A novel substitution reaction of perylene bisimides with Ph2PLi at the α-position†
Xue Wu, Chuanwei Yin, Zhiqiang Shi*, Maoyou Xu, Jin Zhang and Juanjuan Sun
Department of Chemistry, Shandong Normal University, Jinan, 250014, P. R. China. E-mail: zshi@sdnu.edu.cn; Fax: +86 531 82615258; Tel: +86 531 86182540
First published on 23rd October 2009
Abstract
A novel substitution reaction of perylene bisimides has been developed. The Ph2PO group was attached to the PBI core at the α-position instead of the usual bay position. This substitution kept the high fluorescence of the PBI dyes and caused the adjacent proton at the bay position and a tertiary proton at the cyclohexyl group to be shifted significantly in the 1H-NMR spectra.
Introduction
In recent years, functionalized perylene bisimides (PBIs) have received considerable attention for a wide range of applications, for example, as fluorescent dyes for single molecule spectroscopy1 and bio-imaging2 and as organic semiconductors for organic and polymeric light-emitting diodes (OLEDs and PLEDs),3 organic field-effect transistors (OFETs),4 and solar cells.5 They also have been proven to be good building blocks for constructing supramolecular or giant-molecular systems.6 The molecular properties that enable all these applications are the high fluorescent quantum yield, fairly persistent radical anion state, easy accessibility, and outstanding stability of these dyes against environmental influences.7 Due to the intrinsic insolubility of perylene bisimides, two different strategies have been developed to modify the dyes. One is introducing solubilizing substitutes at the imide nitrogen,8 which exhibits usually indistinguishable absorption and emission properties.9 Another is introducing substituents at the carbocyclic scaffold in the so-called bay-area (β-position of imide groups). The latter strategy is more elaborate.10 For example, aryloxy-, cyano-, pyrrolidino, alkoxy and/or alkylthio-substituted PBIs as well as the Suzuki coupling reaction have been investigated. Usually the readily available halogenated PBIs, such as 1,7-dibromo or 1,6,7,12-tetrachloro derivatives, were used as starting materials.11,12 As far as we know, it has seldom been reported that the PBI dye was directly modified at α-position of the aromatic core.13Phosphines have been widely used for organic synthesis. For example, organophosphide anions are considered to be powerful nucleophiles. The diphenylphosphide anion (Ph2P−) readily reacts with haloaromatic compounds involving SRN1 mechanism of nucleophilic substitution with both thermal and photochemical initiation steps.14 And phosphines reduce hydroperoxides, peroxides, disulfides, sulfoxides, epoxides and so on.15 However, the reaction of Ph2P− with PBI dye has not been reported. Herein, a series of diphenylphosphinoyl (Ph2PO) group substituted perylene bisimides at the α-position (1, 2 and 3) were synthesized from corresponding PBIs (1a, 2a and 3a) following the strategy outlined in Scheme 1.
 | ||
| Scheme 1 The synthesis of compounds 1, 2 and 3. | ||
N,N′-Dicyclohexylperylene-3,4:9,10-tetracarboxylic acid bisimide 1a, N,N′-dicyclohexyl-1,7-dibromoperylene-3,4:9,10-tetracarboxylic acid bisimide 2a and N,N′-dicyclohexyl-1,6,7,12-tetrachloroperylene-3,4:9,10-tetracarboxylic acid bisimide 3a were synthesized and purified according to literature procedures.16 A solution of lithium diphenylphosphide (DPPLi) was prepared from triphenylphosphine in THF.17 1,7-Bibromoperylene bisimide 2a was used to carry out this reaction. Under an argon gas stream, DPPLi in THF was added into the THF solution of 2a. Originally we aimed to substitute the bromine atoms of 2a. However, MALDI-TOF mass data suggested that a proton on the perylene core was substituted by a Ph2PO group instead of substitution of the halogen atom. The suggestion was further confirmed by 1H-NMR, 13C-NMR and 31P-NMR spectra. These data indicated that a novel type of substitution reaction of PBIs was developed. This new reaction was also verified by the reactions of DPPLi with 1a (no halogen substituted) and 3a (all of the bay positions halogen substituted).
The substitution of the aromatic core of perylene-3,4:9,10-tetracarboxylic acid bisimides generally takes place at bay (β) positions. For 1,6,7,12-tetrachloroperylene bisimide 3a, four β-positions are occupied by chlorine atoms and no chlorine atom is substituted according to the MS spectrum. Consequently, the Ph2PO group is incorporated at an α-position of the perylene core. All data from the 1H-NMR, 13C-NMR and 31P-NMR spectra are consistent with this result. Due to the shielding effect of the Ph2PO group on compound 3, the tertiary proton on a nearer cyclohexyl group (H1 proton, Scheme 1) shifts upfield 0.99 ppm compared with the tertiary proton on another cyclohexyl group (H2 proton, Table 1). Similar effects are also observed for 1 and 2. Compared with H2 proton, the H1 proton shifts upfield 0.41 and 0.30 ppm, respectively, for 1 and 2. These values are less than that for 3. Because the relatively bulky chlorine atom at the adjacent position makes the Ph2PO group near the H1 proton, and as a result, the shielding effect of the Ph2PO group is reinforced for 3 (Fig. 1). The remarkable chemical shifts of the tertiary protons on cyclohexyl groups reveal that the substitutions also take place at the α-positions of the perylene bisimides 1 and 2.
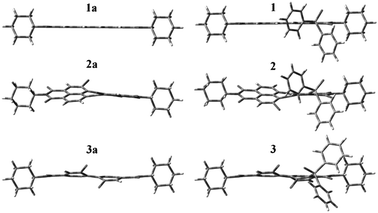 | ||
| Fig. 1 DFT (B3LYP/6-31G*) geometry-optimized structures of 1a, 2a, 3a and 1, 2, 3. The geometry calculations were performed with Gaussian 03 installed on a Windows PC. | ||
| H1 (ppm) | H2 (ppm) | H3 (ppm), J/Hz | |
|---|---|---|---|
| 1 | 4.65 | 5.06 | 9.70, 12.6 |
| 2 | 4.71 | 5.01 | 9.91, 13.7 |
| 2a | 5.06 | 5.06 | 8.63, 8.1 |
| 3 | 4.01 | 5.00 | — |
| 3a | 5.02 | 5.02 | — |
In addition, an unusual change in 1H-NMR spectrum of 1 was noted. The adjacent proton of the substituted group at bay position shifts downfield to 9.70 ppm with a large coupling constant of 12.6 Hz (300 M at 25 °C, Table 1). The remarkable increase in chemical shift mainly results from the deshielding effect of Ph2PO group because this proton locates at a strong induced paramagnetic area of two phenyl groups. The large coupling constant indicates that this proton is split by an adjacent P atom. For compound 2, a doublet signal at low field of 9.91 ppm with a large coupling constant of 13.7 Hz (far more than the coupling constant of 8.1 Hz at 8.63 ppm for 2a) is also observed in the 1H-NMR spectrum, revealing that the adjacent atom of Ph2PO group is also a proton instead of a Br atom.
In the synthesis of the target compounds, lithium diphenylphosphide was used to react with PBIs. However, it was found that the group incorporated was not Ph2P but Ph2PO. In most cases, reactions of alkali metal diphenylphosphide with simple aryl halides produce aryldiphenylphosphines, which are stable in air.18 But the incorporation of the large aromatic PBI moiety caused the phosphines to be three-coordinate structures, which are unstable and readily oxidize to penta-coordinate compounds. The experimental phenomena observed in synthesis were that the resulting solutions remained non-fluorescent until separation. After purification with column chromatography, the target compounds obtained were strongly fluorescent. The Ph2P group can quench the fluorescence of the fluorophore connected due to photo-induced electron transfer (PET) processes. Whereas the PET processes are stopped after the Ph2P group has been oxidized to a Ph2PO group.19 Therefore, it was proposed that non-fluorescent Ph2P incorporated PBIs were yielded firstly. After the Ph2P group has been oxidized to the stable Ph2PO group in the purification procedure,20 the fluorescence of PBIs was restored again due to the hindrance of the PET process.
Spectroscopic grade solvents were used in UV/vis measurements. For the purpose of comparison, the starting materials of perylene bisimides are also included in Fig. 2 and the optical data are summarized in Table 2. Over 400 nm, compounds 1 and 2 show the PBI core absorption band with the same maximum absorption at 534 nm in CHCl3, and compound 3 shows the maximum absorption at 528 nm. The absorption maximum of the derivatives are red-shifted by about 8–9 nm after substitution. In the emission spectra of compounds 1, 2 and 3, the maximum emission locate at 545 nm, 555 nm and 558 nm, which are red-shifted by 8 nm, 7 nm and 8 nm, respectively, compared to their corresponding PBI starting materials. These results mainly are attributed to the extension of the aromatic system after the substitution. From Fig. 2, we know that the modified PBI dyes have similar absorption and emission profiles to their starting materials. Especially, the absorption and emission bands are not broadened. Whereas the substitution at the bay-area usually results in the maximum absorption and emission bands being red-shifted and broadening of the bands due to the electron-donating effect of the substituted group and the steric twisting of the perylene core.6,21
 | ||
| Fig. 2 UV/vis (left axis) and fluorescence (right axis) spectra of the perylene bisimides (10−5 M) in chloroform at room temperature. a: 1 (dashed line) and 1a (solid line), b: 2 (dashed line) and 2a (solid line), c: 3 (dashed line) and 3a (solid line). | ||
From Table 2, we know that the fluorescence quantum yield of 1, 2 and 3 show little difference from the corresponding 1a, 2a and 3a. So the incorporation of the Ph2PO group to the perylene core at α-position keeps the fluorescence property well. These are in agreement with the calculation results. As clearly shown in Fig. 1, the geometries of compound 1, 2 and 3 are kept well after the substitution. This is quite different to the substitution at the bay-area, which usually results in a large PDI core twist.22
In summary, a series of Ph2PO group substituted perylene bisimides have been synthesized firstly by reaction of lithium diphenylphosphide with corresponding PBI dyes. The substituted group is attached to the PBI core at the α-position instead of the usual substitution of a halogen atom at the bay-area. Owing to the shielding or deshielding effect of the Ph2PO group, the adjacent proton on the perylene core and the tertiary proton at a nearer cyclohexyl group shift significantly in the 1H-NMR spectra. The substitution results in a slight red-shift both in absorption and emission spectra and keeps the high fluorescence of the PBI dyes. This reaction provides a new method to modify PBI dyes.
Experimental
1H NMR and 13C NMR were measured with Bruker 300 MHz or 600 MHz spectrometers. Mass spectra were recorded on a Bruker BIFLEXIII spectrometer for MALDI-TOF-MS. Infrared spectra were determined with a Bruker Tensor-27 spectrometer. Electronic absorption spectra were measured on a Beijing Purkinje General Instrument Co. Ltd. TU-190 spectrophotometer. The photoluminescence spectra were recorded on a HITACHI FL-4500 spectrofluorometer. Triphenylphosphine, lithium, and THF were purchased from Sinopharm Chemical Reagent Co. Ltd. The THF used was anhydrous THF which was distilled over sodium and benzophenone. Fluorescence quantum yields were measured by the optically dilute method with N,N′-dicyclohexylperylene-3,4:9,10-tetracarboxylic acid bisimide in chloroform solution as reference (using an excitation wavelength of 527 nm).The solution of lithium diphenylphosphide (DPPLi) was prepared from triphenylphosphine as follows. Triphenylphosphine (5.24 g, 0.02 mol) and lithium (0.84 g, 0.12 mol) which had been washed with hexane and dried carefully with a paper towel were suspended in a three-neck flask containing a magnetic stirring bar and fitted with an argon inlet, and the flask was evacuated and backfilled with argon three times. After the solvent of anhydrous THF (150 ml) had been added, the mixture was stirred vigorously at room temperature for 3 h under an argon gas stream. Then the residual lithium was removed by passing the mixture through a glass tube which was stuffed with glass wool loosely; an argon gas stream was also needed in this step. The red solution of lithium diphenylphosphide (DPPLi) was used directly in the next step of the reaction.
A three-neck flask was charged with N,N′-dicyclohexylperylene-3,4:9,10-tetracarboxylic acid bisimide 1a (280 mg, 0.5 mmol), and the flask was evacuated and backfilled with argon three times. After the solvent of anhydrous THF had been added, the mixture was stirred vigorously at room temperature for 1 h. Subsequently, the solution of lithium diphenylphosphide (DPPLi) was added by syringe with a gentle flow of argon. TLC was used to monitor the reaction. When the reaction was finished, the solvent was removed by rotary evaporation at room temperature. The crude product was purified by silica gel column chromatography with CH2Cl2–THF (50 : 1) as eluent. The main second band was collected, and removal of the solvent yielded 121 mg (32%) as a red-brown solid. MS (MALDI-TOF): m/z 754.2 (M+). 1H-NMR (300 MHz, CDCl3, 25 °C): δ = 9.70 (d, 1H, 3JPH = 12.6), 8.66–8.71 (m, 6H), 7.81–7.89 (m, 4H),7.54–7.56 (m, 2H), 7.28 (m, 4H), 5.06 (m, 1H), 4.65 (m, 1H), 2.57–2.60 (m, 2H), 2.01–2.05 (m, 2H), 1.96–2.01 (m, 2H), 1.76 (m, 4H), 1.47–1.52 (m, 4H), 1.12–1.39 (m, 6H) ppm. 13C-NMR (75 MHz, CDCl3, 25 °C): δ = 163.775, 163.685, 162.982, 162.169, 134.666, 133.983, 133.808, 132.137, 131.772, 131.659, 131.507, 131.406, 131.276, 128.261, 128.096, 124.282, 124.047, 123.990, 123.067, 54.171, 54.089, 52.986, 29.665, 29.336, 29.113, 28.593, 26.528, 26.246, 25.427, 25.159 ppm. 31P-NMR (121 MHz, CDCl3, 25 °C): δ = 34.5403 ppm.
A three-neck flask was charged with N,N′-dicyclohexyl-1,7-dibromoperylene-3,4:9,10-tetracarboxylic acid bisimide 2a (360 mg, 0.5 mmol), and the flask was evacuated and backfilled with argon three times. After the solvent of anhydrous THF had been added, the mixture was stirred vigorously at room temperature for 1 h. Subsequently, the solution of lithium diphenylphosphide (DPPLi) was added by syringe with a gentle flow of argon. TLC was used to monitor the reaction. When the reaction was finished, the solvent was removed by rotary evaporation at room temperature. The crude product was purified by silica gel column chromatography with CH2Cl2–THF (50 : 1) as eluent. The main second band was collected, removal of the solvent yielded 198 mg (43%) as a red-brown solid, which was further purified by silica gel column chromatography with toluene–THF (5 : 1) as eluent. The first band was collected, and the solvent was removed by rotary evaporation to afford 97 mg (49%) of 2 as a red solid. MS (MALDI-TOF): m/z 911.9 (M+). 1H-NMR (300 MHz, CDCl3, 25 °C): δ = 9.91 (d, 1H, 3JPH = 13.7), 9.50 (d, 1H, J = 8.2), 8.94 (s, 1H), 8.82 (s, 1H), 8.69 (d, 1H, J = 8.1), 7.74–7.80 (m, 4H), 7.53–7.57 (m, 2H), 7.46–7.48 (m, 4H), 5.01 (m, 1H), 4.71 (m, 1H), 2.51–2.55 (m, 2H), 2.11–2.16 (m, 2H), 1.90–1.93 (m, 2H), 1.73–1.77 (m, 4H), 1.19–1.60 (m, 10H) ppm. 13C-NMR (75 MHz, CDCl3, 25 °C): δ = 163.201, 162.621, 161.930, 161.824, 138.730, 137.903, 134.188, 134.030, 133.862, 132.741, 132.361, 132.249, 132.167, 131.638, 131.512, 130.474, 129.910, 129.143, 128.600, 128.495, 128.325, 127.462, 126.901, 126.577, 124.014, 123.622, 123.468, 122.103, 121.454, 54.424, 54.275, 29.067, 28.624, 26.471, 26.217, 25.347, 25.126 ppm. 31P-NMR (121 MHz, CDCl3, 25 °C): δ = 2.9746 ppm.
A three-neck flask was charged with 1,6,7,12-tetrachloroperylene bisimide 3a (350 mg, 0.5 mmol), and the flask was evacuated and backfilled with argon three times. After the solvent of anhydrous THF has been added, the mixture was stirred vigorously at room temperature for about 1 h. Subsequently, the solution of lithium diphenylphosphide (DPPLi) was added by syringe with a gentle flow of argon. TLC was used to monitor the reaction. When the reaction was finished, the solvent was removed by rotary evaporation at room temperature. The crude product was purified by silica gel column chromatography with CH2Cl2–THF (50 : 1) as eluent. The main second band was collected, removal of the solvent yielded 190 mg (42%) as a red-brown solid. MS (MALDI-TOF): m/z 891.9 (M+). 1H-NMR (600 MHz, CDCl3, 25 °C): δ = 8.65 (s, 1H), 8.63 (s, 1H), 8.55 (s, 1H), 8.03–8.06 (m, 2H), 7.62–7.65 (m, 1H), 7.57–7.60 (m, 4H), 7.45–7.48 (m, 1H), 7.38–7.40 (m, 2H), 5.00 (m, 1H), 4.01 (m, 1H), 2.49–2.55 (m, 2H), 1.90–1.92 (m, 2H), 1.70–1.75 (m, 6H), 1.43–1.50 (m, 2H), 1.12–1.37 (m, 8H) ppm. 13C-NMR (75 MHz, CDCl3, 25 °C): δ = 162.512, 161.669, 136.342, 135.842, 135.441, 133.317, 132.786, 132.650, 132.098, 131.904, 131.765, 130.933, 130.796, 128.760, 128.586, 128.476, 128.303, 123.987, 123.857, 123.680, 122.675, 55.359, 54.470, 29.068, 29.003, 28.780, 28.328, 26.454, 26.291, 26.062, 25.307, 25.174, 21.382 ppm. 31P-NMR (121 MHz, CDCl3, 25 °C): δ = 33.1312 ppm.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (20771067 and 20531060) and Natural Science Foundation of Shandong Province (2008BS02004).References
- (a) J. Hofkens, T. Vosch, M. Maus, F. Köhn, M. Cotlet, T. Weil, A. Herrmann, K. Müllen and F. De Schryver, Chem. Phys. Lett., 2001, 333, 255 CrossRef CAS; (b) E. Lang, F. Würthner and J. Köhler, ChemPhysChem, 2005, 6, 935 CrossRef CAS; E. Lang, F. Würthner and J. Köhler, ChemPhysChem, 2006, 7, 292 CrossRef CAS; (c) C. Jung, B. Müller, D. Lamb, F. Nolde, K. Müllen and C. Bräuchle, J. Am. Chem. Soc., 2006, 128, 5283 CrossRef CAS.
- (a) C. Kohl, T. Weil, J. Qu and K. Müllen, Chem.–Eur. J., 2004, 10, 5297 CrossRef CAS; (b) S. Krauss, M. Lysetska and F. Würthner, Lett. Org. Chem., 2005, 2, 349 Search PubMed; (c) F. Yukruk, A. Dogan, H. Canpinar, D. Guc and E. Akkaya, Org. Lett., 2005, 7, 2885 CrossRef CAS.
- (a) P. Ranke, B. leyl, J. Simmer, D. Haarer, A. Bacher and H. Schmidt, Appl. Phys. Lett., 1997, 71, 1332 CrossRef CAS; (b) C. Ego, D. Marsitzky, S. Becker, J. Zhang, A. Grimsdale, K. Müllen, J. MacKenzie, C. Silva and R. Friend, J. Am. Chem. Soc., 2003, 125, 437 CrossRef CAS; (c) J. Qu, J. Zhang, A. Grimsdale, K. Müllen, F. Jaiser, X. Yang and D. Neher, Macromolecules, 2004, 37, 8297 CrossRef CAS.
- (a) B. Jones, M. Ahrens, M. Yoon, A. Fachetti, T. Marks and M. Wasielewski, Angew. Chem., Int. Ed., 2004, 43, 6363 CrossRef CAS; (b) W. Shin, H. Jeong, M. Kim, S. Jin, M. Kim, J. Lee, J. Lee and Y. Gal, J. Mater. Chem., 2006, 16, 384 RSC; (c) T. Jung, B. Yoo, S. Wang, A. Dodabalapur, B. Jones, A. Facchetti, M. Wasielewski and T. Marks, Appl. Phys. Lett., 2006, 88, 183102 CrossRef; (d) Y. Wang, Y. Chen, R. Li, S. Wang, W. Su, P. Ma, M. Wasielewski and J. Jiang, Langmuir, 2007, 23, 5863.
- (a) Q. Zhang, A. Cirpan, T. Russell and T. Emrick, Macromolecules, 2009, 42, 1079 CrossRef CAS; (b) T. Edvinsson, C. Li, N. Pschirer, J. Schöneboom, F. Eickemeyer, R. Sens, G. Boschloo, A. Herrmann, K. Müllen and A. Hagfeldt, J. Phys. Chem. C, 2007, 111, 15137 CrossRef CAS; (c) X. Zhan, Z. Tan, B. Domercq, Z. An, X. Zhang, S. Barlow, Y. Li, D. Zhu, B. Kippelen and S. Marder, J. Am. Chem. Soc., 2007, 129, 14319 CrossRef CAS.
- F. Würthner, Chem. Commun., 2004, 1564 RSC.
- (a) A. Grimsdale and K. Müllen, Angew. Chem., Int. Ed., 2005, 44, 5592 CrossRef CAS; (b) M. Wasielewski, J. Org. Chem., 2006, 71, 5051 CrossRef CAS; (c) F. Würthner, Pure Appl. Chem., 2006, 78, 2341 CrossRef.
- H. Langhals, Heterocycles, 1995, 40, 477 CrossRef CAS.
- H. Langhals, S. Demmig and H. Huber, Spectrochim. Acta, Part A, 1988, 44, 1189 CrossRef.
- A. Böhm, H. Arms, G. Henning and P. Blaschka (BASF AG), Ger. Pat. Appl., DE 19
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 547209 A1, 1997.
547209 A1, 1997. - (a) P. Osswald, D. Leusser, D. Stalke and F. Würthner, Angew. Chem., Int. Ed., 2005, 44, 250 CrossRef CAS; (b) M. Ahrens, L. Sinks, B. Rybtchinski, W. Liu, B. Jones, J. Giaimo, A. Gusev, A. Goshe, D. Tiede and M. Wasielewski, J. Am. Chem. Soc., 2004, 126, 8284 CrossRef CAS; (c) Y. Li, N. Wang, H. Gan, H. Liu, H. Li, Y. Li, X. He, C. Huang, S. Cui, S. Wang and D. Zhu, J. Org. Chem., 2005, 70, 9686 CrossRef CAS; (d) C. Zhao, Y. Zhang, R. Li, X. Li and J. Jiang, J. Org. Chem., 2007, 72, 2402 CrossRef CAS.
- (a) T. Korenaga, T. Kosaki, R. Fukumura, T. Ema and T. Sakai, Org. Lett., 2005, 7, 4915 CrossRef CAS; (b) D. Liu, W. Gao, Q. Dai and X. Zhang, Org. Lett., 2005, 7, 4907 CrossRef CAS; (c) C. Chao, M. Leung, Y. Su, K. Chiu, T. Lin, S. Shieh and S. Lin, J. Org. Chem., 2005, 70, 4323 CrossRef; (d) Y. Yamada, Y. Maeda and Y. Uozumi, Org. Lett., 2006, 8, 4259 CrossRef CAS.
- M. Sadrai, G. R. Bird, J. A. Potenza and H. J. Schugar, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1990, 46, 637 CrossRef.
- (a) J. Swartz and J. Bunnett, J. Org. Chem., 1979, 44, 340 CrossRef CAS; (b) C. Galli and J. Bunnett, J. Am. Chem. Soc., 1981, 103, 7140 CrossRef CAS.
- (a) A. Senear, W. Valient and J. Wirth, J. Org. Chem., 1960, 25, 2001 CrossRef CAS; (b) H. Relles and R. Schluenz, J. Am. Chem. Soc., 1974, 20, 6469 CrossRef.
- (a) P. Rajasingh, R. Cohen, E. Shirman, J. Shimon and B. Rybtchinski, J. Org. Chem., 2007, 72, 5973 CrossRef CAS; (b) F. Würthner, V. Stepanenko, Z. Chen, C. Saha-Möller, N. Kocher and D. Stalke, J. Org. Chem., 2004, 69, 7933 CrossRef.
- R. Ireland and D. Walba, Org. Synth., 1997, 56, 44.
- A. Aguiar, H. Greenberg and K. Rubenstein, J. Org. Chem., 1963, 28, 2091 CrossRef CAS.
- (a) N. Soh, T. Ariyoshi, T. Fukaminato, K. Nakano, M. Irie and T. Imato, Bioorg. Med. Chem. Lett., 2006, 16, 2943 CrossRef CAS; (b) N. Soh, T. Ariyoshi, T. Fukaminato, H. Nakajima, K. Nakano and T. Imato, Org. Biomol. Chem., 2007, 5, 3762 RSC; (c) K. Akasaka, H. Ohrui and H. Meguro, J. Chromatogr., B: Biomed. Appl., 1993, 622, 153 CrossRef CAS.
- J. Swartz and J. Bunnett, J. Org. Chem., 1979, 44, 340 CrossRef CAS.
- W. Qiu, S. Chen, X. Sun, Y. Liu and D. Zhu, Org. Lett., 2006, 8, 867 CrossRef CAS.
- (a) M. Ahrens, M. Tauber and M. Wasielewski, J. Org. Chem., 2006, 71, 2107 CrossRef CAS; (b) B. Sun, Y. Zhao, X. Qiu, C. Han, Y. Yu and Z. Shi, Supramol. Chem., 2008, 20, 537 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The MS and 1H-, 13C-, 31P-NMR spectra of 1–3. See DOI: 10.1039/b9nj00364a |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2010 |