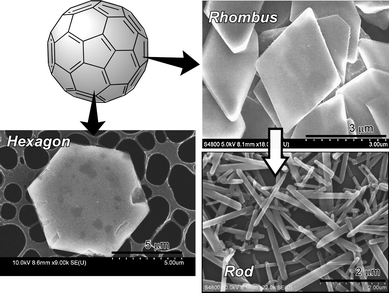 | ||
| Fig. 7 Spontaneous formation of various nanoscale objects through spontaneous assemblies of pure fullerenes. | ||
By human: directed assembly
To fabricate required structures with nanoscale precision, combining self-assembly events with artificial processes has to be considered. Although some examples shown above and recent advanced examples by the Minko,100 Kumacheva,101 and Ulijn102 groups indicated the importance of assemblies guided by molecular components, the contribution by human operation to control of assembled structures cannot be ignored. In the latter methodologies self-assembly is directed by human actions. Therefore, these processes can be regarded as directed assembly. Fabrication techniques defined as bottom-up processes, including various lithographic techniques, microcontact printing (µCP),103 and dip-pen nanolithography (DPN),104 can be applied to self-assembled materials (Fig. 8). Combination of self-assembly phenomena and human action in a series of fabrication processes could be used to create the requisite self-assembled structures. Modulation of self-assembly processes by using external stimuli should be undoubtedly useful for obtaining the desired structures.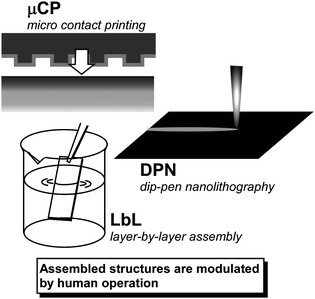 | ||
| Fig. 8 Examples of artificial fabrication methods for modulation of self-assembled structures. | ||
(i) Advanced lithographic techniques
Javey and coworkers reported wafer-scale assembly of highly ordered semiconductor nanowire arrays by contact printing.105 A generic approach was developed to directly transfer regular arrays of semiconductor nanowires from donor to patterned receiver substrates. The transferred nanowires are highly ordered and aligned, enabling the assembly of both single nanowires and densely packed parallel arrays of nanowires with high uniformity across an entire wafer. Wu, Chi and coworkers reported that high-quality CdTe nanoparticles stabilized with thioglycolic acid could be patterned on SiO2/Si surfaces using µCP techniques.106 Either edge printing or homogeneous printing was achieved under optimized conditions. Yan and coworkers reported the fabrication of well-organized arrays of self-assembled functional DNAnanotubes at the sub-millimeter scale by combining the bottom-up and top-down methods.107 DNA-nanotube arrays can be used to efficiently direct the assembly of arrays of quantum dots, proteins, and DNA targets. DNAnanotubes are first self-assembled by a simple annealing process from a pool of single-stranded DNA with rationally designed sequences. Using a surface-patterning technique, including polydimethylsiloxane (PDMS) patterned with micrometer-sized features by soft lithography, the DNAnanotubes were aligned into arrays with high periodicity by taking advantage of the molecular-combing effect of a directional flow. The self-assembled DNAnanotubes display combined stiffness and flexibility allowing them to be easily stretched, aligned, and printed while maintaining their morphological integrity.In another approach, a new tip-based soft lithography technique was created by combining self-assembled monolayer chemistry with atomic force microscopic (AFM) technology. This technology is known as dip-pen nanolithography (DPN). Nanoscale features can be written on metal and semiconductor surfaces using a solution of monolayer-forming materials as ink. Similarly, Huo et al. reported the development of “polymer pen lithography”, a low-cost, cantilever-free lithographic approach that, thus far, allows a digitized pattern to be printed at spot sizes ranging from 90 nm to hundreds of mm simply by changing the force and time over which the ink is delivered.108Polymer pen lithography merges the feature size control of the DPN technique with the large-area capability of contact printing. Because ink delivery is time and force dependent, features on the nanometer, micrometer, and macroscopic length scales can be formed with the same tip array. Arrays with as many as about 11 million pyramid-shaped pens can be brought into contact with substrates and readily leveled optically to ensure uniform pattern development.
Pisignano and coworkers demonstrated the nanopatterning of nanocomposites composed of luminescent zinc oxide nanoparticles and light emitting conjugated polymers by means of soft moldinglithography.109 Once an elastomeric mould is put in contact with the surface of the nanocomposite film, the whole system is heated above the softening temperature of the polymer matrix and the composite starts to wet the lateral walls of the recessed features in the mould up to a filled height depending on the heating time. Afterwards, the system is cooled below the matrix glass transition temperature, and the mould is detached from the surface, which is textured with the same pattern as the starting master template. The results presented opened up new possibilities for the realization of nanopatterned devices based on hybrid organic-inorganic systems. Texturing composites with high (sub-500 nm) spatial resolution will enable tailoring of the device emission resulting from donor–acceptor energy transfer by lithographic filtering of the nanoparticles able to penetrate in the mould template by vertical nanofluidics. Mirkin and coworkers demonstrate how on-wire-lithography-fabricated nanogaps can serve as a new testbed to construct molecular transport junctions through the assembly of thiolated molecular wires across a gap formed between two Au electrodes.110 One can use oil-write lithography to easily characterize a molecular transport junction system and optimize gap size for two molecular wires of different dimensions. Bhiladvala, Mayer and coworkers demonstrated a bottom-up assembly method for integrating nanowires synthesized and chemically functionalized off-chip into arrays of nanoelectromechanical system (NEMS) resonators on a Si chip.111 Three mechanisms were combined to achieve high-yield positioning of over 2000 single nanowires in each array: electric-field forces, capillary forces and nanowire lift-off. Their approach promises to expand the range of nanowire materials, geometries and surface chemistries that can be integrated on a chip, and to enable multiplexed arrays of individually addressable nanowire devices. Frank and coworkers reported a general synthetic strategy for spatially controlling the growth of p-type semiconductors in the nanopores of electrically conducting n-type materials.112 The facile electrochemical deposition of p-CuInSe2 in nanoporous anatase n-TiO2 oriented nanotube arrays and nanoparticle films. p-Type semiconductors can be deposited from the bottom-up by controlling the ambipolar diffusion length, resulting in complete pore filling.
Recently, Bao and coworkers reported the fabrication of single-walled carbon nanotube (SWNT) network field-effect transistors, deposited from solution, possessing controllable topology and an on/off ratio as high as 900000.113 The spin-assisted alignment and density of the SWNTs were tuned by using different surfaces that effectively vary the degree of interaction with surface functionalities in the device channel. Self-sorted SWNT networks in which nanotube chirality separation and simultaneous control of density and alignment occurred in one step during device fabrication were obtained. Ruiz, Nealey and coworkers directed the assembly of defect-free arrays of isolated block copolymer (poly(styrene-block-methyl methacrylate)) domains at densities up to 1 terabit per square inch on chemically patterned surfaces.114 A brush of hydroxyl-terminated polystyrene was deposited on a SiOx substrate. An e-beam resist layer was applied, and the closest possible match to a hexagonal pattern was written using an electron beam. Samples for block copolymer assembly were then subjected to a brief dose of oxygen plasma to generate a chemical contrast on the substrate. Subsequent removal of the bulk of the resist was followed by spin-coating of a block copolymer film, and annealing of the samples in vacuum. The areas of the surface (arrays of spots) exposed to the oxygen plasma were preferentially wetted by the poly(methyl methacrylate) block, and background areas exhibited slight preference toward the poly(styrene) block. The poly(methyl methacrylate) domains were then selectively removed.
(ii) LbL adsorption
The alternate layer-by-layer (LbL) adsorption technique is based on self-assembly processes at interfaces, but adsorption sequences can be artificially modified according to the desired end product.115 Therefore, self-assembled films with predesigned layer sequences can be fabricated. The procedure for assembly is easy to perform and inexpensive. It can be applied to a wide variety of substances, making possible assembly of various materials including organic polymers,116 biomaterials,117inorganic substances,118 and molecular assemblies.119 In addition, application of the LbL technique to colloidal templates can be used to create microscopic hollow capsule structures.120 Therefore, nanofabrication based on the LbL techniques is now widely developed. Several recent examples are described below.Hammond and coworkers proposed a powerful yet economical technique for developing multiple coatings of different morphologies and functions within a single textile membrane, enabling scientists to engineer the properties of a material from the nanoscopic level in commercially viable quantities.121 In order to demonstrate the conformal coating of individual fibres within a material, parallel-plate electrospinning was used to create flexible non-woven mats of microscale nylon 6,6 fibres from hexafluoroisopropanol solutions. Selecting poly(dimethyldiallylammonium chloride) as the cationic species and amphoteric titanium dioxide nanoparticles as the anionic species, a sprayed deposition can be performed. The myriad potential applications of this technology include self-cleaning fabrics, waterpurification and protein functionalization of scaffolds for tissue engineering but were exemplified in this case by the creation of selectively reactive gas purification membranes. van der Boom and coworkers reported solution-based layer-by-layer formation of palladium-organic assemblies.122 In their study, they compared the effect of the nature of the palladium precursors used (i.e., colloids vs. PdCl2) on multilayer structure and optical properties. The colloid -based system has a UV-vis absorption maximum an order of magnitude stronger than that of the PdCl2-based multilayer. The absorption maximum of the PdCl2-based film exhibits a significant red shift of 23 nm with the addition of 12 layers. Remarkably, the structure and physicochemical properties of the submicron scale PdCl2-based structures are determined by the configuration of the ca. 1.5 nm thick template layer. Lee and coworkers reported a novel method to create and pattern conductive polymer-graphite nanoplatelet multilayered nanocomposite films using the intact pattern transfer method.123 Conductive graphite nanoplatelet films were created on PDMS stamps without the use of oxidation/reduction steps. The transferred films exhibited excellent conductivity for electrostatic dissipation applications. Tsukruk and coworkers reported on the preparation of redox-active nanoscale LbL films with poly(amino acid)-decorated surfaces that serve for nucleation and growth of uniformly distributed gold nanoparticles at ambient conditions.124 Introduction of a poly-L-tyrosine was used as a nucleator and stabilizer for gold particles. The flexible nanoparticle-containing LbL films was easily transferred to any solid substrates without multistep processes of solution growth, purification, and casting.
Recently, Ji et al. reported stimuli-free auto-modulated material release from LbL films of mesoporous silica capsules. Hollow mesoporous silica capsules were assembled by the LbL technique into thin films that exhibited special molecular encapsulation and release capabilities (Fig. 9A).125 Stimuli-free auto-modulated stepwise release of water or drug molecules was achieved through the mesopore channels of robust silica capsule containers embedded in the film. Stepwise release of water was reproducibly observed and originates in the non-equilibrated evaporation rates between water and liquid drug from the mesopore channels to the exterior and the capillary penetration of water from the container interior to the mesopore channels. The same research group prepared LbL films of dual-pore carbon capsules with designable selectivity of gas adsorption (Fig. 9B).126 The fabricated dual-porous carbon capsule films were investigated as a sensor substrate for vapors of different organic solvents. The carbon capsule films have much higher adsorption capacities than conventional electrolyte films and even than non-capsular mesoporous carbon films. The dual-pore carbon capsules have greater affinities for aromatic volatiles over their aliphatic counterparts, probably due to stronger π–π interactions. Impregnation of additional recognition components into the carbon capsules permits further control over adsorption selectivity between aromatic and nonaromatic substances and between acids and bases in the prevailing atmosphere. A layered mesoporous carbon sensor was also developed by the same research group.127 The high sensitivity and selectivity of QCM-based sensors coated with CMK-3 LbL film has been demonstrated to originate from the highly cooperative adsorption of tannic acid in its size-comparable nanospaces. The results described therein improved our understanding of molecular interactions within nanospaces, especially nonspecific interactions in aqueous media, a full exploration of which might clarify important phenomena, including those of biological systems.
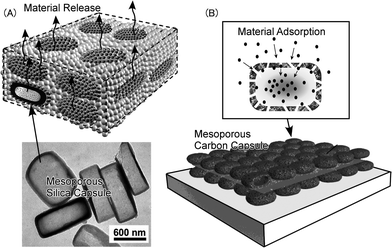 | ||
| Fig. 9 LbL films of mesoporous capsules: (A) LbL film with mesoporous silica capsule for material release; (B) LbL film with mesoporous carbon capsule for material adsorption. | ||
Ito and coworkers fabricated all-solution processed organic solar cells with multilayered structures, the thickness of which was controlled with nanometer precision.128 They employed not only spin-coating but also LbLdeposition. Hole- and electron-transporting layers were prepared by spin-coating with poly(3,4-ethylenedioxythiophene) oxidized with poly(4-styrenesulfonate) and fullerene, respectively. The light-harvesting layer of poly(p-phenylenevinylene) was fabricated by layer-by-layer deposition of the poly(p-phenylenevinylene) precursor cation and poly(sodium 4-styrenesulfonate). Chi and coworkers reported a high-throughput procedure which allows us to generate hierarchical luminescence patterns based on multiscaled self-assembly and specific properties of CdSe nanocrystals and dyes in response to light irradiation.129 The ability to generate hierarchical luminescence patterning with relatively inexpensive instrumentation will facilitate the investigation of other self-assembled systems and should open up ways to fabricate novel advanced functional systems. Whitten, Schanze, and coworkers reported the preparation of novel polyelectrolyte microcapsules consisting of the photoactive charged poly(phenyleneethynylene)-type conjugated polyelectrolytes.130 The microcapsules displayed strong green fluorescence, indicating that the constituent conjugated polymers retain their photophysical properties in the capsule structure. Strong antimicrobial activity was observed upon mixing of polyelectrolyte capsules with Cobetia marina or Pseudomonas aeruginosa followed by white-light irradiation. It was demonstrated that the materials act as highly effective light-activated micro “Roach Motels” with greater than 95% kill after exposure to ∼1 h of white light.
Applying external forces to the self-assembling process can also result in modification to their nanoscale organization. Kato and coworkers designed a fan-shaped molecule with a propylenecarbonate moiety to form nanosegregated columnar liquid-crystalline phases via complexation with lithium triflate.131 The columnar assemblies are macroscopically aligned by applying AC electric fields. This approach to electric-field-responsive columnar liquid crystals that can transport ions anisotropically might be useful for future energy storage or transport devices. Aubry et al. demonstrated micro- and nanoparticleself-assembly for virtually defect-free, adjustable monolayers using an external electric field.132 Capillarity-induced clustering has several limitations: it applies to relatively large particles only, the clustering is usually non-defect-free and lacks long-range order, and the lattice spacing cannot be adjusted. In order to overcome these shortcomings, an external electric field normal to the interface was applied. The lattice spacing dimensions of the resulting self-assembly could be controlled statically or dynamically, forming virtually defect-free monolayers, and was used to manipulate a broad range of particle sizes and types including nanoparticles and electrically neutral particles.
Perspectives
In this review we have described recent examples of fabrication of nano- and micro-objects possessing nanoscale structural features based on self-assembly processes. Self-assembly processes rely essentially on interactions between partner molecules. Therefore, self-assembled structures can be determined by suitable molecular structures and have been attractively termed “molecular programming.” However, molecular programming is only partly effective so that provision of an appropriate environment, such as an interface as well as additional human actions, can modify self-assembled structures. Thus, we can say that the three factors, molecular design, environment, and human intervention, combine to determine the final self-assembled structures.This conclusion should encourage us to bridge functionality between the nanoscale and macroscale size regimes. Function in the nanoscale world can be utilized in bulk processes through appropriate self-assembly and by human intervention. For example, Asaka, Aida and coworkers developed a ‘dry’ actuator composed of single wall carbon nanotubes (SWNTs) and an ionic liquid.133 The bulk actuator was constructed using bucky gel formed by self-assembly between SWNT and an ionic liquid by mechanical grinding (by hand).134 A high-performance dry actuator that shows in air a very large bending motion in rapid response to an applied alternating small square-wave voltage was obtained. Ikeda and coworkers reported bulk rotational motions made by an azobenzene-containing liquid crystalline elastomer and its composite materials.135 Light-driven plastic motors can be fabricated from laminated films composed of a liquid crystalline elastomer film and a flexible polyethylene sheet. Using the opposing concept, Ariga and coworkers recently proposed novel hand-operated molecular machines, in which bulk stimuli such as centimeter-scale compression and expansion of Langmuir films lead to conformational variations and molecular machine activity within the films, permitting reversible molecular capture (Fig. 10).136 This concept can be only achieved through self-assembly of molecular machines at the air-water interface where the macrosized bulk (in the lateral dimension) and nanosized molecules (vertical dimensions) are connected.
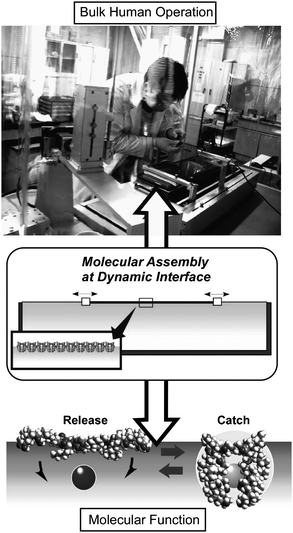 | ||
| Fig. 10 Bridging hand operation and molecular function through dynamic molecular assembly. | ||
To conclude, we propose that materials constructed by appropriate self-assembly processes include nanoscale functionality and properties in their bulk, and such materials can be used to connect nanoscale function operation in the macroscopic regime. It should be one of the best ways to realize and exploit nanoscale science and technology within our everyday lives.
Acknowledgements
The research described in this review was partially supported by World Premier International Research Center (WPI) Initiative on Materials Nanoarchitectonics, MEXT, Japan. XH and SM are grateful for financial support from Japan Society for the Promotion of Science (JSPS).References
- (a) A. T. Bell, Science, 2003, 299, 1688–1691 CrossRef CAS; (b) M. Fujiki, J. R. Koe, K. Terao, T. Sato, A. Teramoto and J. Watanabe, Polym. J., 2003, 35, 297–344 CrossRef CAS; (c) K. Inomata, N. Ikeda, N. Tezuka, R. Goto, S. Sugimoto, M. Wojcik and E. Jedryka, Sci. Technol. Adv. Mater., 2008, 9, 014101 CrossRef; (d) T. Ikeda and J. F. Stoddart, Sci. Technol. Adv. Mater., 2008, 9, 014104 CrossRef; (e) S. Takahashi and S. Maekawa, Sci. Technol. Adv. Mater., 2008, 9, 014105 CrossRef.
- (a) M. Shimomura and T. Sawadaishi, Curr. Opin. Colloid Interface Sci., 2001, 6, 11–16 CrossRef CAS; (b) J. I. Martin, J. Nogues, K. Liu, J. L. Vicent and I. K. Schuller, J. Magn. Magn. Mater., 2003, 256, 449–501 CrossRef CAS; (c) I. W. Hamley, Nanotechnology, 2003, 14, R39–R54 CrossRef CAS; (d) A. del Campo and E. Arzt, Chem. Rev., 2008, 108, 911–945 CrossRef CAS; (e) B. A. Rozenberg and R. Tenne, Prog. Polym. Sci., 2008, 33, 40–112 CrossRef CAS.
- (a) J.-M. Lehn, Angew. Chem., Int. Ed. Engl., 1990, 29, 1304–1319 CrossRef; (b) S. Leininger, B. Olenyuk and P. J. Stang, Chem. Rev., 2000, 100, 853–907 CrossRef CAS; (c) K. Ariga, T. Nakanishi and J. P. Hill, Curr. Opin. Colloid Interface Sci., 2007, 12, 106–120 CrossRef CAS; (d) K. Ariga, J. P. Hill, M. V. Lee, A. Vinu, R. Charvet and S. Acharya, Sci. Technol. Adv. Mater., 2008, 9, 014109 CrossRef.
- (a) A. E. Rowan and R. J. M. Nolte, Angew. Chem., Int. Ed., 1998, 37, 63–68 CrossRef CAS; (b) V. Berl, I. Huc, R. G. Khoury and J.-M. Lehn, Chem.–Eur. J., 2001, 7, 2810–2820 CrossRef CAS.
- S. Yagai, S. Mahesh, Y. Kikkawa, K. Unoike, T. Karatsu, A. Kitamura and A. Ajayaghosh, Angew. Chem., Int. Ed., 2008, 47, 4691–4694 CrossRef CAS.
- (a) K. Katagiri, K. Ariga and J. Kikuchi, Chem. Lett., 1999, 661–662 CrossRef CAS; (b) J. Kikuchi, K. Ariga, T. Miyazaki and K. Ikeda, Chem. Lett., 1999, 253–254 CrossRef CAS; (c) J. Kikuchi, K. Ariga and K. Ikeda, Chem. Commun., 1999, 547–548 RSC; (d) D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2001, 34, 40–48 CrossRef CAS; (e) D. E. Discher and A. Eisenberg, Science, 2002, 297, 967–973 CrossRef CAS; (f) K. Katagiri, M. Hashizume, K. Ariga, T. Terashima and J. Kikuch, Chem.–Eur. J., 2007, 13, 5272–5281 CrossRef CAS; (g) K. Ariga, Q. Ji, J. P. Hill, N. Kawazoe and G. Chen, Expert Opin. Biol. Ther., 2009, 9, 307–320 Search PubMed.
- I. Yoshikawa, J. Sawayama and K. Araki, Angew. Chem., Int. Ed., 2008, 47, 1038–1041 CrossRef CAS.
- J.-K. Kim, E. Lee, Y.-B. Lim and M. Lee, Angew. Chem., Int. Ed., 2008, 47, 4662–4666 CrossRef CAS.
- M. Schappacher and A. Deffieux, Science, 2008, 319, 1512–1515 CrossRef CAS.
- J. A. Hanson, C. B. Chang, S. M. Graves, Z. Li, T. G. Mason and T. J. Deming, Nature, 2008, 455, 85–88 CrossRef CAS.
- T. Gädt, N. S. Ieong, G. Cambridge, M. A. Winnik and I. Manners, Nat. Mater., 2009, 8, 144–150 CrossRef.
- S. Ulrich and J.-M. Lehn, Angew. Chem., Int. Ed., 2008, 47, 2240–2243 CrossRef CAS.
- T. Akutagawa, H. Koshinaka, D. Sato, S. Takeda, S. Noro, H. Takahashi, R. Kumai, Y. Tokura and T. Nakamura, Nat. Mater., 2009, 8, 342–347 CrossRef CAS.
- P. Cordier, F. Tournilhac, C. Soulié-Ziakovic and L. Leibler, Nature, 2008, 451, 977–980 CrossRef CAS.
- S. H. Cho, S. R. White and P. V. Braun, Adv. Mater., 2009, 21, 645–649 CrossRef CAS.
- F. Di Benedetto, E. Mele, A. Camposeo, A. Athanassiou, R. Cingolani and D. Pisignano, Adv. Mater., 2008, 20, 314–318 CrossRef CAS.
- B. Pokroy, S. H. Kang, L. Mahadevan and J. Aizenberg, Science, 2009, 323, 237–240 CrossRef CAS.
- (a) E. Winfree, F. Liu, L. A. Wenzler and N. C. Seeman, Nature, 1998, 394, 539–544 CrossRef CAS; (b) N. C. Seeman, Nature, 2003, 421, 427–431 CrossRef; (c) K. Matsuura, T. Yamashita, Y. Igami and N. Kimizuka, Chem. Commun., 2003, 376–377 RSC.
- P. Yin, H. M. T. Choi1, C. R. Calvert and N. A. Pierce, Nature, 2008, 451, 318–322 CrossRef CAS.
- E. R. Ballister, A. H. Lai, R. N. Zuckermann, Y. Cheng and J. D. Mougous, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 3733–3738 CrossRef CAS.
- (a) R. E. Smalley, Acc. Chem. Res., 1992, 25, 98–105 CrossRef CAS; (b) D. Philp and J. F. Stoddart, Angew. Chem. Int. Ed., 1996, 35, 1155–1196; (c) T. Nakanishi, W. Schmitt, T. Michinobu, D. G. Kurth and K. Ariga, Chem. Commun., 2005, 5982–5984 RSC; (d) T. Michinobu, T. Nakanishi, J. P. Hill, M. Funahashi and K. Ariga, J. Am. Chem. Soc., 2006, 128, 10384–10385 CrossRef CAS.
- (a) T. Nakanishi, K. Ariga, T. Michinobu, K. Yoshida, H. Takahashi, T. Teranishi, H. Möhwald and D. G. Kurth, Small, 2007, 3, 2019–2023 CrossRef CAS; (b) T. Nakanishi, T. Michinobu, K. Yoshida, N. Shirahata, K. Ariga, H. Möhwald and D. G. Kurth, Adv. Mater., 2008, 20, 443–446 CrossRef CAS; (c) Y. Shen, J. Wang, U. Kuhlmann, P. Hildebrandt, K. Ariga, H. Möhwald, D. G. Kurth and T. Nakanishi, Chem.–Eur. J., 2009, 15, 2763–2767 CrossRef CAS.
- S. J. Lee, C. D. Malliakas, M. G. Kanatzidis, J. T. Hupp and S. B. T. Nguyen, Adv. Mater., 2008, 20, 3543–3549 CrossRef CAS.
- (a) C. B. Murray, C. R. Kagan and M. G. Bawendi, Annu. Rev. Mater. Sci., 2000, 30, 545–610 CrossRef CAS; (b) A. N. Shipway, E. Katz and I. Willner, ChemPhysChem, 2000, 1, 18–52 CrossRef CAS; (c) Y. Li, W. Cai and G. Duan, Chem. Mater., 2008, 20, 615–624 CrossRef CAS; (d) Y. Min, M. Akbulut, K. Kristiansen, Y. Golan and J. Israelachvili, Nat. Mater., 2008, 7, 527–538 CrossRef CAS.
- A. R. Tao, D. P. Ceperley, P. Sinsermsuksakul, A. R. Neureuther and P. Yang, Nano Lett., 2008, 8, 4033–4038 CrossRef CAS.
- J. Zhang, A. Kumbhar, J. He, N. C. Das, K. Yang, J.-Q. Wang, H. Wang, K. L. Stokes and J. Fang, J. Am. Chem. Soc., 2008, 130, 15203–15209 CrossRef CAS.
- X. Hu, J. Gong, L. Zhang and J. C. Yu, Adv. Mater., 2008, 20, 4845–4850 CrossRef CAS.
- F. Bai, D. Wang, Z. Huo, W. Chen, L. Liu, X. Liang, C. Chen, X. Wang, Q. Peng and Y. Li, Angew. Chem., Int. Ed., 2007, 46, 6650–6653 CrossRef CAS.
- D. Zerrouki, J. Baudry, D. Pine, P. Chaikin and J. Bibette, Nature, 2008, 455, 380–382 CrossRef CAS.
- Q. Ji, S. Acharya, J. P. Hill, G. J. Richards and K. Ariga, Adv. Mater., 2008, 20, 4027–4032 CrossRef CAS.
- S. H. Joo, J. Y. Park, C.-K. Tsung, Y. Yamada, P. Yang and G. A. Somorjai, Nat. Mater., 2009, 8, 126–131 CrossRef CAS.
- J. Fei, Y. Cui, X. Yan, W. Qi, Y. Yang, K. Wang, Q. He and J. Li, Adv. Mater., 2008, 20, 452–456 CrossRef.
- S. Mandal, A. Shundo, S. Acharya, J. P. Hill, Q. Ji and K. Ariga, Chem. Asian J., 2009, 4, 1055–1058 CrossRef CAS.
- S. Acharya, U. K. Gautam, T. Sasaki, Y. Bando, Y. Golan and K. Ariga, J. Am. Chem. Soc., 2008, 130, 4594–4595 CrossRef CAS.
- J. Zhuang, A. D. Shaller, J. Lynch, H. Wu, O. Chen, A. D. Q. Li and Y. C. Cao, J. Am. Chem. Soc., 2009, 131, 6084–6085 CrossRef CAS.
- W. Zhou, J. Cao, W. Liu and S. Stoyanov, Angew. Chem., Int. Ed., 2009, 48, 378–381 CrossRef CAS.
- J. Yuan, X. Liu, O. Akbulut, J. Hu, S. L. Suib, J. Kong and F. Stellacci, Nat. Nanotechnol., 2008, 3, 332–336 CrossRef CAS.
- J. Sharma, R. Chhabra, A. Cheng, J. Brownell, Y. Liu and H. Yan, Science, 2009, 323, 112–116 CrossRef CAS.
- M. M. Maye, D. Nykypanchuk, M. Cuisinier, D. van der Lelie and O. Gang, Nat. Mater., 2009, 8, 388–391 CrossRef CAS.
- Y. Weizmann, A. B. Braunschweig, O. I. Wilner, Z. Cheglakov and I. Willner, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 5289–5294 CrossRef CAS.
- P. Akcora, H. Liu, S. K. Kumar, J. Moll, Y. Li, B. C. Benicewicz, L. S. Schadler, D. Acehan, A. Z. Panagiotopoulos, V. Pryamitsyn, V. Ganesan, J. Ilavsky, P. Thiyagarajan, R. H. Colby and J. F. Douglas, Nat. Mater., 2009, 8, 354–359 CrossRef CAS.
- C. Yang, Z. Yang, H. Gu, C. K. Chang, P. Gao and B. Xu, Chem. Mater., 2008, 20, 7514–7520 CrossRef CAS.
- Y. Zhou, M. Kogiso, M. Asakawa, S. Dong, R. Kiyama and T. Shimizu, Adv. Mater., 2009, 21, 1742–1745 CrossRef CAS.
- S. Acharya, S. Kundu, J. P. Hill, G. J. Richards and K. Ariga, Adv. Mater., 2009, 21, 989–993 CrossRef CAS.
- (a) B. Moulton and M. J. Zaworotko, Chem. Rev., 2001, 101, 1629–1658 CrossRef CAS; (b) M. Eddaoudi, D. B. Moler, H. Li, B. L. Chen, T. M. Reineke, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2001, 34, 319–330 CrossRef CAS; (c) S. Kitagawa, R. Kitaura and S. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS; (d) S. Bureekaew, S. Shimomura and S. Kitagawa, Sci. Technol. Adv. Mater., 2008, 9, 014108 CrossRef.
- (a) O. M. Yaghi, G. Li and H. Li, Nature, 1995, 378, 703–706 CrossRef CAS; (b) O. M. Yaghi, H. Li, C. Davis, D. Richardson and T. L. Groy, Acc. Chem. Res., 1998, 31, 474–484 CrossRef CAS; (c) O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705–714 CrossRef CAS.
- B. Wang, A. P. Côté, H. Furukawa, M. O'Keeffe and O. M. Yaghi, Nature, 2008, 453, 207–211 CrossRef CAS.
- (a) T. Yanagisawa, T. Shimizu, K. Kuroda and C. Kato, Bull. Chem. Soc. Jpn., 1990, 63, 988–992 CrossRef CAS; (b) C. T. Kresge, M. E. Leonowicz, W. J. Roth, J. C. Vartuli and J. S. Beck, Nature, 1992, 359, 710–712 CrossRef CAS; (c) A. Okabe, T. Fukushima, K. Ariga and T. Aida, Angew. Chem., Int. Ed., 2002, 41, 3414–3417 CrossRef CAS; (d) Q. Zhang, K. Ariga, A. Okabe and T. Aida, J. Am. Chem. Soc., 2004, 126, 988–989 CrossRef CAS; (e) A. Okabe, T. Fukushima, K. Ariga, M. Niki and T. Aida, J. Am. Chem. Soc., 2004, 126, 9013–9016 CrossRef CAS; (f) A. Vinu, K. Z. Hossain and K. Ariga, J. Nanosci. Nanotechnol., 2005, 5, 347–371 CrossRef CAS; (g) H. Jin, Z. Liu, T. Ohsuna, O. Terasaki, Y. Inoue, K. Sakamoto, T. Nakanishi, K. Ariga and S. Che, Adv. Mater., 2006, 18, 593–596 CrossRef CAS; (h) W. Otani, K. Kinbara, Q. Zhang, K. Ariga and T. Aida, Chem.–Eur. J., 2007, 13, 1731–1736 CrossRef CAS; (i) K. Ariga, A. Vinu, J. P. Hill and T. Mori, Coord. Chem. Rev., 2007, 251, 2562–2591 CrossRef CAS.
- (a) R. Ryoo, S. H. Joo and S. Jun, J. Phys. Chem. B, 1999, 103, 7743–7746 CrossRef CAS; (b) J. Lee, S. Yoon, T. Hyeon, S. M. Oh and K. B. Kim, Chem. Commun., 1999, 2177–2178 RSC; (c) A. Vinu, M. Miyahara, V. Sivamurugan, T. Mori and K. Ariga, J. Mater. Chem., 2005, 15, 5122–5127 RSC; (d) A. Vinu, M. Miyahara, T. Mori and K. Ariga, J. Porous Mater., 2006, 13, 379–383 CrossRef CAS; (e) K. Ariga, A. Vinu, M. Miyahara, J. P. Hill and T. Mori, J. Am. Chem. Soc., 2007, 129, 11022–11023 CrossRef CAS.
- (a) A. Vinu, K. Ariga, T. Mori, T. Nakanishi, S. Hishita, D. Golberg and Y. Bando, Adv. Mater., 2005, 17, 1648–1652 CrossRef CAS; (b) A. Vinu, M. Terrones, D. Golberg, S. Hishita, K. Ariga and T. Mori, Chem. Mater., 2005, 17, 5887–5890 CrossRef CAS; (c) A. Vinu, T. Mori and K. Ariga, Sci. Technol. Adv. Mater., 2006, 7, 753–771 CrossRef; (d) A. Vinu, P. Srinivasu, D. P. Sawant, T. Mori, K. Ariga, J.-S. Chang, S.-H. Jhung, V. V. Balasubramanian and Y. K. Hwang, Chem. Mater., 2007, 19, 4367–4372 CrossRef CAS; (e) Y. Yamauchi and K. Kuroda, Chem.–Asian J., 2008, 3, 664–676 CrossRef CAS; (f) Y. Yamauchi, A. Sugiyama, R. Morimoto, A. Takai and K. Kuroda, Angew. Chem., Int. Ed., 2008, 47, 5371–5373 CrossRef CAS.
- (a) K. J. C. van Bommel, A. Friggeri and S. Shinkai, Angew. Chem., Int. Ed., 2003, 42, 980–999 CrossRef; (b) J. H. Jung, H. Kobayashi, M. Masuda, T. Shimizu and S. Shinkai, J. Am. Chem. Soc., 2001, 123, 8785–8789 CrossRef CAS.
- J. M. Szeifert, D. Fattakhova-Rohlfing, D. Georgiadou, V. Kalousek, J. Rathouský, D. Kuang, S. Wenger, S. M. Zakeeruddin, M. Grätzel and T. Bein, Chem. Mater., 2009, 21, 1260–1265 CrossRef CAS.
- T. Brezesinski, J. Wang, J. Polleux, B. Dunn and S. H. Tolbert, J. Am. Chem. Soc., 2009, 131, 1802–1809 CrossRef CAS.
- D. Gottlieb, S. A. Morin, S. Jin and R. T. Raines, J. Mater. Chem., 2008, 18, 3865–3870 RSC.
- S. C. Warren, L. C. Messina, L. S. Slaughter, M. Kamperman, Q. Zhou, S. M. Gruner, F. J. DiSalvo and U. Wiesner, Science, 2008, 320, 1748–1752 CrossRef CAS.
- A. Chen, M. Komura, K. Kamata and T. Iyoda, Adv. Mater., 2008, 20, 763–767 CrossRef CAS.
- M. Kyotani, S. Matsushita, T. Nagai, Y. Matsui, M. Shimomura, A. Kaito and K. Akagi, J. Am. Chem. Soc., 2008, 130, 10880–10881 CrossRef CAS.
- D. Tanaka, K. Nakagawa, M. Higuchi, S. Horike, Y. Kubota, T. C. Kobayashi, M. Takata and S. Kitagawa, Angew. Chem., Int. Ed., 2008, 47, 3914–3918 CrossRef CAS.
- S. K. Ghosh, S. Bureekaew and S. Kitagawa, Angew. Chem., Int. Ed., 2008, 47, 3403–3406 CrossRef CAS.
- Y.-M. Jeon, G. S. Armatas, J. Heo, M. G. Kanatzidis and C. A. Mirkin, Adv. Mater., 2008, 20, 2105–2110 CrossRef CAS.
- C.-F. Lee, D. A. Leigh, R. G. Pritchard, D. Schultz, S. J. Teat, G. A. Timco and R. E. P. Winpenny, Nature, 2009, 458, 314–318 CrossRef CAS.
- M. A. Alam, Y.-Sang Kim, S. Ogawa, A. Tsuda, N. Ishii and T. Aida, Angew. Chem., Int. Ed., 2008, 47, 2070–2073 CrossRef CAS.
- K. Tanaka, G. H. Clever, Y. Takezawa, Y. Yamada, C. Kaul, M. Shionoya and T. Carell, Nat. Nanotechnol., 2006, 1, 190–194 CrossRef CAS.
- J. Yuan, Y. Xu, A. Walther, S. Bolisetty, M. Schumacher, H. Schmalz, M. Ballauff and A. H. E. Muller, Nat. Mater., 2008, 7, 718–722 CrossRef CAS.
- (a) Y. Okahata, T. Tsuruta, K. Ijiro and K. Ariga, Langmuir, 1988, 4, 1373–1375 CrossRef CAS; (b) Y. Okahata, T. Tsuruta, K. Ijiro and K. Ariga, Thin Solid Films, 1989, 180, 65–72 CrossRef CAS; (c) H. Otsuka, Y. Nagasaki and K. Kataoka, Adv. Drug Delivery Rev., 2003, 55, 403–419 CrossRef CAS; (d) A. K. Gupta and M. Gupta, Biomaterials, 2005, 26, 3995–4021 CrossRef CAS; (e) C.-H. Xue, S.-T. Jia, H.-Z. Chen and M. Wang, Sci. Technol. Adv. Mater., 2008, 9, 035001 CrossRef; (f) S. H. Zhang, M. X. Li, J. H. Yoon, T. Y. Cho, Y. Z. He and C. G. Lee, Sci. Technol. Adv. Mater., 2008, 9, 035002 CrossRef; (g) C.-H. Xue, S.-T. Jia, J. Zhang, L.-Q. Tian, H.-Z. Chen and M. Wang, Sci. Technol. Adv. Mater., 2008, 9, 035008 CrossRef; (h) I. Gurrappa and G. Malakondaiah, Sci. Technol. Adv. Mater., 2008, 9, 025005 CrossRef; (i) M. Vukčević, A. Kalijadis, S. Dimitrijević-Branković, Z. Laušević and M. Lauševíc, Sci. Technol. Adv. Mater., 2008, 9, 015006 CrossRef.
- D. Sundrani, S. B. Darling and S. J. Sibener, Langmuir, 2004, 20, 5091–5099 CrossRef CAS.
- J. Ryu and C. B. Park, Adv. Mater., 2008, 20, 3754–3758 CrossRef CAS.
- M. Sofos, J. Goldberger, D. A. Stone, J. E. Allen, Q. Ma, D. J. Herman1, W.-W. Tsai, L. J. Lauhon and S. I. Stupp, Nat. Mater., 2009, 8, 68–75 CrossRef CAS.
- E. Hosono, S. Fujihara, I. Honma and H. Zhou, J. Am. Chem. Soc., 2005, 127, 13458–13459 CrossRef CAS.
- J. Wu and J. L. Coffer, Adv. Mater., 2008, 20, 1571–1575 CrossRef CAS.
- S. A. Morin, Y.-H. La, C.-C. Liu, J. A. Streifer, R. J. Hamers, P. F. Nealey and S. Jin, Angew. Chem., Int. Ed., 2009, 48, 2135–2139 CrossRef CAS.
- (a) T. Yokoyama, S. Yokoyama, T. Kamikado, Y. Okuno and S. Mashiko, Nature, 2001, 413, 619–621 CAS; (b) J. P. Hill, Y. Wakayama, W. Schmitt, T. Tsuruoka, T. Nakanishi, M. L. Zandler, A. L. McCarty, F. D'Souza, L. R. Milgrom and K. Ariga, Chem. Commun., 2006, 2320–2322 RSC; (c) J. P. Hill, Y. Wakayama and K. Ariga, Phys. Chem. Chem. Phys., 2006, 8, 5034–5037 RSC; (d) Y. Wakayama, J. P. Hill and K. Ariga, Surf. Sci., 2007, 601, 3984–3987 CrossRef CAS; (e) J. P. Hill, Y. Wakayama, M. Akada and K. Ariga, J. Phys. Chem. C, 2007, 111, 16174–16180 CrossRef CAS; (f) K. Ariga, J. P. Hill, Y. Wakayama, M. Akada, E. Barrena and D. G. de Oteyza, J. Porphyrins Phthalocyanines, 2009, 13, 22–34 CrossRef CAS; (g) J. P. Hill, Y. Wakayama, M. Akada and K. Ariga, Synth. Met., 2009, 159, 765–768 CrossRef CAS.
- (a) T. Nakanishi, N. Miyashita, T. Michinobu, Y. Wakayama, T. Tsuruoka, K. Ariga and D. G. Kurth, J. Am. Chem. Soc., 2006, 128, 6328–6329 CrossRef CAS; (b) D. Bonifazi, A. Kiebele, M. Stöhr, F. Cheng, T. Jung, F. Diederich and H. Spillmann, Adv. Funct. Mater., 2007, 17, 1051–1062 CrossRef CAS; (c) S. Yoshimoto and K. Itaya, J. Porphyrins Phthalocyanines, 2007, 11, 313–333 CrossRef CAS.
- C. Stadler, S. Hansen, I. Kröger, C. Kumpf and E. Umbach, Nat. Phys., 2009, 5, 153–158 CrossRef CAS.
- J. Puigmartí-Luis, A. Minoia, H. Uji-i, C. Rovira, J. Cornil, S. De Feyter, R. Lazzaroni and D. B. Amabilino, J. Am. Chem. Soc., 2006, 128, 12602–12603 CrossRef CAS.
- S. Lei, J. Puigmartí-Luis, A. Minoia, M. Van der Auweraer, C. Rovira, R. Lazzaroni, D. B. Amabilino and S. De Feyter, Chem. Commun., 2008, 703–705 RSC.
- A. Ciesielski, G. Schaeffer, A. Petitjean, J.-M. Lehn and P. Samor, Angew. Chem., Int. Ed., 2009, 48, 2039–2043 CrossRef CAS.
- P. Gambardella, S. Stepanow, A. Dmitriev, J. Honolka, F. M. F. de Groot, M. Lingenfelder, S. S. Gupta, D. D. Sarma, P. Bencok, S. Stanescu, S. Clair, S. Pons, N. Lin, A. P. Seitsonen, H. Brune, J. V. Barth and K. Kern, Nat. Mater., 2009, 8, 189–193 CrossRef CAS.
- Y. Wei, W. Tong and M. B. Zimmt, J. Am. Chem. Soc., 2008, 130, 3399–3405 CrossRef CAS.
- S. Yoshimoto, Y. Honda, O. Ito and K. Itaya, J. Am. Chem. Soc., 2008, 130, 1085–1092 CrossRef CAS.
- H. L. Zhang, W. Chen, H. Huang, L. Chen and A. T. S. Wee, J. Am. Chem. Soc., 2008, 130, 2720–2721 CrossRef CAS.
- D. G. de Oteyza, I. Silanes, M. Ruiz-Osés, E. Barrena, B. P. Doyle, A. Arnau, H. Dosch, Y. Wakayama and J. E. Ortega, Adv. Funct. Mater., 2009, 19, 259–264 CrossRef CAS.
- R. Madueno, M. T. Räisänen, C. Silien and M. Buck, Nature, 2008, 454, 618–621 CrossRef CAS.
- (a) X. Cha, K. Ariga, M. Onda and T. Kunitake, J. Am. Chem. Soc., 1995, 117, 11833–11838 CrossRef CAS; (b) X. Cha, K. Ariga and T. Kunitake, J. Am. Chem. Soc., 1996, 118, 9545–9551 CrossRef CAS; (c) M. Sakurai, H. Tamagawa, Y. Inoue, K. Ariga and T. Kunitake, J. Phys. Chem. B, 1997, 101, 4810–4816 CrossRef CAS; (d) H. Tamagawa, M. Sakurai, Y. Inoue, K. Ariga and T. Kunitake, J. Phys. Chem. B, 1997, 101, 4817–4825 CrossRef CAS; (e) K. Ariga and T. Kunitake, Acc. Chem. Res., 1998, 31, 371–378 CrossRef CAS; (f) K. Ariga, A. Kamino, X. Cha and T. Kunitake, Langmuir, 1999, 15, 3875–3885 CrossRef CAS.
- (a) Y. Oishi, T. Kato, M. Kuramori, K. Suehiro, K. Ariga, A. Kamino, H. Koyano and T. Kunitake, Chem. Commun., 1997, 1357–1358 RSC; (b) K. Ariga, A. Kamino, H. Koyano and T. Kunitake, J. Mater. Chem., 1997, 7, 1155–1161 RSC; (c) Y. Oishi, Y. Torii, T. Kato, M. Kuramori, K. Suehiro, K. Ariga, K. Taguchi, A. Kamino, H. Koyano and T. Kunitake, Langmuir, 1997, 13, 519–524 CrossRef CAS; (d) K. Ariga, J. Nanosci. Nanotechnol., 2004, 4, 23–34 CrossRef CAS; (e) Y. Oishi, T. Kato, T. Narita, K. Ariga and T. Kunitake, Langmuir, 2008, 24, 1682–1685 CrossRef CAS; (f) K. Ariga, J. P. Hill and Y. Wakayama, Phys. Status Solidi A, 2008, 205, 1249–1257 CrossRef CAS.
- Y. Zhang, P. Chen, L. Jiang, W. Hu and M. Liu, J. Am. Chem. Soc., 2009, 131, 2756–2757 CrossRef CAS.
- R. M. Capito, H. S. Azevedo, Y. S. Velichko, A. Mata and S. I. Stupp, Science, 2008, 319, 1812–1816 CrossRef CAS.
- (a) K. Akatsuka, M. Haga, Y. Ebina, M. Osada, K. Fukuda and T. Sasaki, ACS Nano, 2009, 3, 1097–1106 CrossRef CAS; (b) S. Acharya, J. P. Hill and K. Ariga, Adv. Mater., 2009, 21, 2959–2981 CrossRef CAS.
- N. Lu, X. Chen, D. Molenda, A. Naber, H. Fuchs, D. V. Talapin, H. Weller, J. Mu1ller, J. M. Lupton, J. Feldmann, A. L. Rogach and L. Chi, Nano Lett., 2004, 4, 885–888 CrossRef CAS.
- A. Tao, P. Sinsermsuksakul and P. Yang, Nat. Nanotechnol., 2007, 2, 435–440 CrossRef CAS.
- D. Whang, S. Jin, Y. Wu and C. M. Lieber, Nano Lett., 2003, 3, 1255–1259 CrossRef CAS.
- Y. Li and Y. Wu, J. Am. Chem. Soc., 2009, 131, 5851–5857 CrossRef CAS.
- C. Chen, Q.-H. Yang, Y. Yang, W. Lv, Y. Wen, P.-X. Hou, M. Wang and H.-M. Cheng, Adv. Mater. DOI:10.1002/adma.200803726 , in press.
- L. J. Bonderer, A. R. Studart and L. J. Gauckler, Science, 2008, 319, 1069–1073 CrossRef CAS.
- (a) Y. Okahata, K. Ariga, H. Nakahara and K. Fukuda, J. Chem. Soc., Chem. Commun., 1986, 1069–1071 RSC; (b) K. Ariga and Y. Okahata, J. Am. Chem. Soc., 1989, 111, 5618–5622 CrossRef CAS; (c) K. Ariga, K. Tanaka, K. Katagiri, J. Kikuchi, H. Shimakoshi, E. Ohshima and Y. Hisaeda, Phys. Chem. Chem. Phys., 2001, 3, 3442–3446 RSC.
- M. D. Mager and N. A. Melosh, Adv. Mater., 2008, 20, 4423–4427 CrossRef CAS.
- M. Gleiche, L. F. Chi and H. Fuchs, Nature, 2000, 403, 173–175 CrossRef CAS.
- H. Xia and D. Wang, Adv. Mater., 2008, 20, 4253–4256 CrossRef CAS.
- M. Sathish, K. Miyazawa, J. P. Hill and K. Ariga, J. Am. Chem. Soc., 2009, 131, 6372–6373 CrossRef CAS.
- (a) M. Motornov, R. Sheparovych, R. Lupitskyy, E. MacWilliams, O. Hoy, I. Luzinov and S. Minko, Adv. Funct. Mater., 2007, 17, 2307–2314 CrossRef CAS; (b) M. Motornov, J. Zhou, M. Pita, I. Tokarev, V. Gopishetty and S. Minko, Small, 2009, 5, 817–820 CrossRef CAS.
- (a) Z. Nie, D. Fava, E. Kumacheva, S. Zou, G. C. Walker and M. Rubinstein, Nat. Mater., 2007, 6, 609–614 CrossRef CAS; (b) M. Das, L. Mordoukhovski and E. Kumacheva, Adv. Mater., 2008, 20, 2371–2375 CrossRef CAS.
- (a) A. Laromaine, L. Koh, M. Murugesan, R. V. Ulijn and M. M. Stevens, J. Am. Chem. Soc., 2007, 129, 4156–4157 CrossRef CAS; (b) R. J. Williams, A. M. Smith, R. Collins, N. Hodson, A. K. Das and R. V. Ulijn, Nat. Nanotechnol., 2009, 4, 19–24 CrossRef CAS.
- (a) Y. Xia and G. M. Whitesides, Annu. Rev. Mater. Sci., 1998, 28, 153–184 CrossRef CAS; (b) R. S. Kane, S. Takayama, E. Ostuni, D. E. Ingber and G. M. Whitesides, Biomaterials, 1999, 20, 2363–2376 CrossRef; (c) R. K. Smith, P. A. Lewis and P. S. Weiss, Prog. Surf. Sci., 2004, 75, 1–68 CrossRef CAS.
- (a) R. D. Piner, J. Zhu, F. Xu, S. Hong and C. A. Mirkin, Science, 1999, 283, 661–663 CrossRef CAS; (b) L. M. Demers, D. S. Ginger, S.-J. Park, Z. Li, S.-W. Chung and C. A. Mirkin, Science, 2002, 296, 1836–1838 CrossRef CAS; (c) S. H. Hong, J. Zhu and C. A. Mirkin, Science, 1999, 286, 523–525 CrossRef CAS.
- Z. Fan, J. C. Ho, Z. A. Jacobson, R. Yerushalmi, R. L. Alley, H. Razavi and A. Javey, Nano Lett., 2008, 8, 20–25 CrossRef CAS.
- X. Wu, S. Lenhert, L. Chi and H. Fuchs, Langmuir, 2006, 22, 7807–7811 CrossRef CAS.
- C. Lin, Y. Ke, Y. Liu, M. Mertig, J. Gu and H. Yan, Angew. Chem., Int. Ed., 2007, 46, 6089–6092 CrossRef CAS.
- F. Huo, Z. Zheng, G. Zheng, L. R. Giam, H. Zhang and C. A. Mirkin, Science, 2008, 321, 1658–1660 CrossRef CAS.
- F. D. Benedetto, E. Mele, A. Camposeo, A. Athanassiou, R. Cingolani and D. Pisignano, Adv. Mater., 2008, 20, 314–318 CrossRef CAS.
- X. Chen, Y.-M. Jeon, J.-W. Jang, L. Qin, F. Huo, W. Wei and C. A. Mirkin, J. Am. Chem. Soc., 2008, 130, 8166–8168 CrossRef CAS.
- M. Li, R. B. Bhiladvala, T. J. Morrow, J. A. Sioss, K.-K. Lew, J. M. Redwing, C. D. Kearing and T. S. Mayer, Nat. Nanotechnol., 2008, 3, 88–92 CrossRef CAS.
- Q. Wang, K. Zhu, N. R. Neale and A. J. Frank, Nano Lett., 2009, 9, 806–813 CrossRef CAS.
- M. C. LeMieux, M. Roberts, S. Barman, Y. W. Jin, J. M. Kim and Z. Bao, Science, 2008, 321, 101–104 CrossRef CAS.
- R. Ruiz, H. Kang, F. A. Detcheverry, E. Dobisz, D. S. Kercher, T. R. Albrecht, J. J. de Pablo and P. F. Nealey, Science, 2008, 321, 936–939 CrossRef CAS.
- (a) G. Decher, Science, 1997, 277, 1232–1237 CrossRef CAS; (b) P. T. Hammond, Adv. Mater., 2004, 16, 1271–1293 CrossRef CAS; (c) K. Ariga, J. P. Hill and Q. Ji, Phys. Chem. Chem. Phys., 2007, 9, 2319–2340 RSC; (d) Q. He, Y. Cui, S. Ai, Y. Tian and J. Li, Curr. Opin. Colloid Interface Sci., 2009, 14, 115–125 CrossRef CAS.
- (a) Y. Lvov, G. Decher and H. Möhwald, Langmuir, 1993, 9, 481–486 CrossRef CAS; (b) Y. Lvov, K. Ariga, M. Onda, I. Ichinose and T. Kunitake, Colloids Surf., A, 1999, 146, 337–346 CrossRef CAS; (c) S. S. Shiratori and M. F. Rubner, Macromolecules, 2000, 33, 4213–4219 CrossRef CAS.
- (a) Y. Lvov, K. Ariga and T. Kunitake, Chem. Lett., 1994, 2323–2326 CAS; (b) Y. Lvov, K. Ariga, I. Ichinose and T. Kunitake, J. Am. Chem. Soc., 1995, 117, 6117–6123 CrossRef CAS; (c) M. Onda, Y. Lvov, K. Ariga and T. Kunitake, J. Ferment. Bioeng., 1996, 82, 502–506 CrossRef CAS; (d) M. Onda, Y. Lvov, K. Ariga and T. Kunitake, Biotechnol. Bioeng., 1996, 51, 163–167 CrossRef; (e) F. Caruso, D. N. Furlong, K. Ariga, I. Ichinose and T. Kunitake, Langmuir, 1998, 14, 4559–4565 CrossRef CAS; (f) Y. Lvov, M. Onda, K. Ariga and T. Kunitake, J. Biomater. Sci., Polym. Ed., 1998, 9, 345–355 CAS; (g) M. Onda, K. Ariga and T. Kunitake, J. Biosci. Bioeng., 1999, 87, 69–75 CrossRef CAS; (h) K. Ariga, T. Nakanishi and T. Michinobu, J. Nanosci. Nanotechnol., 2006, 6, 2278–2301 CrossRef CAS; (i) K. Ariga, J. P. Hill and Q. Ji, Macromol. Biosci., 2008, 8, 981–990 CrossRef CAS.
- (a) Y. Lvov, K. Ariga, I. Ichinose and T. Kunitake, Langmuir, 1996, 12, 3038–3044 CrossRef CAS; (b) Y. Lvov, K. Ariga, M. Onda, I. Ichinose and T. Kunitake, Langmuir, 1997, 13, 6195–6203 CrossRef CAS; (c) G. Bantchev, Z. Lu and Y. Lvov, J. Nanosci. Nanotechnol., 2009, 9, 396–403 CrossRef CAS; (d) N. G. Veerabadran, D. Mongayt, V. Torchilin, R. R. Price and Y. M. Lvov, Macromol. Rapid Commun., 2009, 30, 99–103 CrossRef CAS.
- (a) K. Ariga, Y. Lvov and T. Kunitake, J. Am. Chem. Soc., 1997, 119, 2224–2231 CrossRef CAS; (b) K. Katagiri, R. Hamasaki, K. Ariga and J. Kikuchi, J. Am. Chem. Soc., 2002, 124, 7892–7893 CrossRef CAS; (c) K. Katagiri, R. Hamasaki, K. Ariga and J. Kikuchi, Langmuir, 2002, 18, 6709–6711 CrossRef CAS.
- (a) F. Caruso, R. A. Caruso and H. Möhwald, Science, 1998, 282, 1111–1114 CrossRef CAS; (b) Y. Lvov, A. A. Antipov, A. Mamedov, H. Möhwald and G. B. Sukhorukov, Nano Lett., 2001, 1, 125–128 CrossRef CAS; (c) F. Caruso, Adv. Mater., 2001, 13, 11–22 CrossRef CAS; (d) Y. Wang, A. C. Angelatos and F. Caruso, Chem. Mater., 2008, 20, 848–858 CrossRef CAS; (e) Q. He, L. Duan, W. Qi, K. Wang, Y. Cui, X. Yan and J. Li, Adv. Mater., 2008, 20, 2933–2937 CrossRef CAS.
- K. C. Krogman, J. L. Lowery, N. S. Zacharia, G. C. Rutledge and P. T. Hammond, Nat. Mater., 2009, 8, 512–518 CrossRef CAS.
- M. Altman, A. D. Shukla, T. Zubkov, G. Evmenenko, P. Dutta and M. E. van der Boom, J. Am. Chem. Soc., 2006, 128, 7374–7382 CrossRef CAS.
- T. R. Hendricks, J. Lu, L. T. Drzal and I. Lee, Adv. Mater., 2008, 20, 2008–2012 CrossRef CAS.
- E. Kharlampieva, J. M. Slocik, T. Tsukruk, R. R. Naik and V. V. Tsukruk, Chem. Mater., 2008, 20, 5822–5831 CrossRef CAS.
- (a) Q. Ji, M. Miyahara, J. P. Hill, S. Acharya, A. Vinu, S. B. Yoon, J.-S. Yu, K. Sakamoto and K. Ariga, J. Am. Chem. Soc., 2008, 130, 2376–2377 CrossRef CAS; (b) Q. Ji, S. Acharya, J. P. Hill, A. Vinu, S. B. Yoon, J.-S. Yu, K. Sakamoto and K. Ariga, Adv. Funct. Mater., 2009, 19, 1792–1799 CrossRef CAS.
- Q. Ji, S. B. Yoon, J. P. Hill, A. Vinu, J.-S. Yu and K. Ariga, J. Am. Chem. Soc., 2009, 131, 4220–4221 CrossRef CAS.
- K. Ariga, A. Vinu, Q. Ji, O. Ohmori, J. P. Hill, S. Acharya, J. Koike and S. Shiratori, Angew. Chem., Int. Ed., 2008, 47, 7254–7257 CrossRef CAS.
- H. Benten, M. Ogawa, H. Ohkita and S. Ito, Adv. Funct. Mater., 2008, 18, 1563–1572 CrossRef CAS.
- X. Chen, A. L. Rogach, D. V. Talapin, H. Fuchs and L. Chi, J. Am. Chem. Soc., 2006, 128, 9592–9593 CrossRef CAS.
- T. S. Corbitt, J. R. Sommer, S. Chemburu, K. Ogawa, L. K. Ista, G. P. Lopez, D. G. Whitten and K. S. Schanze, ACS Appl. Mater. Interfaces, 2009, 1, 48–52 Search PubMed.
- H. Shimura, M. Yoshio, A. Hamasaki, T. Mukai, H. Ohno and T. Kato, Adv. Mater., 2009, 21, 1591–1594 CrossRef CAS.
- N. Aubry, P. Singh, M. Janjua and S. Nudurupati, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 3711–3714 CrossRef CAS.
- K. Mukai, K. Asaka, T. Sugino, K. Kiyohara, I. Takeuchi, N. Terasawa, D. N. Futaba, K. Hata, T. Fukushima and T. Aida, Adv. Mater., 2009, 21, 1582–1585 CrossRef CAS.
- T. Fukushima, A. Kosaka, Y. Ishimura, T. Yamamoto, T. Takigawa, N. Ishii and T. Aida, Science, 2003, 300, 2072–2074 CrossRef CAS.
- M. Yamada, M. Kondo, J. Mamiya, Y. Yu, M. Kinoshita, C. J. Barrett and T. Ikeda, Angew. Chem., Int. Ed., 2008, 47, 4986–4988 CrossRef CAS.
- (a) K. Ariga, Y. Terasaka, D. Sakai, H. Tsuji and J. Kikuchi, J. Am. Chem. Soc., 2000, 122, 7835–7836 CrossRef CAS; (b) K. Ariga, T. Nakanishi, Y. Terasaka, H. Tsuji, D. Sakai and J. Kukuchi, Langmuir, 2005, 21, 976–981 CrossRef CAS; (c) K. Ariga, T. Nakanishi, H. P. Hill, Y. Terasaka, D. Sakai and J. Kikuchi, Soft Matter, 2005, 1, 132–137 RSC; (d) K. Ariga, T. Nakanishi and J. P. Hill, Soft Matter, 2006, 2, 465–477 RSC; (e) T. Michinobu, S. Shinoda, T. Nakanishi, J. P. Hill, K. Fujii, T. N. Player, H. Tsukube and K. Ariga, J. Am. Chem. Soc., 2006, 128, 14478–14479 CrossRef CAS; (f) K. Ariga, T. Michinobu, T. Nakanishi and J. P. Hill, Curr. Opin. Colloid Interface Sci., 2008, 13, 23–30 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |