Binuclear manganese carbonyl thiocarbonyls: metal–metal multiple bonds versus four-electron donor thiocarbonyl groups†
Zhong
Zhang
a,
Qian-shu
Li
*ab,
Yaoming
Xie
c,
R. Bruce
King
*ac and
Henry F.
Schaefer III
c
aCenter for Computational Quantum Chemistry, South China Normal University, Guangzhou 510631, P. R. China. E-mail: qsli@scnu.edu.cn
bInstitute of Chemical Physics, Beijing Institute of Technology, Beijing 100081, P. R. China
cDepartment of Chemistry and Center for Computational Chemistry, University of Georgia, Athens, Georgia 30602, USA. E-mail: rbking@chem.uga.edu
First published on 15th October 2009
Abstract
Density functional theory (DFT) studies on Mn2(CS)2(CO)8 using the B3LYP and BP86 methods show that no less than eight different unbridged structures are of significantly lower energies than the lowest energy doubly bridged structure. The Mn–Mn single bonds in these Mn2(CS)2(CO)8 structures range from 2.99 ± 0.02 Å for the four structures with staggered equatorial CO/CS groups to 3.12 ± 0.04 Å for the four structures with eclipsed equatorial CO/CS groups. The six lowest energy Mn2(CS)2(CO)7 structures all have four-electron donor bridging η2-μ-CE groups (E = S, O) and formal Mn–Mn single bonds of lengths 2.95 ± 0.01 Å, rather than only two-electron donor CO and CS groups and formal Mn![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Mn double bonds. The Mn2(CS)2(CO)7 structures with an η2-μ-CS group are of lower energy than those with an η2-μ-CO group. These Mn2(CS)2(CO)7 structures are similar to the lowest energy structure for Mn2(CO)9 predicted previously as well as (Ph2PCH2PPh2)2Mn2(CO)4(η2-μ-CO), which has been synthesized and structurally characterized by X-ray diffraction. The lowest energy Mn2(CS)2(CO)6 structures are predicted have a single four-electron donor bridging η2-μ-CS group and a formal MnMn double bond. However, at only slightly higher energies, Mn2(CS)2(CO)6 structures are found with two η2-μ-CS groups and a formal Mn–Mn single bond. A formal Mn
Mn double bonds. The Mn2(CS)2(CO)7 structures with an η2-μ-CS group are of lower energy than those with an η2-μ-CO group. These Mn2(CS)2(CO)7 structures are similar to the lowest energy structure for Mn2(CO)9 predicted previously as well as (Ph2PCH2PPh2)2Mn2(CO)4(η2-μ-CO), which has been synthesized and structurally characterized by X-ray diffraction. The lowest energy Mn2(CS)2(CO)6 structures are predicted have a single four-electron donor bridging η2-μ-CS group and a formal MnMn double bond. However, at only slightly higher energies, Mn2(CS)2(CO)6 structures are found with two η2-μ-CS groups and a formal Mn–Mn single bond. A formal Mn![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Mn triple bond of length 2.36 ± 0.03 Å is found in an even higher energy unbridged Mn2(CS)2(CO)6 structure, similar to the lowest energy Mn2(CO)8 structure found in a previous theoretical study. The lowest energy structures for Mn2(CS)2(CO)5 have two η2-μ-CS groups and a formal MnMn double bond of length 2.57 ± 0.03 Å.
Mn triple bond of length 2.36 ± 0.03 Å is found in an even higher energy unbridged Mn2(CS)2(CO)6 structure, similar to the lowest energy Mn2(CO)8 structure found in a previous theoretical study. The lowest energy structures for Mn2(CS)2(CO)5 have two η2-μ-CS groups and a formal MnMn double bond of length 2.57 ± 0.03 Å.
1. Introduction
Manganese carbonyl chemistry is of interest since the first example of a stable compound containing a four-electron donor bridging carbonyl group was the manganese carbonyl derivative (Ph2PCH2PPh2)2Mn2(CO)4(η2-μ-CO) (Fig. 1), synthesized in 1975 by heating Mn2(CO)10 with two molar equivalents of the small bite chelating diphosphine Ph2PCH2PPh2 in boiling decane.1 Determination of the structure of this manganese complex by X-ray diffraction2 indicated a very unusual bridging CO group with an abnormally short Mn–O distance of 2.29 Å. This suggests some direct manganese–oxygen bonding as well as the usual manganese–carbon bonding to both manganese atoms that is expected for a bridging carbonyl group. The Mn–Mn distance of 2.934 Å in this diphosphine complex is very similar to the unbridged Mn–Mn single bond distance of 2.895 Å in the parent Mn2(CO)10, determined by X-ray diffraction (Fig. 1).3 This suggests a metal–metal single bond in (Ph2PCH2PPh2)2Mn2(CO)4(η2-μ-CO). Therefore, its anomalous bridging CO group needs to be a formal four-electron donor to provide each manganese atom with the favored 18-electron configuration. Such donation of four electrons can occur through one σ-bond from the carbonyl carbon and one π-bond from the CO bond of the anomalous carbonyl group. The π-donation lowers the effective carbon–oxygen bond order, consistent with the very low bridging infrared ν(CO) frequency of 1645 cm−1 found experimentally for (Ph2PCH2PPh2)2Mn2(CO)4(η2-μ-CO).
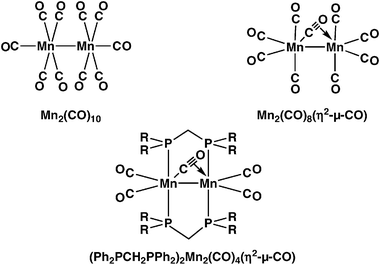 | ||
| Fig. 1 Structures of some manganese carbonyl derivatives. | ||
The above (Ph2PCH2PPh2)2Mn2(CO)4(η2-μ-CO) complex may be regarded as a substitution product of Mn2(CO)9 in which four of the carbonyl groups are replaced by two bidentate phosphine ligands. The parent Mn2(CO)9 is not stable under ambient conditions but has been observed as a photolysis product of Mn2(CO)10 in frozen alkane matrices at 100 K.4 An Mn2(CO)8(η2-μ-CO) structure for Mn2(CO)9 is suggested by the very low ν(CO) frequency of 1760 cm−1 found experimentally in these matrices (Fig. 1). In addition, density functional theory (DFT) studies5 on Mn2(CO)9 indicate the lowest energy structure to be Mn2(CO)8(η2-μ-CO) containing a four-electron donor bridging carbonyl group with a predicted ν(CO) frequency of 1767 cm−1 (BP86), very close to the experimental value.
Despite the occurrence of an unusual four-electron donor η2-μ-CO group in Mn2(CO)9, the limited experimental6 and more extensive theoretical work5 on the more highly unsaturated binuclear manganese carbonyls Mn2(CO)8 and Mn2(CO)7 do not find four-electron donor η2-μ-CO groups in their lowest energy structures. Thus, the lowest energy structure for Mn2(CO)8 is predicted by DFT to have only terminal carbonyl groups and a very short MnMn distance of ∼2.3 Å, suggesting the formal triple bond required to give both manganese atoms the favored 18-electron configuration.
Recent DFT studies7 of Fe2(CS)2(CO)n predicted the thiocarbonyl group to behave quite differently from the carbonyl group in binuclear derivatives, particularly when the thiocarbonyl group bridges two metal atoms in the unsaturated derivatives Fe2(CS)2(CO)n (n = 6, 5, 4). More specifically, four-electron donor thiocarbonyl groups bonded as η2-μ-CS groups through both the sulfur and carbon atoms are frequently preferred energetically over metal–metal multiple bonding. These results suggest that the preferred structures for similar unsaturated binuclear manganese carbonyl thiocarbonyls Mn2(CS)2(CO)n (n = 7, 6, 5) might contain four-electron donor η2-μ-CS groups rather than multiple manganese–manganese bonds. In addition, the relative energies of unbridged (OC)4(SC)Mn–Mn(CS)(CO)4 and bridged Mn2(CO)8(μ-CS)2 structures for the saturated Mn2(CS)2(CO)8 are of interest because of the greater tendency of thiocarbonyl groups to form two-electron bridging groups versus carbonyl groups.
Introduction of the thiocarbonyl ligand into transition metal complexes is often difficult, owing to the instability of carbon monosulfide at temperatures above −100 °C.8,9 Thus, reagents such as carbon disulfide (SCS) and thiophosgene (SCCl2) generally need to be used as sources of the thiocarbonyl ligand. This makes it particularly difficult to synthesize metal thiocarbonyls with several thiocarbonyl ligands. In the case of manganese, the complex Mn2(CS)(CO)9, containing only carbonyl and thiocarbonyl ligands, has been synthesized in low yield and in an impure condition by the reaction of KMn(CO)5 with thiophosgene.10 A manganese complex with a higher CS/Mn ratio, namely Mn(CS)(CO)4Br, has been isolated from the bromination of Mn2(CS)(CO)9/Mn2(CO)10 mixtures generated from the KMn(CO)5/SCCl2 reaction. Examples of more complicated manganese complexes containing a bridging thiocarbonyl group include Cp(CO)Fe(μ-CO)(μ-CS)Mn(CO)4 and Cp(R3P)M(μ-CO)(μ-CS)Mn(CO)Cp (Cp = η5-C5H5; M = Co, Rh).11,12
This paper reports theoretical studies of the binuclear manganese carbonyl thiocarbonyls Mn2(CS)2(CO)n (n = 8, 7, 6, 5). These include the saturated derivative Mn2(CS)2(CO)8, as well as unsaturated derivatives where n = 7, 6, and 5, requiring manganese–manganese double, triple, and quadruple bonds, respectively, to give both manganese atoms the favored 18-electron configurations if all carbonyl and thiocarbonyl groups are two-electron donors. Compounds with a 1 : 1 CS : Mn ratio were chosen for this study in order to allow for possible structures with two bridging thiocarbonyl groups. The parent saturated derivative Mn2(CS)2(CO)8 is currently unknown but is potentially accessible from the reduction of Mn(CS)(CO)4Br with a suitable reducing agent.
2. Theoretical methods
Electron correlation effects were considered using density functional theory (DFT) methods, which have evolved as a practical and effective computational tool, especially for organometallic compounds.13–27 Two DFT methods were used in this study. The first functional is the popular B3LYP method, which is the hybrid HF/DFT method combining the three-parameter Becke exchange functional (B3) with the Lee–Yang–Parr (LYP) generalized gradient correlation functional.28,29 The other DFT method used in the present paper is BP86, which links Becke’s 1988 exchange functional (B) with Perdew’s 1986 gradient corrected correlation functional method (P86).30,31 It has been noted elsewhere that the BP86 method may be somewhat more reliable than B3LYP for the types of organometallic systems considered in this paper.32–34Our DZP basis sets used for carbon, oxygen, and sulfur add one set of pure spherical harmonic d functions with orbital exponents αd(C) = 0.75, αd(O) = 0.85, and αd(S) = 0.70 to the standard Huzinaga–Dunning–Hay contracted DZ sets.35–37 The C and O basis sets are thus designated (9s5p1d/4s2p1d) and the S basis set is similarly described as (12s8p1d/6s4p1d). For Mn, in our loosely contracted DZP basis set, the Wachters primitive set38 was used, augmented by two sets of p functions and one set of d functions, contracted following Hood et al.,39 and designated as (14s11p6d/10s8p3d). For Mn2(CS)2(CO)8, Mn2(CS)2(CO)7, Mn2(CS)2(CO)6 and Mn2(CS)2(CO)5, there are 414, 384, 354 and 324 contracted Gaussian functions, respectively, with the chosen basis sets.
The geometries of all structures were fully optimized using the B3LYP/DZP and BP86/DZP methods. Vibrational frequencies were determined by evaluating analytically the second derivatives of the energy with respect to the nuclear coordinates. The corresponding infrared intensities were also evaluated analytically. All of the computations were carried out with the Gaussian 03 program,40 exercising the fine grid option (75 radial shells, 302 angular points) for evaluating integrals numerically,41 while the tight (10−8 Hartree) designation is the default for the self-consistent field (SCF) convergence.
In the search for minima using all currently implemented DFT methods, low magnitude imaginary vibrational frequencies are suspect because of significant limitations in the numerical integration procedures used in the DFT computations. Some such low magnitude imaginary frequencies arise from numerical errors, and some of them are genuine imaginary frequencies, indicating a transition state or saddle point. In order to resolve this problem, the structures with small imaginary frequencies were further optimized until no significant imaginary frequencies remain. Furthermore, these genuine minimum structures are compared with the precursor structures having small imaginary frequencies. If the two structures are very close in geometry and energy, the imaginary frequencies are considered to be artificial. Otherwise, they are regarded as significant so only the final optimized structures are reported herein.
The optimized structures are listed in Fig. 2–5 and Tables 1–8. A given Mn2(CS)2(CO)a structure is designated as a-b, where a is the number of CO groups, and b orders the structures according to their relative energies. Thus the lowest energy singlet structure of Mn2(CS)2(CO)8 is designated 8-1. In the text, energies and bond distances are listed as 0.7(1.0) kcal mol−1 corresponding to the results from the B3LYP (BP86) methods. The relative energies (ΔE) listed in the tables include the zero point energy corrections.
3. Results
3.1 The coordinately saturated Mn2(CS)2(CO)8 structures isoelectronic with Mn2(CO)10
Nine structures were fully optimized for Mn2(CS)2(CO)8 within 25 kcal mol−1 (Fig. 2 and Table 1). The eight lowest energy structures for Mn2(CS)2(CO)8 have two octahedrally coordinated manganese atoms connected only by a direct Mn–Mn interaction without any bridging CO or CS groups.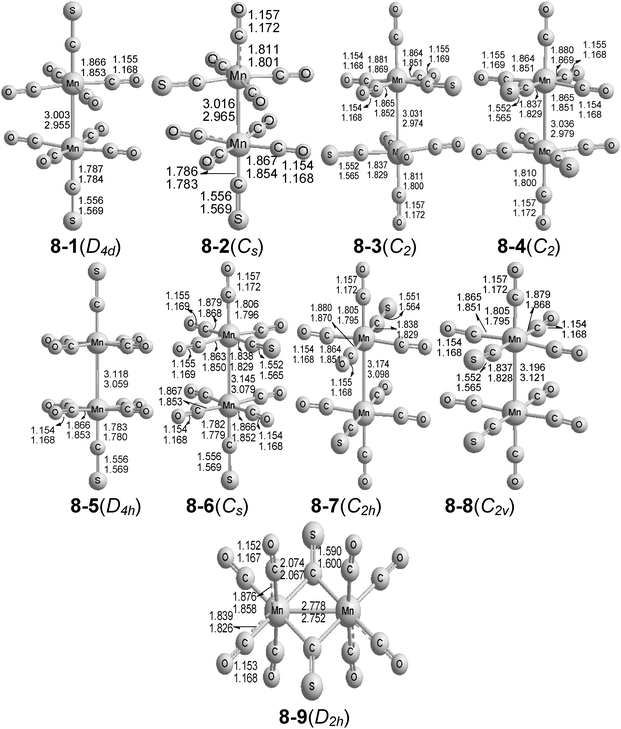 | ||
| Fig. 2 The nine optimized structures of Mn2(CS)2(CO)8. The upper distances were obtained by the B3LYP method and the lower distances by the BP86 method. | ||
| 8-1 (D4d) | 8-2 (Cs) | 8-3 (C2) | 8-4 (C2) | 8-5 (D4h) | 8-6 (Cs) | 8-7 (C2h) | 8-8 (C2v) | 8-9 (D2h) | ||
|---|---|---|---|---|---|---|---|---|---|---|
| B3LYP | −E | 4081.55562 | 4081.55456 | 4081.55341 | 4081.55296 | 4081.55040 | 4081.54857 | 4081.54673 | 4081.54627 | 4081.53354 |
| ZPE | 0.08118 | 0.08109 | 0.08112 | 0.08113 | 0.08147 | 0.08108 | 0.08135 | 0.08137 | 0.08092 | |
| ΔE | 0.0 | 0.6 | 1.3 | 1.6 | 3.5 | 4.4 | 5.7 | 6.0 | 13.7 | |
| N imag | 0 | 0 | 0 | 0 | 13i | 20i | 21i | 18i | 0 | |
| BP86 | −E | 4082.08379 | 4082.05771 | 4082.08055 | 4082.08026 | 4082.07856 | 4082.07636 | 4082.07427 | 4082.07385 | 4082.06621 |
| ZPE | 0.07941 | 0.07927 | 0.07931 | 0.07932 | 0.07973 | 0.07958 | 0.07956 | 0.07960 | 0.07917 | |
| ΔE | 0.0 | 0.9 | 2.0 | 2.2 | 3.5 | 4.8 | 6.1 | 6.4 | 10.9 | |
| N imag | 0 | 0 | 0 | 0 | 4i | 20i | 21i | 16i | 0 |
The global minimum of Mn2(CS)2(CO)8, namely the D4d symmetry structure 8-1, can be derived from Mn2(CO)10 by replacement of the two axial carbonyls with two thiocarbonyl groups. The Mn–Mn distance in 8-1 was predicted to be 3.003(2.955) Å, which is essentially the same as the previously predicted Mn–Mn distance of 3.007(2.954) Å for the lowest energy structure5 of Mn2(CO)10. For comparison, the Mn–Mn distance in Mn2(CO)10 was determined to be 2.98 Å by gas-phase electron diffraction,42 but only 2.895 Å by the most recent X-ray crystallography study.3 The Mn–C(S) bond length in 8-1 was predicted to be 1.787(1.784) Å. Structure 8-2, with an axial and an equatorial CS group, lies 0.6(0.9) kcal mol−1 above the global minimum of 8-1. The Mn–Mn distance was predicted to be 3.016(2.965) Å, which is ∼0.01 Å longer than that of 8-1. The next two Mn2(CS)2(CO)8 structures in terms of energy, namely 8-3 and 8-4 lying 1.5 ± 0.2 (2.1 ± 0.1) kcal mol−1 above 8-1, have an equatorial CS group on each manganese atom in transoid (8-3) or cisoid (8-4) relative positions. The Mn–Mn distances of 3.00 ± 0.03 Å in 8-3 and 8-4 are within 0.03 Å of that for the global minimum of 8-1. The equatorial Mn–C(S) distances of ∼1.83 Å were predicted to be ∼0.05 Å longer than the axial Mn–C(S) distances in 8-1, in accord with a stronger trans effect of a carbonyl group relative to an Mn–Mn bond. The four Mn2(CS)2(CO)8 structures 8-1 to 8-4 were found to be genuine minima, with all real vibrational frequencies.
The four eclipsed structures of Mn2(CS)2(CO)8 (8-5 to 8-8) were all predicted to have small imaginary vibrational frequencies corresponding to internal rotation around the Mn–Mn bond. Following the associated normal modes leads to the three corresponding staggered structures. Their Mn–Mn distances, generally ∼3.1 ± 0.05 Å, are slightly longer than that for 8-1, suggesting more steric hindrance between the eclipsed equatorial CO and/or CS groups in 8-5 to 8-8, relative to the staggered CO and CS groups in 8-1 to 8-4. The Mn–Mn single bonds in these structures, coupled with only two-electron donor CO and CS groups, gives both manganese atoms the favored 18-electron configuration. The closeness of the energies of the corresponding staggered and eclipsed structures (within ∼4 kcal mol−1) suggests that Mn2(CS)2(CO)8 is a fluxional molecule.
The doubly CS-bridged D2h structure 8-9 for Mn2(CS)2(CO)8 lying 13.7(10.9) kcal mol−1 in energy above the global minimum of 8-1 was found to be a genuine minimum without any imaginary vibrational frequencies. The predicted doubly bridged Mn–Mn single bond distance in 8-9 of 2.778(2.752) Å is ∼0.2 Å shorter than the unbridged Mn–Mn single bond distances in 8-1 to 8-8, in accordance with the expected bond-shortening effect of the two bridging groups.7 The computed Mn–C(S) and C–S distances of the bridging CS group in 8-9 were 2.074(2.067) and 1.590(1.600) Å, respectively. Attempts to optimize the doubly bridged Mn2(CS)2(CO)8 structures with one or two bridging CO groups led instead to one of the nine structures in Fig. 2.
The terminal CS groups in the Mn2(CS)2(CO)8 structures 8-1 to 8-8 were predicted to exhibit ν(CS) frequencies in the range 1320–1280 cm−1, similar to the terminal ν(CS) frequencies previously predicted for Fe(CS)(CO)n and Fe2(CS)2(CO)n derivatives.7 The bridging CS groups in the D2h structure 8-9 were predicted to exhibit significantly lower ν(CS) frequencies at 1165 and 1125 cm−1 (Table 2).
| ν(CO)/cm−1 | ν(CS)/cm−1 | |
|---|---|---|
| a Boldface means bridging CO or CS groups. | ||
| 8-1 (D4d) | 2073(0), 2013(175), 2003(0), 2003(0), 2001(2113), 2001(2113), 1970(0), 1970(0) | 1317(0), 1308(1758) |
| 8-2 (Cs) | 2069(225), 2020(765), 2006(49), 2001(1152), 2001(2023), 1985(453), 1977(110), 1969(18) | 1313(752), 1300(684) |
| 8-3 (C2) | 2064(106), 2024(1393), 2004(799), 2003(1451), 1990(222), 1985(156), 1980(825), 1971(85) | 1302(165), 1301(1057) |
| 8-4 (C2) | 2067(455), 2023(1245), 2003(778), 2001(1621), 1989(599), 1988(36), 1975(411), 1972(88) | 1316(859), 1289(228) |
| 8-5 (D4h) | 2074(0), 2016(0), 2014(286), 2004(2085), 2004(2085), 1993(0), 1970(0), 1970(0) | 1319(0), 1310(1753) |
| 8-6 (Cs) | 2069(228), 2021(771), 2012(553), 2004(2069), 1997(848), 1985(301), 1976(66), 1969(11) | 1315(736), 1302(706) |
| 8-7 (C2h) | 2064(0), 2023(1698), 2003(2075), 1998(0), 1995(664), 1986(0), 1980(604), 1969(0) | 1304(0), 1304(1251) |
| 8-8 (C2v) | 2069(440), 2022(1505), 2011(748), 2004(2074), 1989(43), 1986(290), 1976(287), 1970(0) | 1320(923), 1280(75) |
| 8-9 (D2h) | 2075(0), 2044(1422), 2010(0), 2004(2108), 2004(1105), 2001(455), 1999(0), 1975(0) | 1165(0), 1125(852) |
3.2 Coordinately unsaturated Mn2(CS)2(CO)n (n = 7, 6, 5) derivatives
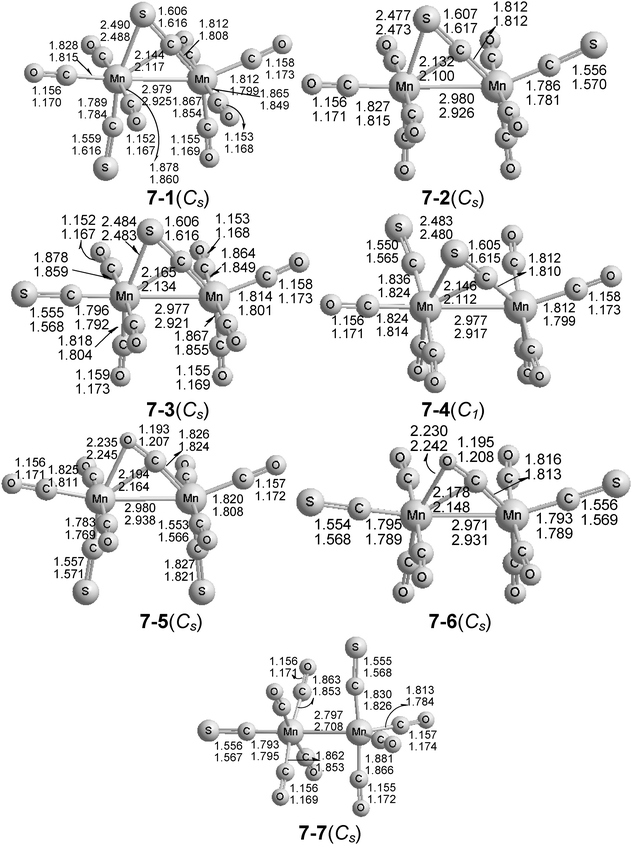 | ||
| Fig. 3 The seven optimized structures of Mn2(CS)2(CO)7. The upper distances were obtained by the B3LYP method and the lower distances by the BP86 method. | ||
| 7-1 (Cs) | 7-2 (Cs) | 7-3 (Cs) | 7-4 (C1) | 7-5 (Cs) | 7-6 (Cs) | 7-7 (Cs) | ||
|---|---|---|---|---|---|---|---|---|
| B3LYP | −E | 3968.20211 | 3968.20207 | 3968.20117 | 3968.19874 | 3968.17717 | 3968.17607 | 3968.16591 |
| ZPE | 0.07298 | 0.07293 | 0.07296 | 0.07295 | 0.07281 | 0.07272 | 0.07214 | |
| ΔE | 0.0 | 0.0 | 0.6 | 2.1 | 15.5 | 16.2 | 22.2 | |
| N imag | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| BP86 | −E | 3968.72488 | 3968.72492 | 3968.72385 | 3968.72197 | 3968.69985 | 3968.69898 | 3968.68563 |
| ZPE | 0.07144 | 0.07144 | 0.07147 | 0.07144 | 0.07124 | 0.07119 | 0.07061 | |
| ΔE | 0.0 | 0.0 | 0.7 | 1.8 | 15.6 | 16.1 | 24.1 | |
| N imag | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| ν(CO)/cm−1 | ν(CS)/cm−1 | |
|---|---|---|
| a Boldface means bridging CO or CS groups; italic bold implies four-electron donor CO or CS bridges. | ||
| 7-1 (Cs) | 2065(68), 2029(1133), 2001(2109), 1998(109), 1990(531), 1976(782), 1972(20) | 1300(515), 1176(320) |
| 7-2 (Cs) | 2066(348), 2027(644), 2004(741), 2002(2003), 1989(499), 1971(143), 1963(163) | 1313(759), 1171(394) |
| 7-3 (Cs) | 2067(420), 2026(493), 2002(597), 2001(2130), 1981(859), 1973(18), 1966(10) | 1311(802), 1180(355) |
| 7-4 (C1) | 2062(415), 2021(1515), 2002(694), 1994(1116), 1983(407), 1978(546), 1964(103) | 1297(595), 1178(334) |
| 7-5 (Cs) | 2059(160), 2025(1001), 2001(2117), 1991(209), 1983(776), 1971(24), 1774(311) | 1322(873), 1286(105) |
| 7-6 (Cs) | 2060(182), 2020(181), 2001(2138), 2000(841), 1973(30), 1970(76), 1766(386) | 1315(138), 1305(1531) |
| 7-7 (Cs) | 2050(374), 2005(937), 1997(181), 1982(1136), 1981(1556), 1962(176), 1957(25) | 1309(875), 1278(591) |
The four lowest energy structures of Mn2(CS)2(CO)7 (7-1 to 7-4 in Fig. 3) all have one four-electron donor bridging CS group, as indicated by short Mn–S distances of ∼2.48 Å and ν(CS) frequencies of 1175 ± 5 cm−1 (Table 4). These four Mn2(CS)2(CO)7 structures differ only in the location of the second CS group, which is a terminal CS group, predicted to exhibit a ν(CS) frequency at 1305 ± 8 cm−1. The Mn–Mn distances of ∼2.98 Å (B3LYP) or ∼2.92 Å (BP86) in each of these four structures correspond to the formal single bonds needed to give both manganese atoms the favored 18-electron configuration. The two lowest energy Mn2(CS)2(CO)7 structures, namely the Cs structures 7-1 and 7-2, have essentially the same energies despite the different location of the terminal CS group. Furthermore, the Mn2(CS)2(CO)7 structure 7-3 lies only 0.6 kcal mol−1 above 7-1/7-2. Even the highest energy of these four structures (7-4) was predicted to lie only ∼2.0 kcal mol−1 above the global minima of 7-1/7-2 by either method. The small energy difference between these four Mn2(CS)2(CO)7 structures again suggests a highly fluxional system. These Mn–Mn distances were reported to be about 2.980(2.920) Å, consistent with the singly bridged Mn–Mn single bond.
The next two Mn2(CS)2(CO)7 structures, 7-5 and 7-6, were predicted to lie 15.8 ± 0.3 kcal mol−1 above 7-1 or 7-2. The Mn–O distances in these two structures were predicted to be ∼2.24 Å with either method, indicating a four-electron donor η2-μ-CO group. This η2-μ-CO group exhibits an unusually low ν(CO) frequency at 1770 ± 4 cm−1. The Mn–Mn distances in 7-5 and 7-6 were predicted to be ∼2.98(2.94) Å, again consistent with the formal single bond needed to give both manganese atoms the favored 18-electron configuration.
A Cs unbridged structure 7-7 was also predicted for Mn2(CS)2(CO)7, but at the relatively high energy of 22.2(24.1) kcal mol−1 above 7-1 or 7-2. The predicted MnMn distance in 7-7 of 2.797(2.708) Å, is ∼0.2 Å shorter than the formal Mn–Mn single bonds in the unbridged Mn2(CS)2(CO)8 structures (Fig. 2), as well as those in the Mn2(CS)2(CO)7 structures 7-1 to 7-6 with four-electron donor bridging CS or CO groups. This is consistent with the formal MnMn double bond in 7-7, needed to give each manganese atom the favored 18-electron configuration in the presence of only two-electron donor CO and CS groups.
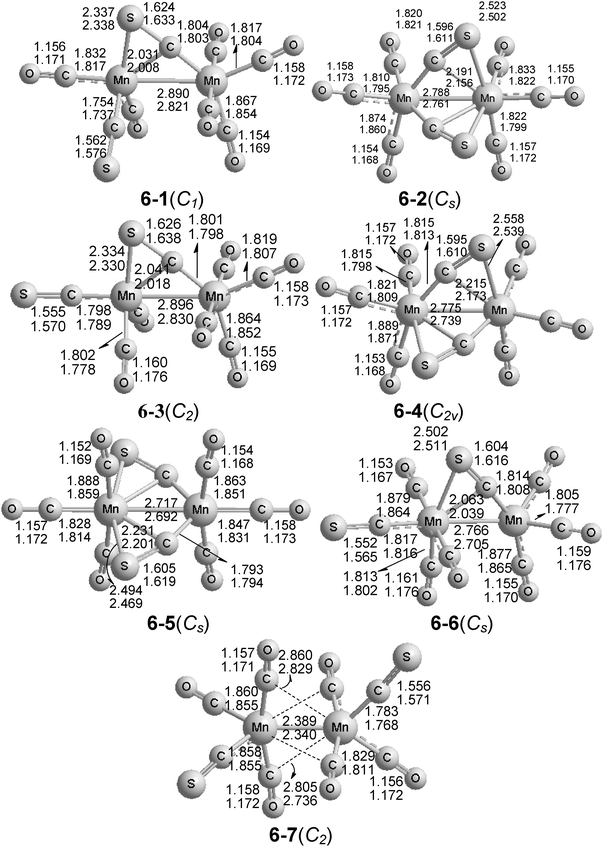 | ||
| Fig. 4 The seven optimized structures of Mn2(CS)2(CO)6. The upper distances were obtained by the B3LYP method and the lower distances by the BP86 method. | ||
| 6-1 (C1) | 6-2 (Cs) | 6-3 (C2) | 6-4 (C2v) | 6-5 (Cs) | 6-6 (Cs) | 6-7 (C2) | ||
|---|---|---|---|---|---|---|---|---|
| B3LYP | −E | 3854.83600 | 3854.83331 | 3854.83202 | 3854.82603 | 3854.81588 | 3854.81495 | 3854.79760 |
| ZPE | 0.06441 | 0.06419 | 0.06426 | 0.06414 | 0.06419 | 0.06377 | 0.06311 | |
| ΔE | 0.0 | 1.8 | 2.6 | 6.4 | 12.8 | 13.6 | 24.9 | |
| N imag | 0 | 0 | 1 (7i) | 0 | 0 | 1 (11i) | 0 | |
| BP86 | −E | 3855.35255 | 3855.35257 | 3855.34728 | 3855.34558 | 3855.33368 | 3855.32982 | 3855.31451 |
| ZPE | 0.06315 | 0.06308 | 0.06300 | 0.06288 | 0.06297 | 0.06256 | 0.06166 | |
| ΔE | 0.0 | 0.0 | 3.4 | 4.5 | 12.0 | 14.6 | 24.8 | |
| N imag | 0 | 0 | 1 (12i) | 0 | 0 | 0 | 0 |
| ν(CO)/cm−1 | ν(CS)/cm−1 | |
|---|---|---|
| a Boldface means bridging CO or CS groups; italic bold implies four-electron CS bridges. | ||
| 6-1 (C1) | 2060(419), 2007(709), 1997(1297), 1993(890), 1981(787), 1951(179) | 1305(515), 1150(316) |
| 6-2 (Cs) | 2047(496), 2010(1894), 1998(847), 1984(708), 1972(366), 1965(261) | 1201(360) , 1146(174) |
| 6-3 (Cs) | 2060(643), 2001(463), 1997(1518), 1985(1081), 1971(67), 1947(294) | 1301(754), 1140(344) |
| 6-4 (C2) | 2045(505), 2008(1760), 1990(1145), 1980(818), 1975(37), 1970(289) | 1181(408) , 1157(194) |
| 6-5 (C2v) | 2055(13), 2021(1092), 1996(1812), 1983(112), 1972(1145), 1958(384) | 1176(303) , 1125(349) |
| 6-6 (Cs) | 2046(212), 2008(1077), 1990(1451), 1975(1077), 1954(431), 1950(12) | 1306(923), 1171(309) |
| 6-7 (C2) | 2035(161), 1995(1322), 1981(874), 1975(235), 1962(922), 1960(559) | 1305(329), 1301(953) |
The Mn2(CS)2(CO)6 structure 6-6 at ∼14.1 ± 0.5 kcal mol−1 above the global minimum of 6-1 (Fig. 4) is closely related to the lowest energy Mn2(CS)2(CO)6 structures 7-1 and 7-2 by having a single four-electron donor bridging η2-μ-CS group, as indicated by a short Mn–S distance of 2.51 Å and a predicted ν(CS) frequency of 1171 cm−1. However, the Mn–S distance in 6-5 is ∼0.2 Å longer than that in 6-1 and 6-3, suggesting a weaker interaction. This may relate to the shorter (by 0.1 Å) MnMn bond distance of 2.766(2.705) Å in 6-5 relative to those in 6-1 and 6-3. This indicates a stronger manganese–manganese interaction corresponding to the formal double bond required to give both manganese atoms the 18-electron configuration.
Three Mn2(CS)2(CO)6 structures containing two four-electron donor bridging η2-μ-CS groups are found, namely 6-2, 6-4 and 6-5 (Fig. 4). Structure 6-2 with two S atoms directly interacting with the “right” manganese atom is calculated to lie 1.7(0.0) kcal mol−1 above 6-1. The two η2-μ-CS groups in 6-2 are characterized by Mn–S distances of 2.51 ± 0.01 Å, and ν(CS) frequencies of 1201 and 1146 cm−1. In the Mn2(CS)2(CO)6 structure 6-4, the Mn–S bonds involve different manganese atoms, whereas in structure 6-5, the Mn–S bonds both involve the same manganese atom. Structure 6-4 is predicted to be a genuine minimum without any imaginary vibrational frequencies and to lie 6.4(4.5) kcal mol−1 in energy above 6-1. The two η2-μ-CS groups in 6-4 are characterized by Mn–S distances of 2.55 ± 0.1 Å, and ν(CS) frequencies of 1181 and 1157 cm−1. The Mn2(CS)2(CO)6 structure 6-5 is predicted to lie 12.8(12.0) kcal mol−1 above 6-1, and also has no imaginary vibrational frequencies. The two η2-μ-CS groups in 6-5 are characterized by Mn–S distances of 2.48 ± 0.02 Å, and ν(CS) frequencies of 1176 and 1125 cm−1. The Mn–Mn distances in structures 6-2, 6-4 and 6-5 are predicted to fall in the range 2.74 ± 0.05 Å corresponding to the formal single bonds, which are shorter than usual because of the two η2-μ-CS bridges.
The unbridged structure 6-7 at 24.8 ± 0.1 kcal mol−1 above the global minimum of 6-1 is the sole Mn2(CS)2(CO)6 structure with only two-electron donor CS and CO groups (Fig. 4). The very short MnMn distance of 2.389(2.340) Å in 6-7 corresponds to the formal triple bond needed to give both manganese atoms the favored 18-electron configuration.
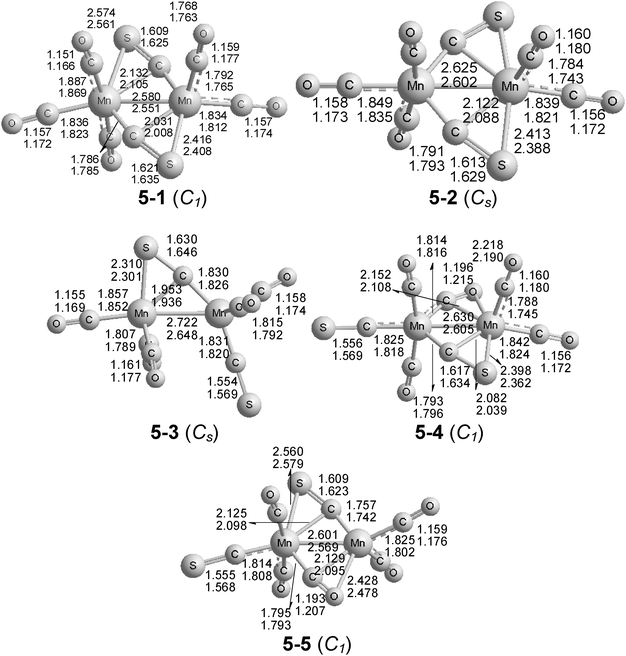 | ||
| Fig. 5 The five optimized structures of Mn2(CS)2(CO)5. The upper distances were obtained by the B3LYP method and the lower distances by the BP86 method. | ||
| 5-1 (C1) | 5-2 (Cs) | 5-3 (C1) | 5-4 (C1) | 5-5 (C1) | ||
|---|---|---|---|---|---|---|
| B3LYP | −E | 3741.46025 | 3741.45400 | 3741.44664 | 3741.42751 | 3741.42609 |
| ZPE | 0.05589 | 0.05579 | 0.05527 | 0.05548 | 0.05525 | |
| ΔE | 0.0 | 3.9 | 8.2 | 20.3 | 21.0 | |
| N imag | 0 | 0 | 0 | 0 | 0 | |
| BP86 | −E | 3741.97305 | 3741.96007 | 3741.93872 | 3741.93980 | 3741.93184 |
| ZPE | 0.05496 | 0.05475 | 0.05425 | 0.05439 | 0.05424 | |
| ΔE | 0.0 | 5.9 | 11.9 | 23.7 | 20.8 | |
| N imag | 0 | 0 | 0 | 0 | 0 |
| ν(CO)/cm−1 | ν(CS)/cm−1 | |
|---|---|---|
| a Boldface means bridging CO or CS groups; italic bold implies four-electron CS bridges. | ||
| 5-1 (C1) | 2047(219), 1992(1120), 1991(970), 1980(1440), 1954(410) | 1168(376) , 1124(125) |
| 5-2 (Cs) | 2045(317), 1997(917), 1993(1448), 1973(961), 1943(409) | 1158(296) , 1098(268) |
| 5-3 (C1) | 2016(175), 1994(1962), 1964(882), 1959(485), 1939(364) | 1281(430), 1103(417) |
| 5-4 (C1) | 2038(72), 1997(1259), 1993(1281), 1942(453), 1717(297) | 1300(796), 1121(410) |
| 5-5 (C1) | 2037(21), 1992(1363), 1984(1314), 1945(536), 1782(384) | 1303(892), 1182(285) |
The two lowest energy Mn2(CS)2(CO)5 structures, namely 5-1 and 5-2 (Fig. 5), both have two four-electron donor bridging η2-μ-CS groups. In the global minimum 5-1, the Mn–S bonds to these η2-μ-CS groups involve different manganese atoms and are of different lengths, namely 2.416(2.408) Å for the shorter Mn–S bond to the “right” manganese atom in Fig. 5, and 2.574(2.561) Å for the longer Mn–S bond to the “left” manganese atom in Fig. 5. Structure 5-2 for Mn2(CS)2(CO)5 lies at 3.9(5.9) kcal mol−1 in energy above 5-1. In 5-2, the Mn–S bonds are equivalent, of length 2.413(2.388) Å to the same manganese atom, namely the “right” manganese atom in Fig. 5. The MnMn bonds of 2.580(2.551) Å for 5-1 and 2.625(2.602) Å are consistent with the formal double bonds required to give both manganese atoms the favored 18-electron configuration in an Mn2(CS)2(CO)5 structure with two four-electron donor bridging CS groups.
The next Mn2(CS)2(CO)5 structure 5-3 (Fig. 5) lies 8.2(11.9) kcal mol−1 above the global minimum of 5-1. Unlike the other four Mn2(CS)2(CO)5 structures, the structure 5-3 has only one bridging group, namely a four-electron donor η2-μ-CS group with an Mn–S distance of 2.310(2.301) Å and a ν(CS) frequency of 1103 cm−1 (Table 8). The Mn–Mn distance of 2.722(2.648) Å is probably best interpreted as a formal single bond, thereby giving both manganese atoms 16-electron configurations.
The remaining two Mn2(CS)2(CO)5 structures lie >20 kcal mol−1 above the global minimum 5-1. Structures 5-4 and 5-5 are closely related to 5-2 and 5-1, respectively, except one of the bridging groups is a four-electron donor carbonyl group rather than a thiocarbonyl group. In 5-4, the Mn–O distance to the η2-μ-CO group is 2.218(2.190) Å and the corresponding ν(CO) frequency is very low at 1717 cm−1 (Table 8). In 5-5, at 21.0(20.8) kcal mol−1 above 5-1, the Mn–O distance to this η2-μ-CO group is 2.428(2.478) Å, and this group exhibits a low ν(CO) frequency at 1782 cm−1 (Table 8). The MnMn distances in these structures, namely 2.630 Å(2.605) for 5-4 and 2.601(2.569) Å for 5-5, are very similar to those in the doubly CS-bridged structures 5-1 and 5-2, and can correspond to the formal double bonds required to give each manganese atom the favored 18-electron configuration.
3.3 Dissociation energies
Table 9 reports the bond dissociation energies (BDEs) in terms of the single carbonyl dissociation steps:| Mn2(CS)2(CO)n → Mn2(CS)2(CO)n−1 + CO (n = 8, 7, 6) | (1) |
| B3LYP | BP86 | |
|---|---|---|
| Mn2(CS)2(CO)8 → Mn2(CS)2(CO)7 + CO | 15.6 | 19.9 |
| Mn2(CS)2(CO)7 → Mn2(CS)2(CO)6 + CO | 23.5 | 28.3 |
| Mn2(CS)2(CO)6 → Mn2(CS)2(CO)5 + CO | 29.6 | 32.2 |
4. Discussion
4.1 Comparison of Mn2(CS)2(CO)n and Mn2(CO)n+2 structures
Previous studies5 have shown that for Mn2(CO)10, the known D4d staggered unbridged global minimum structure, as well as the closely related D4h eclipsed structure, lie far below the doubly bridged structure of Mn2(CO)8(μ-CO)2 in terms of relative energy. Thus, the energy difference between the Mn2(CO)10 global minimum and the lowest energy doubly bridged structure Mn2(CO)8(μ-CO)2 was found to be 21.0 (B3LYP) or 14.8 kcal mol−1 (BP86).5 A similar situation was found for the thiocarbonyls Mn2(CS)2(CO)8, although the energy differences are somewhat smaller. Thus, the doubly bridged structure Mn2(CO)8(μ-CS)2 (8-9 in Fig. 2) lies 13.7 (B3LYP) or 10.9 kcal mol−1 (BP86) above the Mn2(CS)2(CO)8 global minimum of 8-1. The latter, like the global minimum of Mn2(CO)10, also has a staggered arrangement of the equatorial groups. The strong preference for unbridged structures of both Mn2(CO)10 and Mn2(CS)2(CO)8 over doubly bridged structures relates to the fact that the unbridged structures have the favorable octahedral six-coordination of the manganese atoms, whereas the doubly bridged structures have seven-coordinate manganese atoms.A general feature of the structures of the binuclear unsaturated Mn2(CS)2(CO)n (n = 7, 6, 5) derivatives is the absence of two-electron donor bridging CS and/or CO groups in the lowest energy predicted structures. However, four-electron donor bridging η2-μ-CS and η2-μ-CO groups were found in most of the low energy structures. Generally, the structures with such η2-μ-CS groups have lower energies than similar structures with η2-μ-CO groups.
The lowest energy structure predicted for Mn2(CO)9 is an Mn2(CO)8(η2-μ-CO) structure with a four-electron donor bridging CO group and a formal Mn–Mn single bond.5 Such a structure is closely related to the stable small bite chelating ditertiary phosphine complex (Ph2PCH2PPh2)2Mn2(CO)4(η2-μ-CO), which has been synthesized1 and structurally characterized by X-ray crystallography.2
The four lowest energy structures for Mn2(CS)2(CO)7 (7-1 to 7-4 in Fig. 3) all have a single four-electron donor η2-μ-CS group, and differ only in the position of the terminal CS group relative to the central Mn2(η2-μ-CS) system. Their relative energies fall within the narrow range of ∼2 kcal mol−1. The next two structures for Mn2(CS)2(CO)7 (7-5 and 7-6 in Fig. 3) both have a single four-electron donor η2-μ-CO group and lie in the narrow energy range of 16 ± 1 kcal mol−1 above the Mn2(CS)2(CO)7 global minimum of 7-1. All six of these Mn2(CS)2(CO)7 structures can be considered as analogues of the lowest energy Mn2(CO)9 structure with a four-electron donor η2-μ-CO group. The predicted formal Mn–Mn single bond distance in this Mn2(CO)9 [Mn2(CO)8(η2-μ-CO)] structure of 2.94 ± 0.03 Å is very close to the predicted Mn–Mn distances of 2.95 ± 0.03 Å in all six of the Mn2(CS)2(CO)5(η2-μ-CO)2 and Mn2(CO)7(η2-μ-CS)2 structures for Mn2(CS)2(CO)7 (7-1 to 7-6 in Fig. 3).
The next higher energy structure for Mn2(CO)9 is an unbridged structure lying 5.8(7.2) kcal mol−1 above the global minimum.5 An analogous Mn2(CS)2(CO)7 structure 7-7 (Fig. 3) was found, but at the much higher relative energy of 22.2(24.1) kcal mol−1 above 7-1. The MnMn formal double bond distance of 2.75 ± 0.05 Å predicted for the Mn2(CS)2(CO)7 structure 7-7 is very close to the 2.74 ± 0.04 Å MnMn distance predicted for the lowest energy unbridged Mn2(CO)9 structure.
In an earlier study5 of the more highly unsaturated Mn2(CO)8 derivatives, no structures were found containing four-electron donor bridging η2-μ-CO groups. By contrast, the six lowest energy structures for Mn2(CS)2(CO)6, namely 6-1 to 6-6 in Fig. 4, all contain at least one four-electron donor η2-μ-CE groups (E = S, O), and thus have no counterparts in the corresponding Mn2(CO)8 structures. However, the relatively high energy unbridged Mn2(CS)2(CO)6 structure 6-7 (Fig. 4) is very similar to the interesting global minimum structure found for Mn2(CO)8, which consists of two Mn(CO)4 units linked by a very short MnMn formal triple bond. The predicted MnMn formal triple bond distance of 2.33 ± 0.03 Å in this Mn2(CO)8 structure is very similar to the predicted MnMn distance of 2.36 ± 0.03 Å in the Mn2(CS)2(CO)6 structure 6-7.
Re-examination of the Mn2(CO)7 structures5 indicates that one of the structures (7-3 in ref. 5) contains two four-electron bridging η2-μ-CO groups, as indicated by the unusually low ν(CO) frequencies of 1747 and 1718 cm−1. Structures 5-2 and 5-5 for Mn2(CS)2(CO)5 (Fig. 5) are closely related to this Mn2(CO)7 structure, but with one (5-4) or two (5-2) η2-μ-CS groups in place of one or two of the η2-μ-CO groups in the Mn2(CO)7 structure. The formal MnMn double bond length in this Mn2(CO)7 structure of 2.59 ± 0.02 Å is very similar to those of 2.59 ± 0.03 Å in the Mn2(CS)2(CO)5 structures 5-2 and 5-4. None of the other Mn2(CS)2(CO)5 structures in Fig. 5 has any counterpart in the Mn2(CO)7 structures found in the previous work.5
4.2 Manganese–manganese bond lengths
An earlier study on iron carbonyl thiocarbonyls7 related the lengths of Fe–Fe bonds to the number of bridging groups as well as the formal bond order. The Fe–Fe bond lengths were found to decrease, not only with increasing formal bond order, but also with increasing numbers of bridging groups. Similar observations can be made for the Mn–Mn distances in the binuclear Mn2(CS)2(CO)n derivatives discussed in this paper. Thus, for the structures with doubly bridged Mn–Mn single bonds, namely the high energy Mn2(CS)2(CO)8 structure 8-9 (Fig. 2) and the Mn2(CS)2(CO)6 structures 6-2, 6-4 and 6-5 (Fig. 4), the bond lengths fall in the range from 2.70 Å for 6-4 to 2.78 Å for 6-6. The Mn–Mn bonds in the singly bridged Mn2(CS)2(CO)7 structures 7-1 to 7-6 (Fig. 3) are significantly longer, in the narrow range of 2.95 ± 0.01 Å. The unbridged Mn–Mn bonds in the seven lowest energy Mn2(CS)2(CO)8 structures 8-1 to 8-8 are longer still, falling in the wider range of 2.98 Å for 8-1 to 3.16 Å for 8-8. Among these eight Mn2(CS)2(CO)8 structures, the three lowest energy structures with staggered arrangements of the equatorial CO and CS groups, namely 8-1 to 8-4 (Fig. 2), have shorter Mn–Mn distances in the narrow range of 2.98–3.01 Å. However, the next four Mn2(CS)2(CO)8 structures with eclipsed arrangements of the equatorial CO and CS groups, namely 8-5, to 8-8 (Fig. 2), have slightly longer Mn–Mn distances in the range of 3.08–3.20 Å.Similar relationships between the number of bridging groups and the MnMn bond distances were also observed for the Mn2(CS)2(CO)n derivatives with formal double bonds. Thus, the three doubly bridged Mn2(CS)2(CO)5 structures 5-1, 5-2 and 5-4 (Fig. 5) have MnMn bond lengths in the range of 2.55–2.61 Å, which is significantly shorter than the 2.70–2.78 Å range for the doubly bridged Mn–Mn single bonds noted above. The singly bridged Mn2(CS)2(CO)6 structures 6-1, 6-2 and 6-5 exhibit longer MnMn distances in the range of 2.73 Å for 6-5 to 2.86 Å for 6-1 and 6-2. In this case, however, the unbridged Mn2(CS)2(CO)7 structure 7-7 has an MnMn double bond length of 2.75 Å, at the lower end of the range of the MnMn double bond lengths in the singly bridged structures.
The only Mn2(CS)2(CO)n structure with a formal MnMn triple bond found in this research is the unbridged Mn2(CS)2(CO)6 structure 6-7 (Fig. 4). This structure has an MnMn distance of 2.36 Å, which is ∼0.2 Å shorter than the shortest MnMn formal double bond reported here.
Acknowledgements
We are indebted to the National Natural Science Foundation (20873045) of China as well as the U. S. National Science Foundation (Grants CHE-0749868 and CHE-0716718) for support of this research.References
- R. Colton and C. J. Commons, Aust. J. Chem., 1975, 28, 1673 CAS.
- C. J. Commons and B. F. Hoskins, Aust. J. Chem., 1975, 28, 1663 CAS.
- M. Martin, B. Rees and A. Mitschler, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1982, 38, 6 CrossRef.
- A. F. Hepp and M. S. Wrighton, J. Am. Chem. Soc., 1983, 105, 5934 CrossRef CAS.
- Y. Xie, J. H. Jang, R. B. King and H. F. Schaefer, Inorg. Chem., 2003, 42, 5219 CrossRef CAS.
- F. A. Kvietok and B. E. Bursten, Organometallics, 1995, 14, 2395 CrossRef CAS.
- Z. Zhang, Q.-S. Li, Y. Xie, R. B. King and H. F. Schaefer, Inorg. Chem., 2009, 48, 1974 CrossRef CAS.
- M. A. P. Hogg and J. E. Spice, J. Chem. Soc., 1958, 4196 Search PubMed.
- R. Steudel, Z. Anorg. Allg. Chem., 1968, 361, 180 CrossRef CAS.
- W. Petz and D. Rehder, Organometallics, 1990, 9, 856 CrossRef CAS.
- H. Werner, O. Kolb and P. Thometzek, J. Organomet. Chem., 1985, 24, 2209.
- V. G. Albano, M. Monari, L. Busetto, L. Carlucci and V. Zanotti, Gazz. Chim. Ital., 1992, 122, 201 CAS.
- A. W. Ehlers and G. Frenking, J. Am. Chem. Soc., 1994, 116, 1514 CrossRef CAS.
- B. Delley, M. Wrinn and H. P. Lüthi, J. Chem. Phys., 1994, 100, 5785 CrossRef CAS.
- J. Li, G. Schreckenbach and T. Ziegler, J. Am. Chem. Soc., 1995, 117, 486 CrossRef CAS.
- V. Jonas and W. Thiel, J. Chem. Phys., 1995, 102, 8474 CrossRef CAS.
- T. A. Barckholtz and B. E. Bursten, J. Am. Chem. Soc., 1998, 120, 1926 CrossRef CAS.
- S. Niu and M. B. Hall, Chem. Rev., 2000, 100, 353 CrossRef CAS.
- P. Macchi and A. Sironi, Coord. Chem. Rev., 2003, 238–239, 383 CrossRef CAS.
- M. Bühl and H. Kabrede, J. Chem. Theory Comput., 2006, 2, 1282 CrossRef.
- P. Mörschel, J. Janikowski, G. Hilt and G. Frenking, J. Am. Chem. Soc., 2008, 130, 8952 CrossRef.
- T. Ziegler and J. Autschbach, Chem. Rev., 2005, 105, 2695 CrossRef CAS.
- M. P. Waller, M. Bühl, K. R. Geethanakshmi, D. Wang and W. Thiel, Chem.–Eur. J., 2007, 13, 4723 CrossRef CAS.
- P. G. Hayes, C. Beddie, M. B. Hall, R. Waterman and T. D. Tilley, J. Am. Chem. Soc., 2006, 128, 428 CrossRef CAS.
- M. Bühl, C. Reimann, D. A. Pantazis, T. Bredow and F. Neese, J. Chem. Theory Comput., 2008, 4, 1449 CrossRef CAS.
- M. Besora, J.-L. Carreon-Macedo, J. Cowan, M. W. George, J. N. Harvey, P. Portius, K. L. Ronayne, X.-Z. Sun and M. Towrie, J. Am. Chem. Soc., 2009, 131, 3583 CrossRef CAS.
- S. Ye, T. Tuttle, E. Bill, L. Simkhorich, Z. Gross, W. Thiel and F. Neese, Chem.–Eur. J., 2008, 14, 10839 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098 CrossRef CAS.
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822 CrossRef.
- See especially: F. Furche and J. P. Perdew, J. Chem. Phys., 2006, 124, 044103 Search PubMed.
- H. Y. Wang, Y. Xie, R. B. King and H. F. Schaefer, J. Am. Chem. Soc., 2005, 127, 11646 CrossRef CAS.
- H. Y. Wang, Y. Xie, R. B. King and H. F. Schaefer, J. Am. Chem. Soc., 2006, 128, 11376 CrossRef CAS.
- T. H. Dunning, J. Chem. Phys., 1970, 53, 2823 CrossRef CAS.
- T. H. Dunning and P. J. Hay, Methods of Electronic Structure Theory, ed. H. F. Schaefer, Plenum, New York, 1977, pp. 1–27 Search PubMed.
- S. Huzinaga, J. Chem. Phys., 1965, 42, 1293 CrossRef.
- A. J. H. Wachters, J. Chem. Phys., 1970, 52, 1033 CrossRef CAS.
- D. M. Hood, R. M. Pitzer and H. F. Schaefer, J. Chem. Phys., 1979, 71, 705 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. G. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, GAUSSIAN 03 (Revision C.2), Gaussian, Inc., Wallingford, CT, 2004 Search PubMed.
- B. N. Papas and H. F. Schaefer, THEOCHEM, 2006, 768, 175 CrossRef CAS.
- A. Almenningen, G. G. Jacobsen and H. M. Seip, Acta Chem. Scand., 1969, 23, 685 CrossRef CAS.
- L. S. Sunderlin, D. Wang and R. R. Squires, J. Am. Chem. Soc., 1993, 115, 12060 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Tables S1–S4: theoretical harmonic vibrational frequencies for Mn2(CS)2(CO)8 (9 structures), Mn2(CS)2(CO)7 (7 structures), Mn2(CS)2(CO)6 (7 structures) and Mn2(CS)2(CO)5 (5 structures) using the BP86 method; Tables S5–S31: theoretical Cartesian coordinates for Mn2(CS)2(CO)8 (8 structures), Mn2(CS)2(CO)7 (7 structures), Mn2(CS)2(CO)6 (7 structures) and Mn2(CS)2(CO)5 (5 structures) using the B3LYP method; complete Gaussian 03 reference (ref. 40). See DOI: 10.1039/b9nj00340a |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2010 |