DOI:
10.1039/C0MT00021C
(Paper)
Metallomics, 2010,
2, 766-770
Effects of large-scale amino acid substitution in the polypeptide tether connecting the heme and molybdenum domains on catalysis in human sulfite oxidase†
Received
13th June 2010
, Accepted 12th August 2010
First published on
20th September 2010
Abstract
Sulfite oxidase (SO) is a molybdenum-cofactor-dependent enzyme that catalyzes the oxidation of sulfite to sulfate, the final step in the catabolism of the sulfur-containing amino acids, cysteine and methionine. The catalytic mechanism of vertebrate SO involves intramolecular electron transfer (IET) from molybdenum to the integral b-type heme of SO and then to exogenous cytochrome c. However, the crystal structure of chicken sulfite oxidase (CSO) has shown that there is a 32 Å distance between the Fe and Mo atoms of the respective heme and molybdenum domains, which are connected by a flexible polypeptide tether. This distance is too long to be consistent with the measured IET rates. Previous studies have shown that IET is viscosity dependent (Feng et al., Biochemistry, 2002, 41, 5816) and also dependent upon the flexibility and length of the tether (Johnson-Winters et al., Biochemistry, 2010, 49, 1290). Since IET in CSO is more rapid than in human sulfite oxidase (HSO) (Feng et al., Biochemistry, 2003, 42, 12235) the tether sequence of HSO has been mutated into that of CSO, and the resultant chimeric HSO enzyme investigated by laser flash photolysis and steady-state kinetics in order to study the specificity of the tether sequence of SO on the kinetic properties. Surprisingly, the IET kinetics of the chimeric HSO protein with the CSO tether sequence are slower than wildtype HSO. This observation raises the possibility that the composition of the non-conserved tether sequence of animal SOs may be optimized for individual species.
Introduction
Sulfite oxidizing enzymes are molybdenum cofactor dependent enzymes that are found in plants, animals and bacteria. In animals, sulfite oxidase (SO) catalyzes the oxidation of sulfite to sulfate, using ferricytochrome c (cyt cox) as the physiological electron acceptor (eqn (1)):1–4| | | SO32− + H2O + 2(cyt c)ox → SO42− + 2(cyt c)red + 2H+ | (1) |
Oxidation of sulfite is the final step in the catabolism of the sulfur containing amino acids, cysteine and methionine. In humans, inherited defects in SO lead to severe neonatal neurological problems and early death.5–9
The proposed catalytic mechanism of SO includes two intramolecular one-electron transfer (IET) steps from the molybdenum cofactor (Moco) to the integral b5-heme iron. The mechanism also includes two intermolecular one-electron steps to exogenous cytochrome c (cyt c). Laser flash photolysis studies were used to extensively probe the IET kinetics in chicken sulfite oxidase (CSO)10–12 and human sulfite oxidase (HSO).13–16
The only crystal structure for an intact animal SO is that of CSO.17 The structure of CSO is a homodimer, with each subunit containing a b5-type cytochrome heme domain (∼10 kDa) at the N-terminus and a larger C-terminal domain, which contains the Moco. These two domains are linked by a flexible polypeptide tether, and the Mo–Fe distance is 32 Å (Fig. 1). The tether is poorly resolved in the crystal structure,17 suggesting that it has high flexibility and may facilitate conformational changes within SO that alter the distance between the Fe and Mo centers.18 Further support for this comes from flash photolysis studies of the rates of IET between the heme and Mo domains, which occur on the millisecond time scale.10,18–19 Comparison of these IET rates for SO to those from previous studies of electron tunneling kinetics in model systems and in structurally characterized proteins 20–22 imply that the distance between the Fe and Mo centers during IET is much less than the 32 Å separation found in the crystal structure.17 This smaller distance could be due to a conformational change facilitated by the tether that occurs during turnover. Very recently, Mroginski and co-workers have performed steered molecular dynamics and molecular dynamics simulations on CSO.23 These calculations generated a stable 3D structure of SO, which had a distance of less than 20 Å between the heme and molybdenum centers for optimal IET. Their model showed that the heme propionates docked at the substrate binding pocket of the molybdenum domain.
 |
| | Fig. 1 Sequence alignment of the flexible tether regions of CSO and HSO. Amino acids highlighted in red are conserved between the two species, while those in blue are similar. The glutamate highlighted in green was necessary for gene expression of the HSO chimera variant. | |
Previous studies on the effects of the tether on the reactivity of SO include solution viscosity experiments,18 computational modeling24 and steady-state and laser flash photolysis studies.25 All of these investigations are consistent with the proposal of Feng and co-workers that interdomain motion that decreases the Mo to Fe distance is essential for rapid IET and that the flexible tether linking the two domains of SO facilitates this motion.18 The importance of heme mobility for effective electron transfer in multi-domain redox proteins is further underscored by the recent crystal structure of yeast flavocytochrome b2 (Fcb2), which shows unexpected plasticity in the vicinity of the heme cavity.22
We have recently reported the effects of changing the length and flexibility of the polypeptide tether of HSO on its steady-state and IET kinetics.25 The effects of tether flexibility were investigated by preparing single mutations of two tether residues, P105A and P111A, only one of which (P105) is conserved, as well as the P105A/P111A double mutant. Although the P105A variant produced a 3-fold decrease in the IET rate constant, no systematic trends in the kinetic parameters were observed for this series of variants.25 Deletions of other nonconserved amino acids in the HSO tether, thereby shortening its length, resulted in more drastically reduced IET rate constants. The deletion of five amino acid residues (ΔK108V109A110T112V113) (Fig. 1) decreased IET by 70-fold, making it rate-limiting in catalysis.25
Previous laser flash photolysis studies have given an IET rate constant of 1500 s−1 for native CSO ,10 whereas wildtype (wt) recombinant HSO has an IET rate constant of 491 s−1.14,26 One possible reason for the larger IET rate constant for CSO compared to HSO is their differing tether sequences (Fig. 1). For example, the HSO tether has 14 amino acids, whereas CSO has 13 amino acids and lacks a glutamic acid at position 106 (human numbering). The tethers for both HSO and CSO have two conserved proline residues, one near the N-terminal heme domain (P105, human numbering) and another near the C-terminal Moco domain (P118). HSO has another proline residue in the 111 position, which is conserved in other mammalian SO sources. To assess the possibility that the difference in IET rates between CSO and HSO is due to their differing tether sequences, we have made a large scale amino acid substitution of the tether sequence of HSO in order to create a relatively close resemblance to CSO (Fig. 1). Unfortunately, our attempts to express the gene of HSO with a 13-residue tether sequence that is identical to that in CSO were not successful. Therefore, to allow the expression to occur we have mutated the HSO tether sequence into a 14-residue sequence that is a chimera of CSO, but retains the human-like E106 residue (Fig. 1). The steady-state and IET kinetics of this chimeric variant of HSO are described here.
Experimental
Site-directed mutagenesis
Expression plasmids of the HSO chimera mutant were constructed using the pTG918 plasmid27via the Quick Change Site-Directed Mutagenesis protocol (Stratagene). The wt HSO plasmid was used as the template for the first PCR reaction. The primers for the first PCR reaction are as follows: E106f/K108E (ctgaatcctgaagacgaggcaccgg)
E106/K106Er (5′ccggtgcctcgtcttcaggattcag). Once the correct mutant sequence was verified by Sequetech, this mutant plasmid was used as the template for the second PCR reaction. The primers for this second reaction are as follows: V109A/A110P/P111Af (aatcctgacgaggcaccggccaccgtggaga), V109A/A110P/P111Ar (tctccacggtggccggtgcctcgtcaggatt). Lastly, once the sequence of the plasmid was verified by Sequetech, a third PCR reaction was performed, using the doubly mutated plasmid from the second PCR as a template, and the primers for this reaction are as follows: T112A/V113P/E114D/T115A/S116Qf (aggcaccggccgccccggatgcccaagaccctta), T112A/V113P/E114D/T115A/S116Qr (taagggtcttgggcatccggggcggccggtgcct). The mutations were verified by sequence analysis performed at the Sequetech Corporation DNA Analysis facility in Mountain View, California.
Overexpression and purification
The recombinant HSO chimera mutant was purified as previously described27,28 with the following modifications. After the Phenyl Sepharose column (GE Healthcare), fractions exhibiting an A413/A280 ratio of 0.80 or greater were pooled and further purified using a Superdex 200 column (GE Healthcare). Fractions exhibiting an A413/A280 ratio of 0.96 or greater were then pooled and used in the experiments described in this study. The molybdenum content of the purified protein was determined using an IRIS Advantage Inductively Coupled Plasma Emission Spectrometer from the Jarrell Ash Corporation (see the ESI†). Enzyme concentrations were determined by using a molar extinction coefficient of 113![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) 000 M−1 cm−1 at 413 nm for oxidized human SO.16
000 M−1 cm−1 at 413 nm for oxidized human SO.16
Laser flash photolysis
Laser flash photolysis experiments were performed anaerobically on 0.30 mL solutions containing ∼90 μM 5-deazariboflavin (dRF) and 0.5 mM freshly prepared semicarbazide as a sacrificial reductant. The methodologies used to obtain rate and equilibrium constants for IET in SO have been described previously19 and are summarized below. The laser flash photolysis apparatus system has been extensively described29 as has been the basic photochemical process by which 5-deazariboflavin semiquinone (dRFH˙) is generated by reaction between triplet state dRF and the sacrificial reductant and used to reduce redox-active proteins (eqn (2)–(5)).30–32| |  | (2) |
| | | 3dRF + AH2 → dRFH˙ + AH˙ | (3) |
| | | dRFH˙ + Mo(VI)/Fe(III) → dRF + Mo(VI)/Fe(II) | (4) |
| |  | (5) |
The IET rate constant can be calculated by fitting the heme reoxidation curve with an exponential function (eqn (6)) where the IET rate constant is the sum of the forward and the reverse electron transfer rate constants (kf and kr, respectively, in eqn (7)).| |  | (6) |
The equilibrium constant can then be calculated using the parameters a and b, which are determined from the kinetic traces.| |  | (8) |
| | 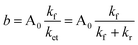 | (9) |
| |  | (10) |
The forward and reverse rate constants (kf and kr, respectively) of IET can then be calculated from the equilibrium constant (eqn (10)), thereby providing quantitative information about interdomain electron transfer in the enzyme. Note that kf in these flash photolysis experiments is actually the reverse of the net physiological IET direction.
Steady-state assays
Steady-state kinetic studies were performed aerobically in a Varian Cary-300 spectrophotometer. Initial velocities were determined by following the reduction of a freshly prepared oxidized cyt c (horse heart, Sigma) solution at 550 nm, using an extinction coefficient change of 19 630 M−1 cm−1.11 SO was routinely assayed at 25 °C in 20 mM Tris pH 7.6, titrated with acetic acid. The steady-state kinetic profile study was conducted using a saturating concentration of cyt c, 400 μM (10-fold greater than Km) and varying the concentration of sulfite, between 2 μM and 200 μM.
Mo EPR spectroscopy experiments
EPR samples of wt and the chimeric HSO variant (0.5–0.7 mM HSO) at pH 5.8 were prepared in 100 mM Bis-Tris buffer containing 100 mM NaCl.33,34 The enzyme samples were reduced with a 20-fold excess of sodium sulfite and immediately frozen in liquid nitrogen. The continuous wave (CW) X-band EPR spectra were obtained on a Bruker ESP-300 spectrometer at 77 K.
Results and discussion
Laser flash photolysis kinetics of HSO chimera
Fig. 2 shows a comparison of ket values between wt HSO and the HSO chimera. Surprisingly, ket for the HSO chimera is approximately 3-fold less than wt HSO (156 ± 14 s−1 for chimeric HSO versus 467 ± 19 s−1 for wt HSO at pH 7.4). Additionally, ket of the HSO chimera shows a pH dependence that is similar to that of wt HSO.25 CSO shows a significant increase in IET between pH 6 (1650 s−1) and 7 (2400 s−1) and then IET decreased with increasing pH (60 s−1 pH 9).10Fig. 3 illustrates the IET equilibrium constants (Keq) for the HSO chimera at various pH values. For wt HSO, Keq increases with increasing pH.25 The chimeric variant shows a slight pH dependence, with Keq values only slightly higher than wt at corresponding pH values.
 |
| | Fig. 2 pH dependence of the IET rate constants for wt HSO and the HSO chimera. The inset shows the pH values for each type of shading. | |
 |
| | Fig. 3 pH dependence of the equilibrium constants for wt HSO and the HSO chimera, calculated from laser flash photolysis experiments. The inset shows the pH values for each type of shading. | |
Steady-state kinetics of the HSO chimera
The steady-state oxidation of sulfite to sulfate as catalyzed by HSO using cyt c as the electron acceptor yields plots of initial velocity versus substrate concentration that display typical saturation kinetics (not shown). The standard Michaelis–Menten parameters at pH 7.6 are given in Table 1. The value of kcat for chimeric HSO is slightly higher than that of wt HSO. However, Km for the chimeric variant shows a 1.6 fold decrease compared to recombinant wt HSO.25 The decrease in Km also affects the catalytic efficiency of the variant, causing kcat/Ksulfitem to increase by a factor of 2.5. We conclude that the chimeric variant results in tighter binding of sulfite to the enzyme. The kcat values in Table 1 are much smaller than ket, and therefore, the IET process is not rate-limiting in catalysis, as is also the case for wt enzymes.
Table 1 Steady-state kinetics data for wt HSO and the HSO chimera varianta
| Enzyme |
k
cat/s−1 |
K
sulfitem/μM |
k
cat/Ksulfitem/M−1s−1 |
|
pH 7.6, 20 mM Tris acetate.
See ref. 16 and 26.
|
|
wt HSOb |
26.9 ± 0.5 |
11.1 ± 0.4 |
2.4 × 106 |
| HSO chimera |
41 ± 5 |
7 ± 3 |
5.9 × 106 |
Electron paramagnetic resonance spectra
At pH 5.8 the chimeric HSO protein shows an EPR spectrum that is characteristic of the blocked Mo(V) form of HSO that is produced in chloride-depleted samples34 (see red trace, Fig. S1 of the ESI†). Addition of 100 mM sodium chloride generates the low pH (lpH) signal (see black trace, Fig. S1 of the ESI†) as has been observed for other HSO variants.34 At pH 9.0, the chimeric HSO variant produces the well known high pH (hpH) signal (not shown). The essential identity of the CW EPR spectra of the chimeric HSO variant to those for wt HSO34–36 indicates that no changes in the Mo environment were caused by the chimeric tether mutations.
Conclusions
The present work continues our investigations of the effects of the tether of HSO on the reaction kinetics and physical properties of the enzyme.25 The present work has explored the effects of changing the sequence of the human tether into that of chicken, but retaining E106. In view of the faster IET kinetics of CSO compared to HSO, we expected that the chimeric HSO protein would show faster IET kinetics compared to wt HSO. Thus, it was surprising to observe slower IET kinetics for the chimeric HSO variant. The value of kcat is similar to wt HSO, and the rate of IET is still much faster than the turnover rate, and consequently IET is not rate limiting. EPR spectroscopy indicates that the molybdenum environment is not affected by the chimeric tether mutation of HSO. These results for this chimeric HSO further support the suggestion that the composition of the interdomain tether of SO affects the rates of IET.25 However, other factors, in addition to the composition of the tether, likely also play a role in the observed IET kinetics. It is possible that the variable tether sequences among species of animals may reflect specialized tuning adaptations, in response to sequence variations in the rest of the SO molecule, which aid the conformational changes needed to optimize docking of the heme domain onto the molybdenum domain surface for effective IET rates and catalysis for specific SOs.
Abbreviations
| SO | sulfite oxidase |
| dRF | 5-deazariboflavin |
| CSO | chicken sulfite oxidase |
| HSO | human sulfite oxidase |
| cyt c | cytochrome c |
| IET | intramolecular electron transfer |
|
k
et
| electron transfer rate constant |
|
K
eq
| equilibrium constant for intramolecular electron transfer |
|
k
f
and kr | microscopic rate constants for the forward and reverse directions, respectively, of intramolecular electron transfer |
| Moco | molybdopterin cofactor |
|
wt
| wildtype |
Acknowledgements
We thank Dr Andrei Astashkin for recording the EPR spectra. We thank Dr Arnold Raitsimring and Ms. Safia Emesh for helpful discussions. We are grateful to Drs Heather Wilson and K. V. Rajagoplalan for the HSO plasmid. This research was supported by NIH Grant GM-037773 (to JHE); Ruth L. Kirchstein-NIH Fellowship 1F32GM082136-01 (to KJW); ACD is a participant in the Undergraduate Biology Research Program, supported in part by a grant to the University of Arizona from the Howard Hughes Medical Institute (52005889).
References
- R. Hille, Chem. Rev., 1996, 96, 2757–2816 CrossRef CAS.
-
K. V. Rajagopalan and J. L. Johnson, Sulfite oxidase, in Wiley Encyclopedia of Molecular Medicine, ed. T. E. Creighton, Wiley, New York, 2002, pp. 3048–3051 Search PubMed.
-
C. Kisker, Sulfite oxidase, in Handbook of Metalloproteins, ed. A. Messerschmidt, R. Huber, T. Poulos, K. Wieghardt, John Wiley and Sons, Ltd, New York, 2001, pp. 1121–1135 Search PubMed.
- H. Schindelin, C. Kisker and K. V. Rajagopalan, Adv. Protein Chem., 2001, 58, 47–94 CAS.
- J. L. Johnson, Prenatal Diagn., 2003, 23, 6–8 Search PubMed.
- A. B. Dublin, J. K. Hald and S. L. Wootton-Gorges, Am. J. Neuroradiol., 2002, 23, 484–485 Search PubMed.
- J. O. Sass, A. Gunduz, C. Araujo, R. Funayama, B. Korkmaz, K. Giselle, D. Pinto, B. Tuysuz, C.-W. Lam, J. Reiss, M. Walter, C. Yalcinkaya and J. S. Camelo Junior, Brain Dev., 2010, 32, 544–549 CrossRef.
- G. Schwarz, J. A. Santamaria-Araujo, S. Wolf, J. H. Lee, I. M. Adham, H. J. Grone, H. Schwegler, J. O. Sass, T. Otte, P. Hanzelmann, R. R. Mendel, W. Engel and J. Reiss, Hum. Mol. Genet., 2004, 13, 1249–1255 CrossRef CAS.
- A. Matthies, K. V. Rajagopalan, R. R. Mendel and S. Leimkuhler, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5946–5951 CrossRef CAS.
- E. P. Sullivan, Jr., J. T. Hazzard, G. Tollin and J. H. Enemark, Biochemistry, 1993, 32, 12465–12470 CrossRef CAS.
- M. S. Brody and R. Hille, Biochemistry, 1999, 38, 6668–6677 CrossRef CAS.
- D. L. Kessler and K. V. Rajagopalan, J. Biol. Chem., 1972, 247, 6566–6573 CAS.
- C. Feng, H. L. Wilson, J. K. Hurley, J. T. Hazzard, G. Tollin, K. V. Rajagopalan and J. H. Enemark, J. Biol. Chem., 2003, 278, 2913–2920 CrossRef CAS.
- C. Feng, H. L. Wilson, J. K. Hurley, J. T. Hazzard, G. Tollin, K. V. Rajagopalan and J. H. Enemark, Biochemistry, 2003, 42, 12235–12242 CrossRef CAS.
- C. Feng, H. L. Wilson, G. Tollin, A. V. Astashkin, J. T. Hazzard, K. V. Rajagopalan and J. H. Enemark, Biochemistry, 2005, 44, 13734–13743 CrossRef CAS.
- H. L. Wilson and K. V. Rajagopalan, J. Biol. Chem., 2004, 279, 15105–15113 CrossRef CAS.
- C. Kisker, H. Schindelin, A. Pacheco, W. A. Wehbi, R. M. Garrett, K. V. Rajagopalan, J. H. Enemark and D. C. Rees, Cell, 1997, 91, 973–983 CrossRef CAS.
- C. Feng, R. V. Kedia, J. T. Hazzard, J. K. Hurley, G. Tollin and J. H. Enemark, Biochemistry, 2002, 41, 5816–5821 CrossRef CAS.
- A. Pacheco, J. T. Hazzard, G. Tollin and J. H. Enemark, JBIC, J. Biol. Inorg. Chem., 1999, 4, 390–401 CrossRef CAS.
- H. B. Gray and J. R. Winkler, Annu. Rev. Biochem., 1996, 65, 537–561 CrossRef CAS.
- J. R. Winkler, Curr. Opin. Chem. Biol., 2000, 4, 192–198 CrossRef CAS.
- K. H. Diêp Lê, A. Boussac, B. Frangioni, C. Léger and F. Lederer, Biochemistry, 2009, 48, 10803–9 CrossRef CAS.
- T. Utesch and M. A. Mroginski, J. Phys. Chem. Lett., 2010, 1, 2159–2164 Search PubMed.
- T. Kawatsu and D. N. Beratan, Chem. Phys., 2006, 326, 259–269 CrossRef CAS.
- K. Johnson-Winters, A. R. Nordstrom, S. Emesh, A. V. Astashkin, A. Rajapakshe, R. E. Berry, G. Tollin and J. H. Enemark, Biochemistry, 2010, 49, 1290–1296 CrossRef CAS.
- H. L. Wilson, S. R. Wilkinson and K. V. Rajagopalan, Biochemistry, 2006, 45, 2149–2160 CrossRef CAS.
- C. A. Temple, T. N. Graf and K. V. Rajagopalan, Arch. Biochem. Biophys., 2000, 383, 281–287 CrossRef CAS.
- R. M. Garrett and K. V. Rajagopalan, J. Biol. Chem., 1996, 271, 7387–7391 CrossRef CAS.
- J. K. Hurley, A. M. Weber-Main, M. T. Stankovich, M. M. Benning, J. B. Thoden, J. L. Vanhooke, H. M. Holden, Y. K. Chae, B. Xia, H. Cheng, J. L. Markleym, M. Martinez-Julvez, C. Gomez-Moreno, J. L. Schmetis and G. Tollin, Biochemistry, 1997, 36, 11100–11117 CrossRef CAS.
- G. Tollin, J. K. Hurley, J. T. Hazzard and T. E. Meyer, Biophys. Chem., 1993, 48, 259–279 CrossRef CAS.
- G. Tollin, J. Bioenerg. Biomembr., 1995, 27, 303–309 CrossRef CAS.
-
G. Tollin, In Electron Trasfer in Chemistry IV, ed. V. Balzani, Wiley-VCH, Weinheim, Germany, 2001, pp. 202–231 Search PubMed.
- E. L. Klein, A. V. Astashkin, D. Ganyushin, C. Riplinger, K. Johnson-Winters, F. Neese and J. H. Enemark, Inorg. Chem., 2009, 48, 4743–4752 CrossRef CAS.
- A. Rajapakshe, K. Johnson-Winters, A. R. Nordstrom, K. T. Meyers, S. Emesh, A. V. Astashkin and J. H. Enemark, Biochemistry, 2010, 49, 5154–5159 CrossRef CAS.
- M. T. Lamy, S. Gutteridge and R. C. Bray, Biochem. J., 1980, 185, 397–403 CAS.
- J. H. Enemark, A. V. Astashkin and A. M. Raitsimring, Dalton Trans., 2006, 3501–3514 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Iron-to-molybdenum ratios determined using ICP, laser flash photolysis results for HSO chimera and CW EPR spectra of HSO chimera. See DOI: 10.1039/c0mt00021c |
|
| This journal is © The Royal Society of Chemistry 2010 |
Click here to see how this site uses Cookies. View our privacy policy here.