Water-soluble tetrapodal N,O ligands incorporating soft N-heterocycles for the selective complexation of Am(III) over Ln(III)
Marie Heitzmanna, Christelle Gateaua, Laurence Chareyreb, Manuel Miguirditchianb, Marie-Christine Charbonnelb and Pascale Delangle*a
aCEA, Inac, Service de Chimie Inorganique et Biologique (UMR_E 3 CEA UJF, FRE CNRS 3200), F-38054 Grenoble, France. E-mail: pascale.delangle@cea.fr; Fax: +33 4 3878 5090; Tel: +33 4 3878 9822
bCEA, DEN, DRCP, SCPS, F-30207, Bagnols-sur-Cèze, France
First published on 15th October 2009
Abstract
A series of four water-soluble N,O-tetrapodal ligands derived from ethylenediamine, bearing hard acetate groups and soft N-heterocycles, either pyridine or pyrazine, was developed to study the impact of the softness of N-donors on the complexation properties with trivalent f ions. Two novel ligands of enhanced soft character, bearing three pyridines (L3py) or three pyrazines (L3pz), were synthesized and the related lanthanide complexes were studied in solution. The ligand containing three pyridylmethyl moieties L3py gives complexes with a coordination similar to EDTA, i.e. a hexadentate coordination mode as indicated by NMR and luminescence decays (q = 3) and stability constants in the range log β110 = 6.99–9.3 (La–Lu). On the other hand, the softest molecule L3pz forms much less stable complexes with log β110 = 4.0–4.4 (La–Eu). The selective back-extraction of Am(III) from organic solutions containing 4f and 5f elements was tested with the four water-soluble complexing agents. The ligand L3pz demonstrates poor stripping ability and selectivity. In contrast, the three ligands Lpy, Lpz and L3py give interesting back-extraction results with Eu/Am separation factors ranging from 36 to 46, which are significantly higher than with HEDTA. This exemplifies the role of the N-heterocycle softness in enhancing the separation between Am(III) and Eu(III). Interestingly, the pyrazine-based ligand, Lpz, demonstrates the best stripping properties, with a distribution factor that approaches that of HEDTA in the same conditions (DAm ≈ 0.3). This molecule is a good compromise between softness and hardness and forms complexes still stable at pH 3 due to its low basicity.
Introduction
The separation of trivalent actinides (An(III)) from trivalent lanthanides (Ln(III)) is a key step in the partitioning and transmutation strategy, which is one of the scenario being seriously considered for the future management of nuclear waste. The aim is to separate long-lived α-emitting minor actinides from spent nuclear fuel and to transmute them by nuclear fission into shorter-lived isotopes. Indeed, the transmutation of the minor actinides will only be possible after separation from the abundant fission products lanthanides having large neutron capture cross sections.1,2This separation is one of the most challenging issues owing to the very similar physicochemical properties of An(III) and Ln(III). Indeed, lanthanides and transplutonium actinides both exist predominantly in their trivalent oxidation state in solution. These cations are hard acids in the Pearson classification (HSAB for hard and soft acids and bases)3 with close ionic radii (e.g. 1.066 for Eu(III) and 1.090 for Am(III), CN = 8).4 Their interactions with inorganic and organic ligands are therefore predominantly determined by electrostatic and steric factors. Even if both An(III) and Ln(III) are considered to be hard acids in HSAB theory, the higher spatial expansion of the 5f actinide orbitals with respect to the 4f lanthanide orbitals opens possibilities to discriminate them through their relative hardness. Therefore, numerous extractants containing nitrogen5 or sulfur6 functionalities which are softer than oxygen donors have been developed to favor An(III) over Ln(III) complexes. Several of these soft molecules extract selectively Am(III) from Am(III)–Ln(III) mixtures.7
Among heterocyclic N-donors, pyridine and pyrazine significantly differ in their soft character and have been inserted in tripodal8,9 or tetrapodal10,11 chemical architectures to test the effect of the softness of the ligand on its extraction ability and selectivity. We have recently reported12 two tetrapodal ligands derived from ethylenediamine bearing both hard and soft donors, Lpy and Lpz (see Scheme 1). These ligands bear (i) two hard acetate groups to provide stability to the trivalent f-element complexes and (ii) two N-heterocyclic soft groups to provide Am(III) versus Eu(III) selectivity. The complexation with Am(III) and Ln(III) is efficient in water: for instance log β110(Eu) = 11.6 (Lpy) and 9.4 (Lpz). Moreover, they show significant selectivities for Am(III) over Eu(III): Kselectivity(Am/Eu) = β110(Am)/β110(Eu) = 60 (Lpy) and 500 (Lpz). These ligands only differ in their N-donor moieties and the related complexes have the same structures in water. Therefore the difference in the stability constants is correlated to the softness of the N-heterocycle and the results indicate an enhancement of the selectivity when the two pyridine groups are replaced by two pyrazine groups.
 | ||
| Scheme 1 Design of the ligands. | ||
In this paper, the synthesis and lanthanide complexation properties of two novel ligands with increased soft character in comparison to Lpy and Lpz are presented. In order to increase the soft character of the latter ligands, one of their acetate arms was replaced by one methylpyridyl group in Lpy or one methylpyrazinyl group in Lpz to obtain the two novel complexing molecules L3py and L3pz (Scheme 1). The four N,O tetrapodal ligands presented in Scheme 1 afford a series of hydrophilic N,O ligands with a range of soft character, which allows us to evaluate the impact of the softness on the complexation of f-elements. Furthermore, they are good candidates for the selective back-extraction of Am(III) from organic solutions containing 4f and 5f elements, as they efficiently complex trivalent f cations in aqueous solution. Back-extraction experiments are presented and show that, whereas the three ligands Lpy, Lpz and L3py are able to selectively strip Am(III) from the organic phase, L3pz which bears three methylpyrazinyl moieties is probably too soft to efficiently complex f-elements in water at pH 3. Moreover it appears that Lpz is a good compromise between hardness and softness because it provides enough affinity for Am(III) at pH 3 to significantly strip this ion from the organic phase and an interesting selectivity (SFAm/Eu = 40–50).
Results and discussion
Synthesis of the N,O ligands
L3py and L3pz are softer analogues of Lpy and Lpz containing one acetate function and three pyridyl or pyrazinyl groups. These functions are introduced on an ethylenediamine bridge. The synthetic procedures are summarized in Schemes 2 and 3. The ligands L3py and L3pz were obtained from N,N-bis(2-pyridylmethyl)N′-acetylethylenediamine and N,N-bis(2-pyrazinylmethyl)N′-acetylethylenediamine, respectively. The syntheses of these two precursors were described in a previous report for the synthesis of Lpy and Lpz, from N-acetylethylenediamine and 2-(chloromethyl)pyridine or 2-(chloromethyl)pyrazine.12 The key step in the synthesis of L3py and L3pz is the monosubstitution of the second aliphatic nitrogen atom, which was achieved by a reductive amination using 2-pyridine- or 2-pyrazinecarbaldehyde and sodium borohydride. Subsequent reaction with ethylchloroacetate in the presence of potassium carbonate followed by the deprotection of the ethyl ester groups gave the expected ligands L3py and L3pz in 19% and 9% yield, respectively. Cleavage of the ethyl ester groups was performed under acidic conditions for L3py and by saponification for L3pz. | ||
| Scheme 2 Synthesis of L3pyH. Reagents and conditions: (i) a. HCl (1 M), b. pyridine-2-carbaldehyde, NaBH4, CH3OH, 55%, (ii) ClCH2COOEt, K2CO3, CH3CN, 70%, (iii) HCl (2 M), H2O, 50%. | ||
 | ||
| Scheme 3 Synthesis of L3pzH. Reagents and conditions: (i) a. HCl (1 M), b. pyrazine-2-carbaldehyde, NaBH4, CH3OH, 27%, (ii) ClCH2COOEt, K2CO3, CH3CN, 49%, (iii) KOH, EtOH–H2O, 81%. | ||
L3py was obtained as the corresponding hydrochloride salt: L3pyH·4HCl·2H2O·EtOH (1 equivalent of ethanol was also detected in the proton NMR spectra). L3pz was obtained as the potassium salt: L3pzK·4.5H2O. The ligands L3py and L3pz were fully characterized by NMR, ESMS and elemental analysis. The proton and carbon NMR spectra of the two ligands in D2O indicate the presence of a unique species in which the two methylpyridine or methylpyrazine arms connected to the same nitrogen atom are equivalent.
Protonation of the ligands
All potentiometric studies were performed in KNO3 0.1 M at 298 K. The protonation constants of L3py and L3pz could be obtained from the titrations of the free ligands with KOH and HNO3. They are defined in eqn (1) and listed in Table 1, together with the values previously reported with their analogues Lpy and Lpz.12| Ki = [HiL]/[Hi−1L][H+] | (1) |
| L3py | Lpya | L3pz | Lpza | |
|---|---|---|---|---|
| a Ref. 12. | ||||
| logK1 | 8.18(7) | 9.60(6) | 7.18(9) | 9.35(8) |
| logK2 | 5.08(6) | 5.46(8) | 2.3(2) | 2.5(1) |
| logK3 | 3.23(9) | 3.4(1) | — | — |
| log β110 (La) | 6.99(9) | 9.86(6) | 4.0(3) | 8.30(7) |
| log β110 (Nd) | 8.1(1) | 11.46(1) | — | 8.90(2) |
| log β110 (Eu) | 8.76(2) | 11.62(4) | 4.4(3) | 9.37(9) |
| log β110 (Dy) | 8.74(6) | 11.89(2) | — | 9.6(1) |
| log β110 (Lu) | 9.3(1) | 13.05(6) | — | 9.77(1) |
| log β110 (Am) | — | 13.4(2) | — | 12.1(1) |
The titration of L3pyH·4HCl is indicative of three acidic sites. The first protonation (logK1 = 8.18) occurs at the most basic nitrogen, i.e. the aliphatic nitrogen atom adjacent to the acetate function. As expected, this logK value is lower than the corresponding value found for Lpy, because of the withdrawing effect of the methylpyridyl group attached to this nitrogen atom. The other two protonations give similar logK values to Lpy and can therefore be assigned to nitrogen atoms of pyridines (logK2 = 5.08 and logK3 = 3.23). Moreover, the logK value of the carboxylate group of the NCH2COO− fragment is expected to be lower than 2.7 like in EDTA or other polyaminocarboxylate ligands. As for Lpy, the second aliphatic nitrogen atom of L3py does not protonate in our experimental conditions (pH > 2.5).
The titration of L3pzH is indicative of only two acidic sites, as observed for LpzH2. The first protonation (logK1 = 7.18) also occurs at the aliphatic nitrogen atom adjacent to the acetate function and is significantly lowered in comparison to L3py, because the electron withdrawing effect of the methylpyrazinyl group is larger than the electron withdrawing effect of the methylpyridyl group. The second logK value can hardly be assigned to the second aliphatic nitrogen atom as in Lpz because the strong electron withdrawing effect of the methylpyrazinyl group directly attached to this nitrogen atom would significantly affect this value to give a much lower logK value than the one found for Lpz (logK2 = 2.5). Therefore, this second logK can be assigned to the protonation of the carboxylate function.
As expected, the ligands containing pyridines are much more basic than the ligands containing pyrazines. Indeed, the pyridine group is much more basic than the pyrazine group: the pKa of 2-methylpyridine is 5.96 whereas the pKa of 2-methylpyrazine is only 1.65.13 Moreover the withdrawing effect of methylpyridyl and methylpyrazinyl groups significantly decreases the basicity of the nitrogen atoms to which they are connected. Furthermore, as this effect is much larger with pyrazine than pyridine, the attachment of one methylpyridyl or one methylpyrazinyl group instead of one methylcarboxylate group decreases the pKa value of the most basic site, i.e. the tertiary amine nitrogen atom adjacent to the acetate function, by 1.4 and 2.2 units, respectively. Consequently the two ligands L3py and L3pz are less basic than their analogues with two N-donors Lpy and Lpz.
Potentiometric studies of the lanthanide complexes
The lanthanide complexes of L3py have been studied by potentiometric titration for five representative cations of the 4f series, namely La(III), Nd(III), Eu(III), Dy(III) and Lu(III).Typical titration curves are shown in Fig. 1. For every cation, these data could be fitted according to the formation of a unique metallic species, namely LnL2+ for pH values inferior to 7. The corresponding stability constants are given in Table 1. Above pH 7, the curves are characteristic of the formation of hydroxo complexes. Nevertheless, a hysteresis exists between the direct titration with KOH and the back titration with HNO3 in this high-pH region and prevented us from determining reliable thermodynamic constants for the hydrolysis reactions. The metal titrations were fitted in the pH-range 2.5–7 (no hysteresis) with the formation of LnL2+.
 | ||
| Fig. 1 Alkalimetric titrations of solutions containing 10−3 M L3pyH·4HCl with 0 or 1 equiv. Ln(NO3)3 in water and 0.1 M KNO3 at 298 K. (A) L3py, (B) LaL3py, (C) EuL3py, (D) LuL3py. | ||
Titrations conducted with the pyrazine-based ligand L3pz indicate the formation of complexes with low stability in comparison to L3py. Furthermore, precipitation occurs for pH > 6 with La and Eu. The titration curve with 1 Eu(III) equiv. is presented in Fig. 2 and could be fitted in the pH-range 2.7–6 (no precipitate) with the formation of the complex (EuL3pz)2+ with log β110 = 4.4. At pH 6, 45% of the complex is formed and 55% of the cation is still free. Therefore the stability constant is calculated with a lower number of points than for L3py, which explains the higher uncertainty associated with this particular value. Other lanthanide complexes of L3pz were not studied because of experimental difficulties due to the precipitation of the complexes.
 | ||
| Fig. 2 Alkalimetric titrations of solutions containing 10−3 M L3pzH + 2HNO3 with 0 or 1 equiv. Eu(NO3)3 in water and 0.1 M KNO3 at 298 K. (A) L3pz, (B) EuL3pz. | ||
The evolution of the stability constants as an inverse function of the cation ionic radii, related to the hardness of the ion, is represented in Fig. 3 for the four N,O ligands. As expected, the series of complexes show an increase of the complex stability when the ionic radius of the cation decreases, according to the well-known electrostatic trend. Indeed the net complexation reaction of a Ln(III) ion with a ligand L is the result of two successive steps corresponding to dehydration followed by the combination of the partially desolved partners. The compensation effect assumes that the free energy for the dehydration process is negligible at room temperature because ΔHdehyd ≈ TΔSdehyd.14 Therefore the global free energy of the complexation process is dominated by the enthalpy-driven combination step, leading to increased formation constants with increasing charge density (or hardness) on the cation.15 For the two pyridine-based ligands, the increase of the stability constants across the 4f series is common and of the same order of magnitude as EDTA: Lpy (log βLu − log βLa ≈ 3.2, 32%); L3py (log βLu − log βLa ≈ 2.3, 33%). The electrostatic trend is less marked for the pyrazine-based ligands, Lpz (log βLu − log βLa ≈ 1.5, 18%) and L3pz (log βEu − log βLa ≈ 0.4, 10%). This tendency can be attributed to their softer character which produces smaller electrostatic interactions and therefore a smoother electrostatic evolution of the stability constants across the 4f series.
 | ||
| Fig. 3 Evolution of the stability constants, log β110 of ML complexes as an inverse function of the cation ionic radii. | ||
As expected the two novel ligands, L3py and L3pz, bearing three N-donor sites and only one O-donor acetate group show lower affinities for the hard lanthanide cations than their analogues with two acetate moieties. The replacement of one acetate arm in Lpy by one methylpyridyl group induces a loss of 2.9 log units in the stability constants, whereas the replacement of one acetate arm in Lpz by one methylpyrazinyl group has an even more dramatic effect, loss of 5 log units in the stability constants.
The three ligands Lpy, Lpz and L3py demonstrate significant affinities for Ln(III) in water (108–1011 for Eu(III)) whereas the softest ligand L3pz forms lanthanide complexes of quite low affinity (104). This indicates that L3pz is too soft to efficiently compete with the water molecules, which have a large affinity for the hard lanthanide cations. For instance, ligands containing only pyrazine groups like tpza8 or tpzen11 have very low affinities for lanthanides preventing the study of their complexation properties in water, whereas their pyridine analogues have larger complexation efficiency even in water.8,16 L3pz has an intermediary behavior between these ligands bearing exclusively N-donors and the three other molecules presented in this paper.
Characterization of LnL3py complexes
The proton NMR spectrum of the diamagnetic complex LaL3py shows the presence of only one set of signals indicating the presence of one isomer without symmetry. The spectra of free L3py and its lanthanum complex, LaL3py, in D2O at pD 7 are presented in Fig. 4. They were assigned with classical 2D NMR experiments. The aromatic protons are slightly shifted with respect to the free ligand. The spectrum of the lanthanum complex evidences the loss of symmetry upon the ion complexation. In particular, the two methylpyridyl arms connected to the same nitrogen atom are no longer equivalent: for instance H6 gives two different doublets in the NMR spectrum of the complex. The methylene signals of protons H2, H8 and H9 are split into well-resolved AB patterns. Each methylene of the ethylenediamine bridge, H1 and H7, is also split into a doublet and a triplet (ABXY system with JAB ≈ JXY ≈ JAX ≈ 13 Hz and JAY ≈ JBY ≈ JBX ≈ 0). The formation of only one complex was confirmed by the spectra obtained either in excess of ligand or in excess of cation. Indeed, the same set of signals was obtained for the complex, in slow exchange with the ligand in default of cation. Exchange correlations between the signals of the protons (especially H6) of the two methylpyridine arms connected to the same nitrogen atom are detected in the EXSY spectrum (mixing time 100 ms). This feature can be explained by the successive steps: decomplexation of the cation followed by the interconversion of these two arms and new coordination of the cation. | ||
| Fig. 4 400 MHz 1H NMR spectra of L3py and LaL3py in D2O (pD = 7.0) at 298 K. | ||
The hydration numbers (q) of the europium and terbium cations in the complexes of L3py were obtained from the measurement of the luminescence lifetimes of the excited-states of the complexes (τ) in H2O and D2O (see Table 2). The numbers of coordinated water molecules were calculated using the equation of Parker and co-workers17 and are 3 within the experimental errors (see Table 2). They are similar to the values obtained previously for Lpy and Lpz.12 These values are very close to the values found for Ln(EDTA)− complexes. Indeed, depending on the empirical equations used, the hydration number of the lanthanide ion in Ln(EDTA)− complexes was found to be between 2.6 and 3.0 for Eu and between 2.8 and 2.9 for Tb.17,18 This indicates that the novel molecule L3py acts as a hexadentate ligand like the parent molecule EDTA, and affords two aliphatic nitrogen atoms, three aromatic nitrogen atoms of pyridines and only one oxygen donor of a carboxylate. Three water molecules complete the Ln(III) coordination sphere to obtain an overall coordination number of 9.
Unfortunately, the lanthanide complexes of L3pz could not be studied in solution because of the occurrence of precipitation at low concentration. Therefore the coordination behavior of this ligand could not be compared with its analogues. As pyrazine has only a very low contribution to the stability of the lanthanide complexes,12 other types of coordination in which more than one aminoacetate moiety is coordinated to the cation may be favored leading to insoluble polymolecular systems.
Back-extraction of Am–Ln mixtures with the ligand series
The selectivity for Am(III) over Eu(III) of the two previously reported ligands Lpy and Lpz has been demonstrated in water solution by the values of the stability constants in aqueous 0.1 M KNO3 at 298 K and makes them interesting candidates for the separation of the two families of f-elements. Indeed, soft-donor reagents can be used to separate actinides from lanthanides either as lipophilic extractant molecules or as water-soluble complexing agents.2 For instance, the TALSPEAK process (trivalent actinide–lanthanide separation by phosphorus reagent extraction from aqueous komplexes)19 and the DIAMEX-SANEX process20 are based on the partitioning of lanthanides and actinides between an acidic phosphorus extractant organic solution and an aqueous phase containing a high concentration of a carboxylic acid buffer and a polyaminopolycarboxylate complexant. The four water-soluble molecules Lpy, Lpz, L3py and L3pz were subjected to back-extraction experiments to test their ability to selectively strip Am(III) from an organic metal solution containing Am(III) and Eu(III). The ligand HEDTA, used as a selective complexing agent in the DIAMEX-SANEX partitioning process,20 was tested in the same conditions for comparison.The metal-containing organic solution was obtained by equilibrating an aqueous solution of lanthanides (25 mM) spiked with 152Eu(III) and 241Am(III) in HNO3 3 M, with an organic solution containing the malonamide derivative DMDOHEMA (0.6 M) and the dialkyl phosphoric acid HDEHP (0.3 M) in TPH, which have been demonstrated to extract these cations in the organic phase.21 Then, this metal-containing organic solution was contacted with 0.5 M water solutions of the ligands at pH 3 buffered with 0.4 M citric acid. The results are reported in Table 3. The distribution ratios, DM, were calculated as the ratio of the analytical concentrations of the relevant cation in the organic and aqueous phase (eqn (2)). Therefore, the smaller DM is, the more efficient the back-extraction is
| DM = [M]org/[M]aq | (2) |
 | (3) |
| SFAm/Eu = DEu/DAm | (4) |
| Lpz | Lpy | L3py | L3pz | HEDTA | |
|---|---|---|---|---|---|
| a Aqueous phase: ligand 0.5 M, citric acid 0.4 M, pH = 3. Organic phase: 0.3 M HDEHP, 0.6 M DMDOHEMA, 152Eu and 241Am trace level, TPH.b The conditional stability constants at pH 3 were calculated from the pK and stability constants given in Table 1. | |||||
| DEu | 13 | 49 | 53 | 130 | 1.6 |
| DAm | 0.31 | 1.37 | 1.16 | 8.3 | 0.13 |
| %stripped Eu | 7 | 2 | 2 | 1 | 38 |
| %stripped Am | 76 | 42 | 46 | 11 | 88 |
| log β110(Eu) at pH 3 | 2.9 | 2.0 | 1.5 | 0.1 | 6.1 |
| log β110(Am) at pH 3 | 5.6 | 3.8 | — | — | 6.5 |
| SFAm/Eu | 41 | 36 | 46 | 15 | 12 |
Most of the distribution ratios given in Table 3 are above 1 indicating that a small percentage of the cations are back-extracted in the water phase. In particular, it appears that the softest molecule L3pz has too low an affinity to perform significant cation back-extraction. This ligand gives the largest distribution ratios but also a poor selectivity factor, which may indicate that the pyrazine N-donors play no role in the coordination of the cations. The four other ligands give DAm below 1 (Lpz and HEDTA) or very close to 1 (Lpy and L3py) and are thus able to strip Am(III) from the organic solution in significant amounts. The conditional stability constants at pH 3 are also reported in Table 3. These values have been calculated from the known stability constants of the Eu(III) and possibly Am(III) complexes and the logK values of the ligands. As expected, large conditional stability constants give efficient back-extraction and small distribution ratios DEu or DAm. The high conditional stability constant values calculated for the two ligands HEDTA and Lpz explain their better efficiency to strip Am(III) at pH 3. Indeed, Lpz shows lower affinity constants than Lpy but is less basic, therefore, its conditional stability constants at pH 3 are higher than those of Lpy, making the former a more efficient complexant. For the same reason, the two pyridine-based ligands Lpy and L3py give nearly the same distribution ratios, even though the stability constants of their complexes are quite different, because L3py is less basic than Lpy. As the DAm values of these two ligands are very similar, this allows us to roughly estimate the conditional stability constant of L3py at pH 3: log β110pH3(AmL3py) ≈ log β110pH3(AmLpy) ≈ 4, which gives an estimation of the stability constant: log β110(AmL3py) ≈ 11. As expected, this indicates that L3py, which is softer than Lpy, has a greater selectivity for Am(III) over Eu(III) in water: Kselectivity(Am/Eu) = β110(Am)/β110(Eu) ≈ 102.24 = 170.
The three ligands Lpy, Lpz and L3py give interesting back-extraction results which are illustrated in Fig. 5, with separation factors ranging from 36 to 46. The SF values do not reflect the thermodynamic selectivity, Kselectivity(Am/Eu), determined in homogeneous water solutions. This may be due to the different media used to determine the affinity constants (homogeneous water, KNO3 0.1 M) and to perform the back-extraction experiments (different ionic strength, presence of citric acid, extractants DMDOHEMA and HDEHP in the organic phase). In particular, the formation of ternary complexes with citric acid cannot be excluded. Nevertheless, the separation factors obtained with the three ligands containing soft N-donors, Lpy, Lpz and L3py, give higher SF than the ligand HEDTA, exemplifying the role of the ligand’s softness in enhancing the separation between Am(III) and Eu(III).
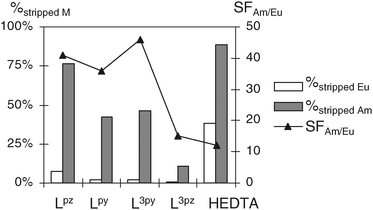 | ||
| Fig. 5 Percentage of stripped ions and separation factors for the back-extraction of Eu(III) and Am(III) by 0.5 M water solutions of Lpz, Lpy, L3py, L3pz and HEDTA at pH 3. | ||
The most interesting molecule in the series of N,O tetrapodal ligands presented here is Lpz which combines several advantages. Lpz exhibits a good selectivity for Am(III) over Eu(III). Despite its soft character it has a significant affinity for f ions due to the presence of two acetate hard donors. Finally, it is less basic than the pyridine-based ligands due to the strong withdrawing effect of the methylpyrazinyl moiety that significantly lowers the highest pKa. Therefore Lpz still has an efficient conditional stability constant at pH 3 leading to a distribution ratio for Am(III) approaching the value measured with HEDTA. In this series, Lpz is the only molecule able to slightly strip Am(III) from Am–Eu mixtures with a good selectivity under more drastic conditions, i.e. at lower pH or lower ligand concentration. These results are reported in Table 4. Of course, the distribution ratios are higher than those presented in Table 3 and the back-extraction is less efficient, but the selectivity factors are in the same range: 40–50. Moreover, it can be added that working at higher pH, e.g. pH 4, should increase the stripping ability of this selective ligand.
Conclusion
To conclude, a series of N,O-tetrapodal ligands containing soft N-heterocycles, either pyridine or pyrazine, was developed to study the impact of the softness of the N-donor on the complexation properties with trivalent f ions. As expected, as the softness of the ligand increases, by replacing either a pyridine with a pyrazine or an acetate with a N-heterocycle, the affinity for f-elements decreases. Indeed, trivalent f-element cations are hard acids which interact mainly via electrostatic bonds with their ligands. The scale of affinity is the following: Lpy > Lpz ≥ L3py ≫ L3pz. The ligand containing three pyridylmethyl moieties L3py gives similar complexes to Lpy and Lpz with a hexadentate coordination mode as indicated by NMR and luminescence decays (q = 3). The coordination behavior of the softest molecule L3pz is less clear as the complexes precipitate in the mM range, which prevents detailed solution studies. Nevertheless, potentiometric titration shows that L3pz complexes are much less stable than those obtained with the other three ligands.Selective back-extraction is a promising process for the separation of minor actinides from fission products.19 The series of water-soluble complexing agents presented in this paper was subjected to back-extraction to test their stripping ability in regard to their complexation properties for trivalent f-elements. The softest ligand L3pz appears not to be an efficient stripping agent as its complexing ability is too low. Moreover, its selectivity factor is similar to that obtained with HEDTA which suggests that the pyrazine groups may not coordinate the cations in these low-affinity complexes. In contrast, the other three molecules give similar selectivity factors (∼40) which are higher than that obtained with HEDTA and evidence the role of the N-heterocycles in enhancing the Am(III)–Eu(III) selectivity. The second pyrazine-based ligand, Lpz, demonstrates the best stripping properties. This molecule is indeed a good compromise between softness and hardness. The presence of two hard O-donors gives f-element complexes of significant stability; therefore the Am distribution ratio at pH 3 approaches that of HEDTA under the same conditions (DAm ≈ 0.3). Moreover, the two soft pyrazine groups provide a significant selectivity for Am(III) with respect to Eu(III). To obtain selective and efficient water-soluble complexing agents, soft ligands with sufficient affinity and also with a low basicity should be developed in order to maintain the stripping ability at low pH. Therefore a compromise has to be found between softness and affinity as in the pyrazine-based ligand Lpz.
Experimental
General details
Solvents and starting materials were purchased from Aldrich, Acros, Fluka and Alfa Aesar and used without further purification. Lanthanide salts were purchased from Aldrich and deuterium oxide (99.9 atom% D) from Euriso-Top. All water solutions were prepared from ultrapure laboratory grade water that had been filtered and purified by reverse osmosis using a Millipore MilliQ reverse-osmosis cartridge system (resistivity 18 MΩ cm). Thin layer chromatography (TLC) was performed on aluminium oxide 60 F254 neutral (Merck). Flash chromatography was performed on aluminium oxide 90 active neutral (+4.9% water wt, 63–200 μm, Merck). 1H NMR and 13C NMR spectra were recorded at 298 K on a Mercury Varian 400 spectrometer. Chemical shifts are reported in ppm with the solvent as the internal reference and coupling constants are given in Hz. Mass spectra were acquired with a Finigan LCQ-ion trap equipped with an electrospray source. Elemental analyses were performed by the Service Central d′Analyse (Solaize, France).Lpy, Lpz and 2-pyrazinecarbaldehyde were obtained according to published procedures.12,22
Synthesis
Potentiometry
Carbonate-free 0.1 mol L−1 KOH and 0.1 mol L−1 HNO3 were prepared from Fisher Chemicals concentrates. Potentiometric titrations were performed in 0.1 mol L−1 aqueous KNO3 under an argon atmosphere; the temperature was controlled to ±0.1 °C with a circulating water bath. The p[H] (p[H] = −log [H+], concentration in molarity) was measured in each titration with a combined pH glass electrode (Metrohm) filled with 3 mol L−1 KCl and the titrant addition was automated by use of a 751 GPD titrino (Metrohm). The electrode was calibrated in hydrogen ion concentration by titration of HNO3 with KOH in 0.1 mol L−1 KNO3.23 A plot of meter reading versus p[H] allows the determination of the electrode standard potential (E°) and the slope factor (f).Ligand concentration was determined by potentiometric titrations and was in accordance with the elemental analysis of the molecules. Lanthanide salt solutions were prepared by dissolving the appropriate amount of lanthanide nitrate in water. The exact metal ion concentration was determined by colorimetric titration using standardized EDTA solutions (Fisher Chemicals) and xylenol orange as indicator. Continuous potentiometric titrations with KOH 0.1 mol L−1 were conducted on 20 mL of aqueous solutions containing 10−3 mol L−1 of the ligand and 0, 0.5, 1 and 2 equiv. of the desired metallic cation. Back titrations with HNO3 0.1 mol L−1 were systematically performed after each experiment to check whether equilibration had been achieved. In a typical experiment, 100 points were measured with a 2 min delay between the measurements for the free ligand, and a 5 min delay for metallic complexes.
Experimental data were refined using the computer program Hyperquad 2000.24 All equilibrium apparent constants are expressed as concentration ratios and not activities. The ionic product of water at 25 °C and 0.1 mol L−1 ionic strength is pKw = 13.78.13 The initial concentrations of ligand, metal and proton were fixed, as well as the ligand pK values for the metallic complex stability constant determination. All values and errors (one standard deviation) reported represent the average of at least three independent experiments.
NMR spectroscopy
Samples for NMR spectroscopy were prepared by mixing appropriate volumes of stock solutions of the ligand (∼0.005 mol L−1) and the lanthanide triflate salt in deuterium oxide. Ligand concentrations were determined by potentiometric titration, and the metal concentrations by EDTA titrations using xylenol orange indicator. The pD of the samples were adjusted to the desired value by adding stock solutions of DCl or NaOD in D2O. The pD were measured according to pD = pHread + 0.41.25Luminescence
Terbium and europium luminescence lifetimes were measured on a Perkin-Elmer LS50B spectrofluorimeter by recording the decay of the emission intensity at 545 nm for Tb and 616 nm for Eu (excitation at 274 nm). The decays of luminescence intensities followed systematically monoexponential laws and were analyzed as single-exponential decays. The instrument settings were as follows: a gate time of 1 ms, a flash count of 1, excitation and emission slit widths of 10 nm, and a varied delay time. The complexes were prepared in situ by mixing 0.9 equivalents of the metal solution with one equivalent of ligand. The concentration selected was 1 mM for europium complexes and 0.5 mM for terbium complexes either in H2O or D2O. The pH (or pD) was then adjusted to 7 with a NaOH (or NaOD) solution. Reported lifetimes, τ, are average of three separate measurements calculated by monitoring the emission intensity after at least 20 different delay times covering two or more lifetimes.Back-extraction experiments
Abbreviations: HDEHP: bis(2-ethylhexyl)phosphoric acid, DMDOHEMA: N,N′-dimethyl-N,N′-dioctylhexylethoxy malonamide, TPH (hydrogenated tetrapropylene) is a mixture of highly branched alkanes containing 11 to 13 carbon atoms, HEDTA: N-hydroxyethylethylenediaminetriacetic acid.All extraction experiments were performed at 25 °C. First, to prepare the organic phase containing the metallic ions, 4 mL of an organic solution (0.3 M HDEHP, 0.6 M DMDOHEMA, TPH) and 4 mL of an aqueous solution (HNO3 3 M, [Ln]tot = 25 mM spiked with 241Am(III) and 152Eu(III)) were equilibrated by vortexing for 10 min. Preliminary experiments showed that a 10 min vortexing time is adequate to reach equilibrium. The organic and aqueous phases were separated by centrifugation. The organic phase was scrubbed by an aqueous solution (citric acid 0.5 M, pH 3) and used in the following for the back-extraction experiments.
The aqueous solutions used in the back-extraction experiments were prepared with a ligand concentration of 0.5 M (or 0.05 M), citric acid 0.4 M, and pH adjusted to 3 with sodium hydroxide (Lpy and L3py) or nitric acid (Lpz and L3pz). For the back-extraction experiment performed at pH 2, the pH of the aqueous phase was adjusted to 2 with HNO3. 0.5 mL of the aqueous solution of ligand and 0.5 mL of the organic phase containing the metal ions were equilibrated by vortexing for 60 min at 25 °C. After separation by centrifugation aliquots of both phases were analyzed using a gamma counting spectrometer (Hyper pure Ge detector, CANBERRA). 152Eu and 241Am tracers were used for the measurement of the Eu–Am distribution. In the range of 0.1–10 the error in D values is about 5%, while in the ranges of 0.01–0.1 and 10–100 the error is greater, about 10%.
Acknowledgements
We thank Elodie Bosso for participation in this study as an undergraduate trainee and Colette Lebrun for the mass spectrometry experiments.Notes and references
- W. H. Runde and W. W. Schultz, in The Chemistry of Actinide and Transactinide Elements, ed. L. R. Morss, N. M. Edelstein, J. Fuger and J. J. Katz, Springer, Dordrecht, 2006, pp. 1265–1395 Search PubMed; K. L. Nash, in Handbook on the Physics and Chemistry of Rare Earths, ed. J. K. A. Gschneidner, L. Eyring, G. R. Choppin and H. H. Lander, Elsevier Science, Amsterdam, 1994, pp. 197–238 Search PubMed.
- K. L. Nash, C. Madic, J. N. Mathur and J. Lacquement, in The Chemistry of the Actinide and Transactinide Elements, ed. L. R. Morss, N. M. Edelstein, J. Fuger and J. J. Katz, Springer, Dordrecht, 2006, pp. 2623–2798 Search PubMed.
- R. G. Pearson, J. Am. Chem. Soc., 1963, 85, 3533–3539 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1969, 25, 925 CrossRef CAS.
- C. Musikas, Proceedings of Symposium on Americium and Curium Chemistry and Technology; International Chemical Congress of Pacific Basin Societies, Honolulu, 1985 Search PubMed; Z. Kolarik, U. Müllich and F. Gassner, Solvent. Extr. Ion Exch., 1999, 17, 23–32 Search PubMed; M. G. B. Drew, D. Guillaneux, M. J. Hudson, P. B. Iveson, M. L. Russel and C. Madic, Inorg. Chem. Commun., 2001, 4, 12–15 CrossRef CAS; P. B. Iveson, C. Rivière, D. Guillaneux, M. Nierlich, P. Thuéry, M. Ephritikhine and C. Madic, Chem. Commun., 2001, 1512–1513 CrossRef CAS; M. A. Denecke, A. Rossberg, P. J. Panak, M. Weigl, B. Schimmelpfennig and A. Geist, Inorg. Chem., 2005, 44, 8418–8425 RSC; M. J. Hudson, C. E. Boucher, D. Braekers, J.-F. Desreux, M. G. B. Drew, M. R. S. J. Foreman, L. M. Harwood, C. Hill, C. Madic, F. Marken and T. G. A. Youngs, New J. Chem., 2006, 30, 1171–1183 CrossRef CAS.
- Y. Zhu, Radiochim. Acta, 1995, 68, 95–98 CAS.
- Z. Kolarik, Chem. Rev., 2008, 108, 4208–4252 CrossRef CAS; H. H. Dam, D. N. Reinhoudt and W. Verboom, Chem. Soc. Rev., 2007, 36, 367–377 RSC.
- R. Wietzke, M. Mazzanti, J.-M. Latour, J. Pécaut, P.-Y. Cordier and C. Madic, Inorg. Chem., 1998, 37, 6690–6697 CrossRef CAS.
- K. Ishimori, M. Watanabe, T. Kimura, T. Yaita, T. Yamada, Y. Kataoka, S. Shinoda and H. Tsukube, Chem. Lett., 2005, 1112–1113 CrossRef CAS.
- R. Mirvaliev, M. Watanabe, T. Matsumura, S. Tachimori and K. Takeshita, J. Nucl. Sci. Technol., 2004, 41, 1122–1224 Search PubMed; M. Watanabe, R. Mirvaliev, S. Tachimori, K. Takeshita, Y. Nakano, K. Morikawa and R. Mori, Chem. Lett., 2002, 1230–1231 CrossRef CAS; M. Watanabe, R. Mirvaliev, S. Tachimori, K. Takeshita, Y. Nakato, K. Morikawa, T. Chikazawa and R. Mori, Solvent. Extr. Ion Exch., 2004, 22, 377–390 CrossRef CAS.
- L. Karmazin, M. Mazzanti, C. Gateau, C. Hill and J. Pécaut, Chem. Commun., 2002, 2892–2893 RSC.
- M. Heitzmann, F. Bravard, C. Gateau, N. Boubals, C. Berthon, J. Pecaut, M. C. Charbonnel and P. Delangle, Inorg. Chem., 2009, 48, 246–256 CrossRef CAS.
- R. M. Smith, A. E. Martell and R. J. Motekaitis, NIST Critically Selected Stability Constants of Metal Complexes Database, NIST Standard Reference Database 46, 2001 Search PubMed.
- G. R. Choppin, Pure Appl. Chem., 1971, 27, 23–41 CAS.
- J. C. G. Bunzli and C. Piguet, Chem. Rev., 2002, 102, 1897–1928 CrossRef.
- M. P. Jensen, L. R. Morss, J. V. Beitz and D. D. Ensor, J. Alloys Compd., 2000, 303–304, 137–141 CrossRef CAS; F. Bravard, C. Rosset and P. Delangle, Dalton Trans., 2004, 2012–2018 RSC.
- A. Beeby, I. M. Clarkson, R. S. Dickins, S. Faulkner, D. Parker, L. Royle, A. S. d. Sousa, J. A. G. Williams and M. Woods, J. Chem. Soc., Perkin Trans. 2, 1999, 493–503 RSC.
- W. D. Horrocks and D. R. Sudnick, J. Am. Chem. Soc., 1979, 101, 334–340 CrossRef CAS; R. M. Supkowski and W. D. Horrocks, Inorg. Chim. Acta, 2002, 340, 44–48 CrossRef CAS.
- M. Nilsson and K. L. Nash, Solvent. Extr. Ion Exch., 2007, 25, 665–701 CrossRef CAS.
- P. Baron, X. Hérès, M. Lecomte and M. Masson, Proceedings of International Conference on Future Nuclear Systems, GLOBAL’01, Paris, France, 2001 Search PubMed; M. Miguirditchian, L. Chareyre, X. Hérès, P. Baron and M. Masson, Proceedings of Advanced Nuclear Fuel Cycles and Systems, GLOBAL’07, Boise, Idaho, USA, 2007 Search PubMed; M. Miguirditchian, L. Chareyre, C. Sorel, I. Bisel, P. Baron and M. Masson, Proceedings of ATALANTE 2008, Montpellier, France, 2008 Search PubMed.
- B. Gannaz, R. Chiarizia, M. R. Antonio, C. Hill and G. Cote, Solvent. Extr. Ion Exch., 2007, 25, 313–337 CrossRef CAS.
- H. Rutner and P. E. Spoerri, J. Org. Chem., 1963, 28, 1898–1899 CAS.
- A. E. Martell and R. J. Motekaitis, Determination and Use of Stability Constants, VCH, New York, 1992 Search PubMed.
- P. Gans, A. Sabatini and A. Vacca, Talanta, 1996, 43, 1739–1753 CrossRef CAS; L. Alderighi, P. Gans, A. Ienco, D. Peters, A. Sabatini and A. Vacca, Coord. Chem. Rev., 1999, 184, 311–318 CrossRef CAS.
- P. K. Glasoe and F. A. Long, J. Phys. Chem., 1960, 64, 188–190 CrossRef CAS.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2010 |