Copper handling machinery of the brain
Svetlana
Lutsenko
*a,
Ashima
Bhattacharjee
a and
Ann L.
Hubbard
b
aDepartment of Physiology, Johns Hopkins University, 725N Wolfe St., Baltimore, MD 21205, USA. E-mail: lutsenko@jhmi.edu; Tel: +1 410-614-4661
bDepartment of Cell Biology, Johns Hopkins University, 725N Wolfe St., Baltimore, MD 21205, USA
First published on 17th August 2010
Abstract
Copper plays an indispensable role in the physiology of the human central nervous system (CNS). As a cofactor of dopamine-β-hydroxylase, peptidyl-α-monooxygenase, superoxide dismutases, and many other enzymes, copper is a critical contributor to catecholamine biosynthesis, activation of neuropeptides and hormones, protection against reactive oxygen species, respiration and other processes essential for normal CNS function. Copper content in the CNS is tightly regulated, and changes in copper levels in the brain are associated with a wide spectrum of pathologies. However, the mechanistic understanding of copper transport in the CNS is still in its infancy. Little is known about copper distribution among various cell types or cell-specific regulation of copper homeostasis, despite the fact that the molecules mediating copper transport and distribution in the brain (CTR1, Atox1, CCS, ScoI/II, ATP7A and ATP7B) have been identified and their importance in CNS function increasingly understood. In this review, we summarize current knowledge about copper levels and uses in the CNS and describe the molecules involved in maintaining copper homeostasis in the brain.
 Svetlana Lutsenko | Dr Svetlana Lutsenko is a faculty member in the Department of Physiology at the Johns Hopkins Medical School in Baltimore, USA. Her laboratory is working on dissecting the mechanisms of ATP driven ion transport, regulation of human copper homeostasis, as well as developing tractable models for human disorders of copper metabolism. |
 Ashima Bhattacharjee | Dr Ashima Bhattacharjee is a postdoctoral fellow in the Department of Physiology at the Johns Hopkins Medical School in Baltimore, USA. She is interested in the pathophysiology of human genetics disorders. Her current research focuses on determining the role of mutations and polymorphisms in the Wilson's disease gene on stability, function, and the intracellular behavior of copper-transporting ATPase ATP7B. |
 Ann Hubbard | Dr Ann Hubbard is a professor in the Department of Cell Biology at the Johns Hopkins Medical School in Baltimore, USA. Her research has been focused generally on pathways and mechanisms of membrane protein trafficking in polarized epithelial cells, specifically hepatocytes and intestinal cells. Currently, her lab is determining the structural signals that direct the copper-dependent movements of the copper-transporting ATPases, ATP7B and ATP7A, within epithelial cells. |
Copper content in the central nervous system
The human brain requires copper for its normal development and function. Compared to other organs in the body, copper concentration in the brain is one of the highest, second only to the liver,1 which is the major organ for copper uptake and excretion. The estimated copper content of human brain ranges from 2.9 to 10.7 μg Cu/g wet weight, while rat brains appear to have a lower copper content-1–2.3 μg Cu/g wet weight.2,3 Fractionation of brain homogenates demonstrates that copper is present in all major cell compartments, with nuclei and mitochondria containing 1.12–1.3 μg Cu/g, microsomes-0.49–0.65 μg Cu/g, and the cytosol-2.56 ± 1.02 μg Cu/g.4 Disease states alter copper concentrations in the CNS. Copper elevation (in the range of 7.8–37.8 μg/l) was observed in patients with neurological symptoms related to dementia;5 in Alzheimer's-like dementia CNS copper was reported to be 2-fold higher than in age matched controls.6,7Copper is not uniformly distributed in the CNS. Experiments in adult rats (6–12 months old) using laser ablation inductively coupled plasma mass spectrometry (LA-ICP-MS) have yielded two-dimensional maps of copper distribution in brain sections.8 These maps show 63Cu enrichment in the medial geniculate nucleus (the center processing visual information), superior colliculus (a component of the midbrain associated with motor functions) and periaqueductal grey (the gray matter located around the cerebral aqueduct of the midbrain that modulates pain and defensive behavior). Copper is also high in the lateral amygdala (groups of nuclei involved in memory and emotional reactions) and in the dorsomedial aspect of the diencephalon (the part of the forebrain containing thalamus, hypothalamus and the posterior portion of the pituitary gland). It is notable that copper distribution within the CNS varies between species. For example, copper levels in rat hippocampus are moderate. In contrast, the human hippocampus has the highest copper concentration (up to 15 μg g−1) compared to other regions.9 Since human brains are considerably larger than rodent brains, the LA-ICP-MS methodology has only been applied to certain regions, such as the insular, central and hippocampal areas.9,10 Analysis of these areas further confirmed the non-uniform distribution of copper. Thus, in the hippocampus, copper is highly enriched in the pyramidal and lacunosum molecular layers of cornu ammonis.10
Higher copper concentrations in certain regions of the brain most likely reflect higher metabolic demands for copper in these regions. The metabolic demands for copper within the CNS change during brain development. In humans and in rats, copper concentration in the CNS increases with age.11,12 Detailed studies by Tarohda and colleagues12 demonstrated that the rat neonatal brain has low copper levels, which rapidly increase during the first two weeks after birth (Fig. 1). A particularly striking increase is observed between the postnatal day 7 and 14, when the striatum, thalamus, and superior colliculus show marked and preferential accumulation of copper.12 The proper copper delivery to the brain during this developmental period can be literally a matter of life and death. Severely copper-deficient mice, which otherwise die, can be rescued by a single copper injection administered before postnatal day 7.13–15 This copper supplementation results in a significant restoration of morphology and function of the CNS. The sufficiency of a one-dose injection suggests that copper in the brain is largely recyclable, and only a small additional amount of copper is needed during an animal's life. In humans, very early treatment with copper-(histidine)2 complex produces beneficial effects in copper deficient Menkes disease patients and corrects some neurological symptoms.16,17
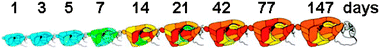 | ||
| Fig. 1 Time-dependent changes in copper concentration in different regions of rat brain (reproduced with modifications from Tarohda et al., 2004, Springer permission #2414301072545) Pseudo-colors indicate minimal amounts of copper (blue) and maximum amount of copper (red). The scale (0–3.95 μg g−1 tissue). | ||
Copper-requiring enzymes play essential roles in brain development and function
In the CNS, copper is utilized for general cellular metabolism such as respiration and radical defense, as well as for specialized processes such as production of neuroendocrine peptides and hormones. The mitochondria function and protection against reactive oxygen species in the cytosol rely on the activities of copper-dependent enzymes cytochrome C oxidase (CCO) and Cu,Zn-dependent superoxide dismutase 1 (SOD1), respectively. Superoxides at the cell surface are neutralized by extracellular membrane-bound SOD3, which also maintains cerebral vascular tone and regulates neurogenesis.18,19 Many peptide hormones and neurotransmitters such as oxytocin, vasopressin, gastrin, corticotropin-releasing factor, thyotropin-releasing hormone and others require the presence of an α-amide moiety for their activity.20 Amidation of neuropeptides and the production of norepinephrine is mediated by two homologous cuproenzymes: peptidyl-α-amidating monooxygenase (PAM) and dopamine-β-hydroxylase (DBH), respectively. Another DBH-like monooxygenase MOXD1 is expressed throughout the brain (olfactory bulb, pituitary, cerebellum, parietal cortex), but its physiological role is unclear.21Recent studies indicate that several enzymes well known for their roles in peripheral tissues are also present in the brain and therefore contribute to normal brain function. For example, the copper-requiring enzymes, tyrosinase and tyrosinase-related protein, are best known as key players in melanin synthesis in melanocytes. These two proteins have also been found in developing and adult brain with particularly high expression in the cortex, olfactory system, hippocampus, epithalamus and substantia nigra.22–24 The role of tyrosinase in the CNS has yet to be fully understood; one suggestion is that it oxidizes excess dopamine and thus prevents damage of catecholaminergic neurons caused by dopamine auto-oxidation.25 However, increased susceptibility of neurons to dopamine in response to tyrosinase overexpression has also been reported,24 illustrating the need for better understanding of the function of this copper-dependent enzyme in the CNS. An interesting signaling role for tyrosinase was proposed in studies on the development of the visual system where tyrosinase expression was shown to determine the pathway taken by ganglion cell axons.26
The lysyl oxidase (LOX) protein family, another class of copper dependent enzymes with a well-characterized peripheral function, is also found in the CNS. These proteins catalyze the oxidation of the side chain of a lysine as the first step of a cross-linking process that leads to the formation of collagen and elastin. In the CNS of normal rats and mice, expression of LOX was observed in the choroid plexus, blood vessel walls, brain matrix, and neurons.27 The LOX activity is needed for the formation and remodeling of the extracellular matrix during development and for maintaining normal structural organization of tissue.28 In pathological states, the involvement of LOX is also apparent; recent data suggest that LOX is upregulated in malignant astrocytes and its activity might be necessary for the migratory behavior of these cells.29
The multicopper oxidase ceruloplasmin and the blood coagulation factor fVIII share significant structural homology but have very different properties and functions. Factor fVIII is a component of a blood clotting cascade; copper binding to fVIII is required for stable association of protein chains, i.e. the major role for copper in fVIII is structural.30 In contrast, ceruloplasmin uses copper for electron transfer. By oxidizing ferrous ion into ferric ion, ceruloplasmin facilitates iron efflux from tissues.31 The neuronal degeneration caused by the loss of ceruloplasmin function in the CNS becomes particularly apparent with age.32–34 In brains of patients with aceruloplasminemia and in mice lacking ceruloplasmin, astrocytes show a notable accumulation of iron associated with a significant deformity and cell loss. The number of Purkinje neurons is also reduced and their iron metabolism is altered. However, in contrast to astrocytes, neurons of ceruloplasmin-deficient mice appear iron-deprived.34 Since the activity of ceruloplasmin strictly depends on copper availability, an inadequate supply of copper to CNS is likely to trigger cell-specific pathological responses similar to those observed in aceruloplasminemia.
There is accumulating evidence linking changes in cellular copper levels to changes in the biochemical and cell biological properties of amyloid precursor proteins (APP and APLP), which are associated with the pathology of Alzheimer's disease (AD).35,36 The abnormal proteolytic processing of APP is known to result in the accumulation of Aβ fragment and plaque formation in the brains of AD patients. Copper is found in high concentration in these deposits; drugs that decrease copper levels in the CNS decrease the rate of plaque formation.37–39In vitro, complexes of Aβ with copper oxidize cholesterol producing 4-cholesten-3-one; coincidentally brain tissue from AD subjects has almost 100% more 4-cholesten-3-one than tissue from age-matched controls.40 Copper was also shown to bind to the full-length APP and regulate APP endocytosis.41 In animal models of AD, modulation of copper levels significantly affects the course of the disease.42,43 Reciprocally, copper accumulation in cultured fibroblasts from Menkes disease patients shows significant upregulation of APP mRNA and protein, while copper depletion down-regulates APP.44,45 The connection between copper and the aggregation state of α-synuclein (in Parkinson’sisease) and prion protein (transmissible spongiform encephalopathy) has also emerged (for recent detailed review see ref. 46).
Since many, if not all, cells in the CNS express more than one copper-dependent enzyme, the complexity of cellular responses to changing copper levels is bound to be very high. The shift in distribution of copper between cuproenzymes and/or between different cell compartments, as well as cell-specific loss (or accumulation) of the metal may trigger/enhance CNS pathology, even if total copper levels in the brain do not change dramatically. The apparent involvement of copper in the pathology of Alzheimer's disease47–49 and Parkinson's disease50–52 is likely to reflect not only the direct interaction of copper with APP53–55 or its role in the catecholamine production,56–58 but an overall change in copper metabolism in the aging brain. The next important frontier of copper neurobiology is to provide mechanistic understanding of copper metabolism in the CNS and its regulation in different cell types as well as the entire brain.
Copper imbalance has severe consequences for CNS function
Given numerous physiological processes that require copper (see above), it is not surprising that insufficient copper supply during brain development has long-lasting consequences.59 Severe copper deprivation is associated with marked developmental defects in humans and animals. Inactivation of copper-dependent enzymes can result in altered cell morphology,28 increased inflammation,60 and even embryonic death. In fish, genetic or dietary copper limitation is associated with abnormal notochord development and impaired neurogenesis affecting both the midbrain-hindbrain region as well as primary motor neurons.61 In rodents, copper deficiency is associated with vacuolization of neurons, necrosis, edema in the cerebral cortex and corpus striatum and convulsive seizures.3,62Copper deficiency in humans can be caused by several factors, such as genetic defects resulting in lower copper absorption and abnormal copper distribution (as in Menkes disease), or limited absorption due to an inadequate diet. Neuropathology due to copper malabsorption has also been reported as a complication of bariatric surgeries or other operations affecting the gastrointestinal tract.63 Overuse of dietary supplements such as zinc and iron, and certain drugs (such as D-penicillamine) may greatly decrease copper availability and, if used in pregnancy, result in congenital malformations in infants.64 In adults, copper deficiency results in myeloneuropathy, anemia65 and motor neuron disease that can have manifestations similar to those of amyotrophic lateral sclerosis.66 Either genetic or acquired copper deficiency leads to demyelination and microcavitation of the neuropil in the white matter of the spinal cord and brainstem.67 There has been a growing appreciation that neurological manifestations caused by copper deficiency may represent a distinct syndrome; early recognition of this syndrome may prevent neurologic disability.65
Angiogenesis, an important physiological process of blood vessel growth, is essential for formation of the circulatory system. On the other hand, angiogenic blood vessels are the major source of sustenance for neoplastic tumors and facilitators of metastatic tumor migration. There is a large body of literature indicating that copper sequestration diminishes angiogenesis and that tumors are more significantly affected by such treatment compared to surrounding tissue.68,69 Copper chelators D-penicillamine and tetrathiomolybdate have been repeatedly shown to decrease the microvascular density and tumor volume in murine models.70–72 However, the application of D-penicillamine to treatment of glioblastoma in clinical trials did not improve patient survival73 illustrating the need for a better mechanistic understanding of the role of copper in angiogenesis and tumor development.
Another process in the CNS with a well-established link to copper homeostasis is a myelination of neurons. Copper deficiency is associated with the loss of myelin; this process can be reversed by copper supplementation. Delayed myelination has been observed in infants suffering from Menkes disease,74 a disorder associated with severe copper deficiency in the CNS. Similar to angiogenesis, the specific role of copper in myelination is not well understood; the reversible nature of copper effects suggests a regulatory role. One suggested mechanism by which Cu deficiency might lead to a delayed or decreased rate of myelination involves AMP-activated protein kinase (AMPK) inhibiting acetyl-CoA carboxylase (ACC), the latter being involved in fatty acid biosynthesis.75 In addition, studies of copper-dependent myelination in rats have demonstrated that copper imbalance is associated with changes in mRNA levels of proteins forming myelin sheath.76,77 Similarly, the microarray experiments using mRNA from a post-mortem cerebral cortex and cerebellum of a Menkes disease patient showed preferential dysregulation of genes involved in myelination, energy metabolism, and translation/ribosomal function.78 Thus, copper levels appear to influence transcription and/or translation of protein components of myelin.
Two human genetic disorders illustrate harmful consequences of either copper deficiency or copper overload. Menkes disease, caused by mutations in the gene encoding copper-transporting ATPase ATP7A, is an X-linked, fatal disorder of copper metabolism. Impaired copper delivery to the brain in patients with Menkes disease results in marked metabolic and developmental changes, progressive neuro-degeneration and death of a majority of patients in early childhood.74,79 Patients exhibit a loss of neurons in the granular layer of the cerebellum, cortical atrophy, and extensive degeneration of grey matter. Insufficient copper supply is a likely cause of low dopamine-β-hydroxylase activity and a resultant abnormal ratio of DOPA to catechol, which is used as a diagnostic test. Among cuproenzymes in the CNS, lysyl oxidase appears particularly sensitive to a diminished copper supply. In Menkes disease patients, vascular tortuosity is common and is ascribed to the loss of lysyl oxidase function.80,81
Wilson's disease (WD) is an example of severe consequences for CNS function caused by copper overload. WD is caused by mutations inactivating the copper transporting ATPase ATP7B.82 In WD patients, copper accumulates in the liver, brain and kidneys; the CSF copper concentration is also elevated (76.25 ± 5.95 μg L−1).83 A large proportion of WD patients (about 40%) display neurological and/or psychiatric symptoms84 that are ascribed to copper imbalance in the CNS. In advanced WD, several regions of the brain can be affected, including the thalamus, subthalamic nuclei, brainstem, and frontal cortex. The pathology manifests as atrophy, spongy degeneration, increased ventricular size, cavitation and cyst formation in the putamen.85
Copper transport in the CNS
The molecular mechanism through which copper is distributed within the brain among different cell populations is not clear. The concentration and form(s) in which copper exists in the interstitial fluid bathing cells are also unknown. Copper concentration in cerebrospinal fluid (CSF), largely produced by the choroid plexus, was estimated to be 0.3–0.5 μM or 22.3 ± 2.23 μg L−1. These levels are considerably lower than the total copper concentration in serum (1129 ± 124 μg L−1,5,86,87), although the amounts of exchangeable copper in the blood and CSF have not been measured/compared. Nevertheless, the ∼50-fold difference between copper in the blood and CSF suggests that the blood brain barrier effectively limits copper's access to the brain. Analysis of CSF by gel filtration produced a complex profile of copper-containing molecules with masses ranging from small ligands to proteins with apparent molecular weights of 15–66 kDa.86 It remains unknown whether these protein components represent carriers of exchangeable copper or copper-containing enzymes. The in vitro studies using hypothalamic slices and the clinical data indicate that the copper-His2 complex can be used by the cells of the CNS;17,88,89 whether such a complex exists endogenously is unclear.Recently, brain perfusion with 64CuCl2, 64Cu-albumin, and 64Cu-ceruloplasmin was employed to compare the rates of copper uptake into brain capillaries, parenchyma, choroid plexus, and CSF.90 Very little protein-bound copper was absorbed compared to “free” copper (from 64CuCl2); the major accumulation of 64Cu was in the choroid plexus. Comparison of the initial rates of copper transport demonstrated that the entry of copper into cells of the choroid plexus was 3–4 times faster than that into brain capillaries, and 9–10 times faster than copper entry into the brain parenchyma. Copper entry into the CSF was negligible in these short-term experiments.90 These observations suggest that copper efflux from the cells of brain barriers rather than uptake is the rate limiting step for copper delivery to brain parenchyma. It was suggested that choroid plexus may tightly regulate the movement of Cu into the CSF.90 The data also suggest that parenchymal cells receive copper from the blood and not from CSF.90 The machinery involved in copper uptake, distribution and export in the CNS is described in sections below.
At the individual cell level, the uptake of copper by astrocytes was examined using enriched primary cultures. These studies showed transport with an apparent Km of 9.7 ± 2.4 μM (5–30 min).91 This Km value is similar to the Km of copper transporter hCTR1 (see below for details) heterologously expressed in insect cells (8.9 μM)92 and slightly higher than that of hCTR1 expressed in HEK293 cells (2.56 ± 1.04 μM).93 Similar uptake rates and basal copper content of astocytes (1.1 nmol copper per mg protein) and liver parenchymal cells (1.4 nmol mg−1)94 or hepatoblastoma HepG2 cells (1.3 nmol mg−1)95 suggest that copper handling in the CNS and peripheral cells could be similar. At the same time, some pharmacologic differences were observed between astrocytes and HEK293 cells pointing to additional components contributing to copper regulation in the CNS. For example, a strong inhibition of copper accumulation by zinc was seen in cultured astrocytes but not in HEK293 cells, suggesting that additional zinc-sensitive copper transporter(s) may be present in astrocytes. The molecular identity of these other transporters is unknown; two other proteins with proposed roles in copper uptake are DMT96 and CTR2.97
Molecules regulating copper homeostasis in the brain
Presently, our knowledge of copper handling machinery in different cell types of the CNS is rudimentary. Nevertheless, it is clear that all the key copper handling proteins mediating copper homeostasis in peripheral tissues are also present in the brain (Fig. 2). These proteins can be divided into three major groups. Copper uptake transporters transfer copper into the cytosol; copper chaperones facilitate copper distribution to intracellular protein targets; whereas Cu-transporting ATPases translocate copper from the cytosol to the lumen of the secretory pathway and small vesicles for the delivery of copper to newly synthesized cuproenzymes and export into extracellular fluids, respectively (Fig. 2). Cu-ATPase may also facilitate copper efflux into the synaptic cleft,98,99 although this has not been formally shown (the in vitro measurements using synaptosomes and the copper-dependent quenching of tetrakis-(4-sulfophenyl)porphine estimated copper concentration in synaptic cleft as 2–4 μM100). The physiologic significance of synaptic release of copper is not very clear; one suggestion is that copper protects against excitotoxic cell death by regulating the activities of extra-synaptic NMDA receptors.98,99 Overall, the complexity of CNS architecture and homeostasis implies sophisticated regulation of copper metabolism. Mechanistic understanding of this regulation on the molecular and cellular levels is a wide-open area of study. | ||
| Fig. 2 Copper distribution in a generalized cell in the CNS. In extracellular fluids copper (green balls) is bound to either specific copper carriers (exchangeable copper) or to enzymes that use copper as a cofactor (cuproenzymes). Copper enters the cell via the high affinity copper transporter CTR1, located at the plasma membrane. The levels of CTR1 at the membrane can be regulated via recycling mechanism. Copper binds to cytosolic copper chaperones CCS and Atox1, which facilitate copper delivery to SOD1 and Cu-ATPases ATP7A and ATP7B, respectively. ATP7A and ATP7B transfer copper into the lumen of the TGN for incorporation into secreted and plasma membrane-bound cuproenzymes. When Cu is elevated or in response to other signals (such as activation of NMDA receptor), ATP7A moves from the TGN and facilitates copper excretion. Whether or not ATP7B traffics in the CNS is presently unknown. | ||
High affinity copper transporter CTR1
Copper transporter 1 (CTR1) is the major pathway for copper entry into cells. CTR1 function is essential for organism growth and development; the whole-body knockout of CTR1 is associated with embryonic lethality in mice and fish.101–103 This phenotype is likely due to severe copper deficiency in tissues, although a copper-independent role for CTR1 in development has also been reported.104 Experiments in zebrafish revealed that neural tissue is particularly sensitive to the loss of CTR1 function,103 as indicated by marked cell death in the brain and spinal cord in response to CTR1 down-regulation. In mice lacking CTR1, neuroepithelial layering is aberrant, and the embryo exhibits impaired neural tube closure.101,102In mammals, CTR1 mRNA is uniformly expressed in the brain and is higher in the choroid plexus.105 The CTR1 protein levels appear to parallel the mRNA levels with the most intense immunostaining observed in the choroid plexus and in the endothelial cells of small blood vessels.106 Astrocytes and neurons do not show marked differences in CTR1 levels, at least in culture. For example, human CCF-STTG1 astrocytoma and SY5Y neuroblastoma cells express comparable amounts of CTR1 mRNA and show similar uptake of copper.107 The level of CTR1 in the choroid plexus can be altered (upregulated) by dietary copper deficiency.106
By analogy with other tissues, CTR1 is expected to be present at the plasma membrane of cells forming the CNS (Fig. 2). However, the polarity of CTR1's distribution needs to be determined, particularly for cells of brain barriers. In peripheral tissues, the abundance of CTR1 at the plasma membrane is regulated in response to changes in copper levels.106,108,109 High copper facilitates endocytosis of CTR1 to intracellular vesicles, whereas lowering copper recruits CTR1 to the plasma membrane. Whether CTR1 is “silent” in the intracellular vesicles or is able to release the luminal copper content is presently unknown. Also, despite significant effort, the biochemical mechanism of copper transport by CTR1 remains largely enigmatic. Current data suggest that CTR1-mediated permeation of copper does not require ATP hydrolysis or co-transport of another ion.92,102,110 Reducing agents facilitate transport suggesting that CTR1 transports Cu(I); this conclusion is further supported by the Ag+-mediated inhibition of CTR1. Zinc and other divalent metals do not affect the CTR1 transport function.102
The structure of human CTR1 has been solved by electron microscopy with 6–7 A resolution.111 CTR1 is a stable trimer of identical monomers with a conducting pore for copper located in the center of the molecule between the monomers. Each monomer of CTR1 is a 190 amino-acid membrane protein with three trans-membrane segments. The N-terminus of CTR1 is oriented extracellularly and is N- and O-glycosylated; the glycosylation protects the protein against proteolysis.112 Structural data in combination with extensive site-directed mutagenesis92,93,113 have demonstrated that the functionally and structurally important residues are predominantly clustered around the pore entry and exit or contribute to helix packing.113 At the same time, the vast majority of residues lining the “pore” are not critical for copper transport.92 This observation points to the lack of tight interaction between copper and the walls of the permeation pathway during transport. The conserved MxxxM motif at the pore entrance is a notable exception; mutation of this motif abolishes copper transport.92,93 The structure of CTR1 suggests that it may act as a gated channel in which copper binding to the extracellular surface triggers opening of the cytosolic gate and allowing copper release. Copper that exits CTR1 is thought to bind to cytosolic copper chaperones that act as specific shuttles delivering copper to various intracellular destinations (Fig. 2).
Copper chaperone CCS
Human Copper Chaperone for Superoxide dismutase 1 (CCS) has two important functions: it facilitates the formation of an essential disulfide bond in its target protein superoxide dismutase 1 (SOD1) and delivers copper to the SOD1 catalytic site. CCS is a soluble protein with a predominant localization in the cytosol. The 274 amino-acid residues that compose CCS are folded into three distinct domains (Fig. 3). Domain I is a structural homologue of another copper chaperone, Atox1 (see below). This domain has a ferredoxin fold with a MxCxxC copper binding motif in the exposed loop and is thought to participate in copper binding, although the CxxC motif can be mutated without a negative effect on copper transfer or disulfide formation in SOD1. In rats, Domain I was shown to link CCS to an aspartic protease BACE1 allowing co-transport of the two proteins along the axon.114 The physiological consequences of this interesting partnership, which can be disrupted by RNA aptamers,115 is unknown. Domain II of CCS is highly homologous to SOD1 and is responsible for hetero-dimerization of CCS and SOD1 during copper transfer. Domain III is small and has a large extended loop with the highly conserved CXC motif (Fig. 3). This domain is required for both functional activities of CCS: it participates in the disulfide bond formation and in copper transfer to SOD1. The proposed mechanism of CCS-mediated activation has been described in a recent excellent review.116 Briefly, CCS interacts with SOD1 via Domain II; the heterodimerization facilitates the formation of a disulfide bond between Cys229 of Domain III in CCS and Cys57 of SOD1. Under aerobic conditions this bond is rapidly converted into the intramolecular disulfide in SOD1.117 At which step Cu is being inserted and the precise copper coordination during transfer remain controversial.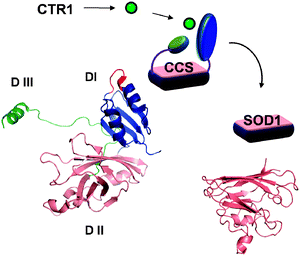 | ||
| Fig. 3 Cytosolic copper chaperone CCS transfers copper to its target SOD1 in the cytosol and in the inter-membrane space of mitochondria. CCS has three structural domains (DI-DIII), which have distinct function in binding of copper, docking to SOD1, and incorporating copper and disulfide bond. The MxCxxC motif is shown in red. The critical CxC motif is located in the extended part of DIII (green). | ||
In addition to its primary location and function in the cytosol, a small fraction of CCS is found in mitochondria and peroxisomes. Hypoxia facilitates the entry of SOD1, the downstream target of CCS, into mitochondria. CCS is thought to aid this process by incorporating copper and disulfide bond into SOD1 that enters mitochondria, thus increasing retention of the latter in the inter-membrane space. While the role of CCS in the maturation of SOD1 is without dispute, the function of CCS at the organism level appears to be critical only under stress conditions. Genetic inactivation of CCS in mice does not lead to an obvious phenotype. In contrast, the overexpression of CCS markedly accelerates neurological deficits and death of the transgenic mice bearing the G93A-SOD1 (a mutant SOD1 associated with amyotropic lateral sclerosis), apparently by facilitating mitochondria targeting of mutant SOD1. The CCS/wild-type-SOD1 dual transgenic mice are neurologically normal.118
Copper chaperone Atox1
Human Atox1 is a small (68 amino acids) cytosolic protein, which is highly conserved (≥85% sequence identity) in mammalian species and has orthologues in lower eukaryotes and all other phyla. In solution, Atox1 exists as a monomer, which likely represents the functional form. However, in the presence of metals Atox1 can form dimers;119 whether such dimers have a distinct functional role in the cell is currently unknown. The crystallographic and spectroscopic studies of Atox1 have demonstrated that the protein folds into a compact ferredoxin-like structure (Fig. 4). Copper binds to Atox1 within the surface-exposed loop that connects the first β-strand and the first α-helix. The loop contains a highly conserved motif MxCxxC, in which two cysteines coordinate copper, whereas methionine is buried in the hydrophobic core of Atox1 and stabilizes protein structure.120 Copper binding to Atox1 does not change protein structure but makes it more rigid.121 | ||
| Fig. 4 Copper chaperone Atox1 transfers copper to the N-terminal metal binding sites of Cu-transporting ATPases ATP7A and ATP7B. The Cu-ATPases transport copper across membrane using the energy of ATP hydrolysis. The structure of Atox1 (shown on the right) is similar to the structure of individual metal binding domains of Cu-ATPases (shown in blue and numbered 1–6). The copper-binding loop MxCxxC is shown in red; copper is shown as a green ball. | ||
In vitro, the binding of copper by Atox1 has been explored in detail;121–123 however the mechanism by which Atox1 and other copper chaperones acquire copper in cells remains poorly understood. Several possibilities exist that are not mutually exclusive: (i) Atox1 may directly interact with CTR1 and retrieve copper at the “exit gate” of the transporter; (ii) it may directly or indirectly exchange copper with other copper carrying proteins (such as copper chaperone CCS, which has an Atox1-like domain, see above); and/or (iii) Atox1 can bind copper from a copper-glutathione complex. The last possibility has been shown to occur in vitro in experiments where Cu-glutathione was used to produce a functional Atox1-Cu(I) complex.124 Glutathione also weakly binds to Cu-Atox1 as a third copper-coordinating ligand with a Kd of 10–25 mM.122 In CNS, the intracellular levels of glutathione are unlikely to reach sufficiently high concentrations for it to form stable adducts with Atox1-Cu and interfere with the subsequent transfer of copper, although this has not been formally investigated. (The estimate for glutathione levels in cultured cells lines ranges from 1–40 nmol mg−1 protein, with neurons containing less glutathione than astroglia.125,126)
The expression of Atox1 in a normal adult brain has been characterized for humans and rats using Northern blotting and in situ hybridization histochemistry.127,128 Widespread expression was observed,127 with the highest levels of Atox1 mRNA found in the pyramidal neurons of cerebral cortex and hippocampus, as well as in neurons of the locus coeruleus.128 Atox1 is moderately expressed throughout the olfactory bulb and is low in cerebellum with the notable exception of Purkinje neurons, where the expression was reported as very high. It is thought that the function of Atox1 is common for all cell types, including cells of the CNS, and involves primarily the delivery of copper to the copper transporting ATPases (Cu-ATPases) ATP7A and ATP7B (Fig. 4; for details on Cu-ATPases see below). There is a strong evidence for direct interaction between Atox1 and the N-terminal region of Cu-ATPases as well as the transfer of copper from Atox1 to the metal-binding sites of Cu-ATPases.124,129–132 Atox1-mediated copper transfer is accompanied by upregulation of the Cu-ATPase's activity.124 Reciprocally, apo-Atox1 can retrieve copper from the ATPases, at least in vitro, and down-regulate their activity.124 Thus, in cells Atox1 may function as a sensor that controls a proper delivery of copper to the secretory pathway. In fact, recent studies demonstrate a marked change in the intracellular distribution of copper in cells lacking functional Atox1.133,134
In addition to a copper-transfer function, Atox1 was proposed to play an antioxidant role, because its over-expression in yeast cells lacking SOD1 was shown to suppress growth defects caused by the loss of SOD1 activity.135 The intracellular levels of functional Atox1 have indeed significant consequences for neuronal cell survival, although the mechanism of this phenomenon remains unexplored. Experiments using different neuronal cells (SKNMC, a neuroblastoma cell line; NT-2, a teratocarcinoma cell line and GT-1, hypothalamic neuronal cells) showed higher viability of cells over-expressing Atox1 under conditions of serum deprivation.136 Furthermore, cells expressing the Atox1 variant with mutated cysteines in the MxCxxC motif were less metabolically active and showed decreased viability under either regular conditions or serum deprivation.136 It seems that the over-expressed active Atox1 may provide protection during stress, whereas the over-expressed mutant may compete with the endogenous Atox1 for its docking sites at the N-terminus of ATP7A and/or ATP7B and thus diminish copper delivery to the secretory pathway. In addition, if Atox1 receives copper directly through interactions with CTR1, then the excess of mutant Atox1 may decrease normal copper uptake into cells.
Recent studies demonstrated the role of Atox1 in cell growth and proliferation, and it was proposed that Atox1 may act as a transcription regulator.137 It is also interesting that the changes in the Atox1 levels appear to induce remodeling of the entire copper-metabolic network. An increase in copper-transporting ATPase ATP7A levels was observed in the immortalized Atox1−/− cells,138 whereas in a separate study, the protein and mRNA levels of SOD3, a downstream target of ATP7A, were dramatically decreased in cultured Atox1−/− fibroblasts.139 Complete Atox1 inactivation in mice has grave consequences for their development and function. The Atox1−/− mice display growth retardation and significant mortality (up to 45%); in some cases death can be a result of prolonged seizure activity.140 Analysis of copper metabolism revealed that newborn Atox1−/− mice have markedly reduced levels of copper in the liver and brain due to decreased placental copper transport.140 They also exhibit low activity of several copper-dependent enzymes including cytochrome C oxidase (CCO). Some animals display severe congenital eye defects and microphthalmia;140 the precise molecular mechanisms behind these pathologies remain unknown.
The cytochrome C oxidase assembly factors
Copper delivery to mitochondria is essential for cell survival, because copper is required for the activity of CCO. CCO is a terminal enzyme of the mitochondria respiratory chain; it catalyzes electron transfer to molecular oxygen and contributes to formation of the electrochemical potential that is used for the synthesis of ATP. Human CCO consists of 13 subunits; the assembly of the entire complex and copper delivery to the catalytic sites of CCO is a fascinating and complex process, which remains poorly understood. Studies in yeast have suggested that the cytosolic protein Cox17 may play an important role in copper delivery to mitochondria,141 however subsequent studies showed that tethering Cox17 to the mitochondria inner membrane does not disrupt copper delivery to CCO,142i.e. other cytosolic molecules must accept copper from Ctr1 and deliver it to the organelle. Nevertheless, Cox17 plays an important role in copper transfer to CCO; genetic inactivation of Cox17 in mice markedly reduces the CCO activity and is embryonic lethal.143 The siRNA knock-down of COX17 in human cells has demonstrated that the down-regulation of Cox17 is accompanied by abnormal formation of a CCO supercomplex.144 Human Cox17 is a 62-residue protein with a hairpin-like structure (Fig. 5) and one copper(I) binding site formed by two consecutive Cys residues.145 Recent studies also revealed the fascinating ability of Cox17 to transfer copper to other CCO chaperones (Cox11 and SCOI) along with the transfer of electrons.146 It was suggested that such coupled transfer may ensure the efficient delivery of the metal to proteins when conditions are oxidizing.146 | ||
| Fig. 5 Several proteins facilitate the maturation and assembly of CCO in mitochondria. Cox17 is a soluble protein found in the cytosol and, preferentially, in the intermembarne space. Cox17 can transfer copper (green ball) to Cox11, which is important for the CCO Cu(B) site formation, and to Sco1. Sco2 acts as oxidoreductase for Sco1; the activity of both Sco1 and Sco2 are required for the CCO assembly. In addition to copper-binding proteins, mitochondria have labile copper pool in the matrix. | ||
Other chaperones for CCO implicated in copper delivery (Cox11, Sco1 and Sco2) are membrane proteins (Fig. 5). Cox 11 is required for the formation of Cu(B) site in CCO.147 The structure of the soluble domain of Cox11 from bacterial homologue has been solved.148 These studies revealed that Cox11 has a novel β-immunoglobulin-like fold, shows propensity to dimerize in the absence of reductants, and binds copper via Cys residues at the dimerization interface.148 Cox11p is needed in catalytic amounts and is thought to act at the final step of CCO assembly.147 In contrast, Sco1 and Sco2 are more central to CCO assembly, as evidenced by grave consequences of Sco1/2 inactivation. In humans, mutations in Sco2 are associated with neonatal encephalocardiomyopathy,149,150 while Sco1 mutations cause neonatal hepatic failure and encephalopathy.151 Analysis of Sco2 and Sco1 structures revealed that both proteins have a thioredoxin fold, which led to the suggestion that Scos function as “redox signaling molecules”152 rather than copper carriers. The redox function for Sco2 has recently been directly demonstrated by Leary and colleagues.153 These authors have found that Sco2 acts as a thiol–disulfide oxidoreductase to oxidize the copper-binding cysteines of Sco1 during maturation of the CuA site of CCO.153 Interestingly, mutations in Sco1 and Sco2 are also associated with the reduction of cellular copper content, suggesting that the assembly of CCO and/or mitochondria function have a profound effect on overall cellular copper homeostasis. The important role of mitochondria in regulating cellular copper homeostasis is also suggested by the presence in the mitochondria matrix of exchangeable pool of copper (for review see ref. 154).
Copper-transporting ATPases ATP7A and ATP7B (Cu-ATPases)
Human Cu-ATPases play a particularly important role in CNS physiology. Genetic mutations in either ATP7A or ATP7B are associated with neuronal degeneration, demyelination of neurons, severe neurological, developmental and psychiatric problems (see Menkes disease and Wilson's disease, above). Decreased expression of mouse ATP7A in Purkinje cells was shown to be associated with impaired synaptogenesis and dramatic cytoskeletal dysfunction.155 Such severe consequences of Cu-ATPase inactivation illustrate not only the functional significance of these transporters but also the requirement for precise balancing of copper in the CNS. Although ATP7A and ATP7B are often co-expressed in same cells, permanently or during a certain stage of development (as in Purkinje neurons155–157), they do not fully compensate for each other's function when one is lost.155 The precise reason for this phenomenon is not clear, however differences in regulation and trafficking properties of Cu-ATPases are likely to contribute to the lack of complete functional complementation.ATP7A and ATP7B are large (175 kDa and 160 kDa) membrane proteins (Fig. 4). They belong to the diverse family of P-type ATPases and use the energy of ATP hydrolysis to transport copper from the cytosol into the lumen of the secretory pathway where copper is incorporated as a co-factor into various copper requiring enzymes (dopamine-β-hydroxylase, PAM, ceruloplasmin, SOD3, lysyl oxidase, etc., see above). It is thought that the biosynthetic incorporation of copper into these enzymes occurs largely in the trans-Golgi network (TGN). Consistent with this idea, ATP7A was found predominantly targeted to the TGN. Similarly, the presence of ATP7B in the TGN was shown for Purkinje neurons in the cerebellum. Interestingly, ATP7A was also found co-localized with its target PAM in secretory vesicles. This localization suggests that the function of ATP7A might be needed downstream of the TGN to maintain the copper levels (and thus the metallation state of PHM) in vesicles. In peripheral cells, both Cu-ATPases were shown to traffic from the TGN in response to changing copper levels and other stimuli. Regulated trafficking was also observed in the CNS for ATP7A (Fig. 2, see below section on trafficking).
ATP7A and ATP7B exhibit distinct expression patterns in the CNS. ATP7A is expressed throughout the brain both in embryonic and postnatal periods;105,158–160 this pattern suggests a housekeeping role for this copper transporter in the CNS. In the early postnatal period, ATP7A expression is most abundant in neocortex and cerebellum,158 whereas in the developing and adult brain, ATP7A levels are greatest in the choroid plexus/ependymal cells of the lateral and third ventricles.158 ATP7A expression decreases in most neuronal subpopulations from birth to adulthood, with the exception of CA2 hippocampal pyramidal layer where the levels of ATP7A increase.158 An age-dependent increase in the staining of ATP7A was also detected in Bergman glia;156 and in cerebellar Purkinje neurons, both up- and down-regulation were reported.105,156,158–160 The ATP7B mRNA and protein are present at high levels in the postnatal cerebellum.156 Tissue blotting using rat brains suggested the presence of ATP7B in neuronal cells of the hippocampus, olfactory bulbs, cerebellum, cerebral cortex and nuclei in the brainstem in which high amounts of dopamine-β-hydroxylase and SOD1 were also detected.161
In recent years, there has been a significant progress in describing the biochemical properties and domain structure of Cu-ATPases (for reviews see ref. 162–164). The Cu-ATPases show high sequence similarity, common domain structure and both hydrolyze ATP in a copper dependent manner with the formation of a transient phospho-intermediate. Unlike ATP7B, ATP7A is glycosylated, which increases its mass and allows separation of the two Cu-ATPases in gels. The membrane portion of human Cu-ATPases consists of 8 transmembrane segments that form the copper translocation pathway and copper-binding sites with a high selectivity for Cu(I). The bulk of the Cu-ATPases is exposed to the cytosol (Fig. 4). The cytosolic domains include: the N-terminal domain with 6 copper binding sites; the nucleotide-binding domain where ATP docks; the P (phosphorylation domain) with an invariant Asp (the site of catalytic phosphorylation); the A-domain that regulates the catalytic cycle and conformational transitions in the Cu-ATPases; and the C-terminal tail that is needed for protein stability and trafficking. In addition, both proteins have unique sequences that are thought to be responsible for differences in their regulation.164
Regulation of copper balance
Available data indicate that copper balance in the CNS is maintained under very tight control, and compensatory mechanisms are activated to achieve copper balance in disease situations. For example, in MoBr/y mice (an animal model of Menkes disease) the decreased expression of ATP7A protein in Purkinje cells is associated with impaired synaptogenesis and marked decrease in overall copper levels.155 These events are accompanied by compensatory upregulation of ATP7A in endothelial cells, as well as increased association of MoBr/y astrocytes and microglia with the blood–brain barrier. Interestingly, ATP7B is not upregulated.155Very little is known about specific molecular mechanisms that respond to changing copper levels in the CNS. The available data indicate that copper handling machinery is regulated in a cell-specific manner at many levels, including transcriptional, translational, and posttranslational control. For example, the mRNA and protein levels of copper chaperone CCS are sensitive to intracellular copper levels, and the response is cell-specific.165 In mice, copper deficiency is accompanied by an increase in the CCS levels in the cerebellum, whereas CCS in choroid plexus is essentially unchanged.165 In another example, the levels of ATP7A mRNA and protein change during development, as does the intracellular localization of ATP7A.156,158,166 In developing axons, ATP7A is initially expressed in cell bodies, whereas later it moves to extending axons, peaking in its amounts prior to synaptogenesis. Similarly, injury-stimulated neurogenesis is associated with the increase of ATP7A in neurons and axons prior to synaptogenesis.166
Transcriptional control was reported for both ATP7A and ATP7B. In neuroblastoma cells, ATP7A expression is triggered by retinoic acid receptor β (RAR β2).167 The factors regulating expression of the full-length ATP7B in the brain are unknown. However, the production of a shorter, pineal gland specific form of ATP7B (PINA) appears to be under control of a pineal/retina specific nuclear factor, cone rod homeobox CRX,168 and follows a circadian rhythm.52 In cultured pineal cells, expression of PINA can be stimulated by activating the cAMP signal transduction pathway.52 In addition to transcriptional control, ATP7A and ATP7B mRNA are subjects of alternative splicing, both in normal and disease states.169–176 The level of expression, developmental regulation, and trafficking behavior of such variants, when different from the full-length form, may have important effects on CNS copper homeostasis.
Regulation of copper balance through trafficking of Cu-ATPases
Although ATP7A and ATP7B are ∼65% identical and share a common transport function in the secretory pathway of cells, they exhibit several important differences.157,177,178 Perhaps most dramatic is the distinct trafficking behavior of each protein when intracellular Cu(I) levels increase and the enzymes initiate their Cu(I) export function. ATP7A disperses from the TGN in small vesicles, which move toward/to the basolateral region of polarized cells.178,179 In contrast, ATP7B in slightly larger vesicles moves toward the apical region of polarized cells (e.g., liver hepatocytes180–182). Several molecular signals necessary for the polarity of the trafficking have been identified on each protein183–188 but the recognition and transport machinery are still unknown (although relevant interacting partners such as dynactin p62189 and the PDZ-domain containing PICP/AIPP1190 were identified). Recent interesting study indicates that glutathionylation (and the removal of glutathione by glutaredoxin) may regulate the activity and trafficking of Cu-ATPases.191While it is thought that Cu-ATPases in vesicles actively sequester Cu(I) for export during vesicle fusion, the possibility remains that they act as vehicles for delivery of the specific Cu-ATPase to its appropriate plasma membrane destination. The composition of the ATP7A and ATP7B carrying vesicles needs to be determined to better understand the driving forces behind cell specific regulation of ATP7A and ATP7B (see below).
So far, work on Cu-ATPase trafficking has focused almost exclusively on tissues/cells outside of the CNS. To our knowledge, little is known of the Cu-responsive trafficking behavior of either Cu-ATPase in any region of the brain. Since in some tissues, where both Cu-ATPAses are expressed, there is evidence for one or the other protein not responding to changes in Cu levels,192–194 it is formally possible that ATP7A and ATP7B in the some cells of CNS function exclusively at the TGN. However, Cu-independent trafficking of ATP7A clearly occurs in hippocampal neurons in response to glutamate excitation of NMDA receptors.99 The ATP7A-mediated release of Cu appears to modulate the excitotoxicity of the NMDA receptor through nitrosylation.99 The signal(s) recruiting ATP7A from the cell body to the periphery are not known but are likely to be distinct from those responding to Cu(I) levels. Such novel results emphasize the need for more study of the CNS Cu-ATPases.
Conclusions
The above discussion has aimed to illustrate two major points. First, the available data leave little doubt that copper homeostasis plays a very important role in normal function and development of the mammalian CNS. Second, despite its importance, our knowledge of copper homeostasis on the brain is rudimentary, particularly considering the complexity of the CNS architecture and the abundance of regulatory mechanisms. With the major components of copper handling machinery now identified, the field is ripe for studies directed on understanding the mechanisms that control copper levels within the CNS, the role of different copper transporters and chaperones in CNS development and their adaptation to pathological changes. Such studies would contribute to better understanding of neuropathology of many disorders including Menkes disease, Wilson's disease, Alzheimer's disease, Parkinson's disease, and cancer.Acknowledgements
This work was supported by the NIH Program Project Grant P01 GM067166.References
- T. Lech and J. K. Sadlik, Biol. Trace Elem. Res., 2007, 118(1), 10–15 CrossRef CAS.
- L. Rongzhu, W. Suhua, X. Guangwei, R. Chunlan, H. Fangan, J. Junjie and M. Aschner, Biol. Trace Elem. Res., 2009, 130(1), 39–47 CrossRef.
- A. A. Gybina, I. Tkac and J. R. Prohaska, Nutr. Neurosci., 2009, 12(3), 114–122 Search PubMed.
- D. J. Waggoner, B. Drisaldi, T. B. Bartnikas, R. L. Casareno, J. R. Prohaska, J. D. Gitlin and D. A. Harris, J. Biol. Chem., 2000, 275(11), 7455–7458 CrossRef CAS.
- V. Nischwitz, A. Berthele and B. Michalke, Anal. Chim. Acta, 2008, 627(2), 258–269 CrossRef CAS.
- C. O. Hershey, L. A. Hershey, A. Varnes, S. D. Vibhakar, P. Lavin and W. H. Strain, Neurology, 1983, 33(10), 1350–1353 CAS.
- H. Basun, L. G. Forssell, L. Wetterberg and B. Winblad, J. Neural. Transm. Park. Dis. Dement. Sect., 1991, 3(4), 231–258 Search PubMed.
- B. Jackson, S. Harper, L. Smith and J. Flinn, Anal. Bioanal. Chem., 2006, 384(4), 951–957 CrossRef CAS.
- J. Dobrowolska, M. Dehnhardt, A. Matusch, M. Zoriy, N. Palomero-Gallagher, P. Koscielniak, K. Zilles and J. S. Becker, Talanta, 2008, 74(4), 717–723 CrossRef CAS.
- J. S. Becker, M. V. Zoriy, C. Pickhardt, N. Palomero-Gallagher and K. Zilles, Anal. Chem., 2005, 77(10), 3208–3216 CrossRef CAS.
- P. Zatta, D. Drago, P. Zambenedetti, S. Bolognin, E. Nogara, A. Peruffo and B. Cozzi, J. Chem. Neuroanat., 2008, 36(1), 1–5 CrossRef CAS.
- T. Tarohda, Y. Ishida, K. Kawai, M. Yamamoto and R. Amano, Anal. Bioanal. Chem., 2005, 383(2), 224–234 CrossRef CAS.
- Y. Nose, B. E. Kim and D. J. Thiele, Cell Metab., 2006, 4(3), 235–244 CrossRef CAS.
- T. Yamano, M. Shimada, A. Onaga, H. Kawasaki, S. Iwane, K. Ono and M. Nishimura, Acta Neuropathol., 1988, 76(6), 574–580 CrossRef CAS.
- H. Kawasaki, T. Yamano, S. Iwane and M. Shimada, Acta Neuropathol., 1988, 76(6), 606–612 CrossRef CAS.
- J. Kreuder, A. Otten, H. Fuder, Z. Tumer, T. Tonnesen, N. Horn and D. Dralle, Eur. J. Pediatr., 1993, 152(10), 828–832 CrossRef CAS.
- Z. Tumer and L. B. Moller, Eur. J. Hum. Genet., 2010, 18(5), 511–518.
- I. T. Demchenko, T. D. Oury, J. D. Crapo and C. A. Piantadosi, Circ. Res., 2002, 91(11), 1031–1037 CrossRef CAS.
- R. Rola, Y. Zou, T. T. Huang, K. Fishman, J. Baure, S. Rosi, H. Milliken, C. L. Limoli and J. R. Fike, Free Radical Biol. Med., 2007, 42(8), 1133–1145 CrossRef CAS ; discussion 1131–1132.
- B. A. Eipper, D. A. Stoffers and R. E. Mains, Annu. Rev. Neurosci., 1992, 15, 57–85 CrossRef CAS.
- X. Xin, R. E. Mains and B. A. Eipper, J. Biol. Chem., 2004, 279(46), 48159–48167 CrossRef CAS.
- K. Tief, A. Schmidt and F. Beermann, Mol. Brain Res., 1998, 53(1–2), 307–310 CrossRef CAS.
- K. Tief, M. Hahne, A. Schmidt and F. Beermann, Eur. J. Biochem., 1996, 241(1), 12–16 CrossRef CAS.
- E. Greggio, E. Bergantino, D. Carter, R. Ahmad, G. E. Costin, V. J. Hearing, J. Clarimon, A. Singleton, J. Eerola, O. Hellstrom, P. J. Tienari, D. W. Miller, A. Beilina, L. Bubacco and M. R. Cookson, J. Neurochem., 2005, 93(1), 246–256 CrossRef CAS.
- Y. Higashi, M. Asanuma, I. Miyazaki and N. Ogawa, J. Neurochem., 2000, 75(4), 1771–1774 CAS.
- C. A. Cronin, A. B. Ryan, E. M. Talley and H. Scrable, J. Neurosci., 2003, 23(37), 11692–11697 CAS.
- P. A. Li, Q. He, T. Cao, G. Yong, K. M. Szauter, K. S. Fong, J. Karlsson, M. F. Keep and K. Csiszar, Mol. Brain Res., 2004, 120(2), 115–122 CrossRef CAS.
- N. W. Bronson, J. S. Hamilton, M. Han, P. A. Li, I. Hornstra, J. M. Horowitz and B. A. Horwitz, Neurosci. Lett., 2005, 390(2), 118–122 CrossRef CAS.
- R. Laczko, K. M. Szauter, M. K. Jansen, P. Hollosi, M. Muranyi, J. Molnar, K. S. Fong, A. Hinek and K. Csiszar, Neuropathol. Appl. Neurobiol., 2007, 33(6), 631–643 CrossRef CAS.
- N. Bihoreau, S. Pin, A. M. de Kersabiec, F. Vidot and M. P. Fontaine-Aupart, Eur. J. Biochem., 1994, 222(1), 41–48 CAS.
- Z. L. Harris, A. P. Durley, T. K. Man and J. D. Gitlin, Proc. Natl. Acad. Sci. U. S. A., 1999, 96(19), 10812–10817 CrossRef CAS.
- K. Kaneko, K. Yoshida, K. Arima, S. Ohara, H. Miyajima, T. Kato, M. Ohta and S. I. Ikeda, J. Neuropathol. Exp. Neurol., 2002, 61(12), 1069–1077.
- H. Miyajima, S. Kono, Y. Takahashi, M. Sugimoto, M. Sakamoto and N. Sakai, Neurology, 2001, 57(12), 2205–2210 CAS.
- S. Y. Jeong and S. David, J. Neurosci., 2006, 26(38), 9810–9819 CrossRef CAS.
- N. C. Inestrosa, W. Cerpa and L. Varela-Nallar, IUBMB Life, 2005, 57(9), 645–650 Search PubMed.
- N. C. Inestrosa, A. E. Reyes, M. A. Chacon, W. Cerpa, A. Villalon, J. Montiel, G. Merabachvili, R. Aldunate, F. Bozinovic and F. Aboitiz, Neurobiol. Aging, 2005, 26(7), 1023–1028 CrossRef CAS.
- C. S. Atwood, R. C. Scarpa, X. Huang, R. D. Moir, W. D. Jones, D. P. Fairlie, R. E. Tanzi and A. I. Bush, J. Neurochem., 2000, 75(3), 1219–1233 CrossRef CAS.
- K. A. Price, P. J. Crouch and A. R. White, Recent Pat. CNS Drug Discovery, 2007, 2(3), 180–187 Search PubMed.
- S. Schafer, F. G. Pajonk, G. Multhaup and T. A. Bayer, J. Mol. Med., 2007, 85(4), 405–413 CrossRef.
- L. Puglielli, A. L. Friedlich, K. D. Setchell, S. Nagano, C. Opazo, R. A. Cherny, K. J. Barnham, J. D. Wade, S. Melov, D. M. Kovacs and A. I. Bush, J. Clin. Invest., 2005, 115(9), 2556–2563 CrossRef CAS.
- Y. H. Hung, E. L. Robb, I. Volitakis, M. Ho, G. Evin, Q. X. Li, J. G. Culvenor, C. L. Masters, R. A. Cherny and A. I. Bush, J. Biol. Chem., 2009, 284(33), 21899–21907 CrossRef CAS.
- D. L. Sparks, J. Alzheimers. Dis., 2008, 15(4), 641–656 CAS.
- A. L. Phinney, B. Drisaldi, S. D. Schmidt, S. Lugowski, V. Coronado, Y. Liang, P. Horne, J. Yang, J. Sekoulidis, J. Coomaraswamy, M. A. Chishti, D. W. Cox, P. M. Mathews, R. A. Nixon, G. A. Carlson, P. St George-Hyslop and D. Westaway, Proc. Natl. Acad. Sci. U. S. A., 2003, 100(24), 14193–14198 CrossRef CAS.
- A. D. Armendariz, M. Gonzalez, A. V. Loguinov and C. D. Vulpe, Physiol. Genomics, 2004, 20(1), 45–54 Search PubMed.
- S. A. Bellingham, D. K. Lahiri, B. Maloney, S. La Fontaine, G. Multhaup and J. Camakaris, J. Biol. Chem., 2004, 279(19), 20378–20386 CrossRef CAS.
- D. R. Brown, Metallomics, 2010, 2, 186–194 RSC.
- Y. H. Hung, A. I. Bush and R. A. Cherny, JBIC, J. Biol. Inorg. Chem., 2010, 15(1), 61–76 CrossRef CAS.
- D. R. Brown, Dalton Trans., 2009,(21), 4069–4076 RSC.
- J. F. Quinn, S. Crane, C. Harris and T. L. Wadsworth, Expert Rev. Neurother., 2009, 9(5), 631–637 CrossRef CAS.
- K. J. Barnham and A. I. Bush, Curr. Opin. Chem. Biol., 2008, 12(2), 222–228 CrossRef CAS.
- M. Alcaraz-Zubeldia, M. C. Boll-Woehrlen, S. Montes-Lopez, F. Perez-Severiano, J. C. Martinez-Lazcano, A. Diaz-Ruiz and C. Rios, Rev. Invest. Clin., 2009, 61(5), 405–411 Search PubMed.
- J. Borjigin, A. S. Payne, J. Deng, X. Li, M. M. Wang, B. Ovodenko, J. D. Gitlin and S. H. Snyder, J. Neurosci., 1999, 19(3), 1018–1026 CAS.
- M. Suazo, C. Hodar, C. Morgan, W. Cerpa, V. Cambiazo, N. C. Inestrosa and M. Gonzalez, Biochem. Biophys. Res. Commun., 2009, 382(4), 740–744 CrossRef CAS.
- G. K. Kong, L. A. Miles, G. A. Crespi, C. J. Morton, H. L. Ng, K. J. Barnham, W. J. McKinstry, R. Cappai and M. W. Parker, Eur. Biophys. J., 2008, 37(3), 269–279 CrossRef CAS.
- K. J. Barnham, W. J. McKinstry, G. Multhaup, D. Galatis, C. J. Morton, C. C. Curtain, N. A. Williamson, A. R. White, M. G. Hinds, R. S. Norton, K. Beyreuther, C. L. Masters, M. W. Parker and R. Cappai, J. Biol. Chem., 2003, 278(19), 17401–17407 CrossRef CAS.
- D. S. Goldstein, C. S. Holmes and S. G. Kaler, Neurochem. Res., 2009, 34(8), 1464–1468 CrossRef CAS.
- T. Saito, T. Nagao, M. Okabe and K. Saito, Neurosci. Lett., 1996, 216(3), 195–198 CAS.
- V. Gerbasi, S. Lutsenko and E. J. Lewis, Neurochem. Res., 2003, 28(6), 867–873 CrossRef CAS.
- J. R. Prohaska, Nutrition, 2000, 16(7–8), 502–504 CrossRef CAS.
- H. E. Lob, P. J. Marvar, T. J. Guzik, S. Sharma, L. A. McCann, C. Weyand, F. J. Gordon and D. G. Harrison, Hypertension, 2010, 55(2), 277–283 CrossRef CAS , 276p following 283.
- E. C. Madsen and J. D. Gitlin, PLoS Genet., 2008, 4(11), e1000261 Search PubMed.
- J. Y. Uriu-Adams, R. E. Scherr, L. Lanoue and C. L. Keen, Biofactors, 36(2), 136–152 Search PubMed.
- K. Juhasz-Pocsine, S. A. Rudnicki, R. L. Archer and S. I. Harik, Neurology, 2007, 68(21), 1843–1850 CrossRef.
- M. L. Martinez-Frias, E. Rodriguez-Pinilla, E. Bermejo and M. Blanco, Am. J. Med. Genet., 1998, 76(3), 274–275 CrossRef CAS.
- N. Kumar and P. A. Low, J. Neurol., 2004, 251(6), 747–749.
- C. C. Weihl and G. Lopate, Muscle Nerve, 2006, 34(6), 789–793 CrossRef CAS.
- B. Schleper and H. J. Stuerenburg, J. Neurol., 2001, 248(8), 705–706 CrossRef CAS.
- X. Z. Wu, Neoplasma, 2008, 55(6), 472–481 Search PubMed.
- D. Yoshida, Y. Ikeda and S. Nakazawa, Neurosurgery, 1995, 37(2), 287–292 CrossRef CAS , discussion 292–283.
- C. Cox, S. D. Merajver, S. Yoo, R. D. Dick, G. J. Brewer, J. S. Lee and T. N. Teknos, Arch. Otolaryngol., Head Neck Surg., 2003, 129(7), 781–785 CrossRef.
- S. S. Brem, D. Zagzag, A. M. Tsanaclis, S. Gately, M. P. Elkouby and S. E. Brien, Am. J. Pathol., 1990, 137(5), 1121–1142 Search PubMed.
- B. Hassouneh, M. Islam, T. Nagel, Q. Pan, S. D. Merajver and T. N. Teknos, Mol. Cancer Ther., 2007, 6(3), 1039–1045 CrossRef CAS.
- S. Brem, S. A. Grossman, K. A. Carson, P. New, S. Phuphanich, J. B. Alavi, T. Mikkelsen and J. D. Fisher, Neuro-Oncology, 2005, 7(3), 246–253 Search PubMed.
- S. G. Kaler, C. S. Holmes, D. S. Goldstein, J. Tang, S. C. Godwin, A. Donsante, C. J. Liew, S. Sato and N. Patronas, N. Engl. J. Med., 2008, 358(6), 605–614 CrossRef CAS.
- A. A. Gybina and J. R. Prohaska, Brain Res., 2008, 1204, 69–76 CrossRef CAS.
- N. Franco-Pons, J. Tomas, B. Roig, C. Auladell, L. Martorell and E. Vilella, J. Mol. Neurosci., 2009, 38(1), 2–11 Search PubMed.
- P. Morell, C. V. Barrett, J. L. Mason, A. D. Toews, J. D. Hostettler, G. W. Knapp and G. K. Matsushima, Mol. Cell. Neurosci., 1998, 12(4–5), 220–227 CrossRef CAS.
- P. C. Liu, Y. W. Chen, J. A. Centeno, M. Quezado, K. Lem and S. G. Kaler, Mol. Genet. Metab., 2005, 85(4), 291–300 CrossRef CAS.
- L. B. Moller, M. Mogensen and N. Horn, Biochimie, 2009, 91(10), 1273–1277 CrossRef CAS.
- S. C. Godwin, T. Shawker, B. Chang and S. G. Kaler, J. Pediatr., 2006, 149(3), 412–415 CrossRef.
- P. M. Royce and B. Steinmann, Pediatr. Res., 1990, 28(2), 137–141 CAS.
- S. K. Das and K. Ray, Nat. Clin. Pract. Neurol., 2006, 2(9), 482–493 Search PubMed.
- H. J. Stuerenburg, J. Neural Transm., 2000, 107(3), 321–329 CrossRef CAS.
- M. T. Lorincz, Ann. N. Y. Acad. Sci., 2010, 1184, 173–187 CrossRef CAS.
- S. Sinha, A. B. Taly, S. Ravishankar, L. K. Prashanth, K. S. Venugopal, G. R. Arunodaya, M. K. Vasudev and H. S. Swamy, Neuroradiology, 2006, 48(9), 613–621 CrossRef CAS.
- K. Gellein, P. M. Roos, L. Evje, O. Vesterberg, T. P. Flaten, M. Nordberg and T. Syversen, Brain Res., 2007, 1174, 136–142 CrossRef CAS.
- G. Forte, B. Bocca, O. Senofonte, F. Petrucci, L. Brusa, P. Stanzione, S. Zannino, N. Violante, A. Alimonti and G. Sancesario, J. Neural Transm., 2004, 111(8), 1031–1040 CAS.
- B. M. Katz and A. Barnea, J. Biol. Chem., 1990, 265(4), 2017–2021 CAS.
- A. Barnea and B. M. Katz, J. Inorg. Biochem., 1990, 40(1), 81–93 CrossRef CAS.
- B. S. Choi and W. Zheng, Brain Res., 2009, 1248, 14–21 CrossRef CAS.
- I. F. Scheiber, J. F. Mercer and R. Dringen, Neurochem. Int., 2010, 56(3), 451–460 CrossRef CAS.
- J. F. Eisses and J. H. Kaplan, J. Biol. Chem., 2005, 280(44), 37159–37168 CrossRef CAS.
- J. Lee, M. M. Pena, Y. Nose and D. J. Thiele, J. Biol. Chem., 2002, 277(6), 4380–4387 CrossRef CAS.
- G. J. Van den Berg, J. J. de Goeij, I. Bock, M. J. Gijbels, A. Brouwer, K. Y. Lei and H. F. Hendriks, J. Nutr., 1991, 121(8), 1228–1235 CAS.
- L. Tapia, M. Suazo, C. Hodar, V. Cambiazo and M. Gonzalez, BioMetals, 2003, 16(1), 169–174 Search PubMed.
- W. Zheng, N. Xin, Z. H. Chi, B. L. Zhao, J. Zhang, J. Y. Li and Z. Y. Wang, FASEB J., 2009, 23(12), 4207–4217 CrossRef CAS.
- B. Zhou and J. Gitschier, Proc. Natl. Acad. Sci. U. S. A., 1997, 94(14), 7481–7486 CrossRef CAS.
- M. L. Schlief and J. D. Gitlin, Mol. Neurobiol., 2006, 33(2), 81–90 Search PubMed.
- M. L. Schlief, T. West, A. M. Craig, D. M. Holtzman and J. D. Gitlin, Proc. Natl. Acad. Sci. U. S. A., 2006, 103(40), 14919–14924 CrossRef CAS.
- A. Hopt, S. Korte, H. Fink, U. Panne, R. Niessner, R. Jahn, H. Kretzschmar and J. Herms, J. Neurosci. Methods, 2003, 128(1–2), 159–172 CrossRef CAS.
- J. Lee, J. R. Prohaska and D. J. Thiele, Proc. Natl. Acad. Sci. U. S. A., 2001, 98(12), 6842–6847 CrossRef CAS.
- Y. M. Kuo, B. Zhou, D. Cosco and J. Gitschier, Proc. Natl. Acad. Sci. U. S. A., 2001, 98(12), 6836–6841 CrossRef CAS.
- N. C. Mackenzie, M. Brito, A. E. Reyes and M. L. Allende, Gene, 2004, 328, 113–120 CrossRef CAS.
- T. Haremaki, S. T. Fraser, Y. M. Kuo, M. H. Baron and D. C. Weinstein, Proc. Natl. Acad. Sci. U. S. A., 2007, 104(29), 12029–12034 CrossRef CAS.
- N. A. Platonova, S. A. Barabanova, R. G. Povalikhin, N. V. Tsymbalenko, A. D. Nozdrachev and L. V. Puchkova, Dokl. Biol. Sci., 2005, 401, 88–91 Search PubMed.
- Y. M. Kuo, A. A. Gybina, J. W. Pyatskowit, J. Gitschier and J. R. Prohaska, J. Nutr., 2006, 136(1), 21–26 CAS.
- Y. Qian, Y. Zheng, L. Abraham, K. S. Ramos and E. Tiffany-Castiglioni, Mol. Brain Res., 2005, 134(2), 323–332 CrossRef CAS.
- S. A. Molloy and J. H. Kaplan, J. Biol. Chem., 2009, 284(43), 29704–29713 CrossRef CAS.
- Y. Guo, K. Smith, J. Lee, D. J. Thiele and M. J. Petris, J. Biol. Chem., 2004, 279(17), 17428–17433 CrossRef CAS.
- J. F. Eisses and J. H. Kaplan, J. Biol. Chem., 2002, 277(32), 29162–29171 CrossRef CAS.
- C. J. De Feo, S. G. Aller, G. S. Siluvai, N. J. Blackburn and V. M. Unger, Proc. Natl. Acad. Sci. U. S. A., 2009, 106(11), 4237–4242 CrossRef CAS.
- E. B. Maryon, S. A. Molloy and J. H. Kaplan, J. Biol. Chem., 2007, 282(28), 20376–20387 CrossRef CAS.
- C. J. De Feo, S. Mootien and V. M. Unger, J. Membr. Biol., 234(2), 113–123 Search PubMed.
- B. Angeletti, K. J. Waldron, K. B. Freeman, H. Bawagan, I. Hussain, C. C. Miller, K. F. Lau, M. E. Tennant, C. Dennison, N. J. Robinson and C. Dingwall, J. Biol. Chem., 2004, 280(18), 17930–17937 CrossRef.
- A. Rentmeister, A. Bill, T. Wahle, J. Walter and M. Famulok, RNA, 2006, 12(9), 1650–1660 CrossRef CAS.
- J. M. Leitch, P. J. Yick and V. C. Culotta, J. Biol. Chem., 2009, 284(37), 24679–24683 CrossRef CAS.
- Y. Furukawa and T. V. O'Halloran, Antioxid. Redox Signaling, 2006, 8(5–6), 847–867 Search PubMed.
- M. Son, K. Puttaparthi, H. Kawamata, B. Rajendran, P. J. Boyer, G. Manfredi and J. L. Elliott, Proc. Natl. Acad. Sci. U. S. A., 2007, 104(14), 6072–6077 CrossRef CAS.
- V. Tanchou, F. Gas, A. Urvoas, F. Cougouluegne, S. Ruat, O. Averseng and E. Quemeneur, Biochem. Biophys. Res. Commun., 2004, 325(2), 388–394 CrossRef CAS.
- A. Rodriguez-Granillo and P. Wittung-Stafshede, Biochemistry, 2009, 48(5), 960–972 CrossRef CAS.
- I. Anastassopoulou, L. Banci, I. Bertini, F. Cantini, E. Katsari and A. Rosato, Biochemistry, 2004, 43(41), 13046–13053 CrossRef CAS.
- M. Ralle, S. Lutsenko and N. J. Blackburn, J. Biol. Chem., 2003, 278(25), 23163–23170 CrossRef CAS.
- A. K. Wernimont, L. A. Yatsunyk and A. C. Rosenzweig, J. Biol. Chem., 2003, 279(13), 12269–12276 CrossRef.
- J. M. Walker, R. Tsivkovskii and S. Lutsenko, J. Biol. Chem., 2002, 277(31), 27953–27959 CrossRef CAS.
- S. P. Raps, J. C. Lai, L. Hertz and A. J. Cooper, Brain Res., 1989, 493(2), 398–401 CrossRef CAS.
- E. Pileblad, P. S. Eriksson and E. Hansson, J. Neural Transm., 1991, 86(1), 43–49 CrossRef CAS.
- L. W. Klomp, S. J. Lin, D. S. Yuan, R. D. Klausner, V. C. Culotta and J. D. Gitlin, J. Biol. Chem., 1997, 272(14), 9221–9226 CrossRef CAS.
- G. S. Naeve, A. M. Vana, J. R. Eggold, G. S. Kelner, R. Maki, E. B. Desouza and A. C. Foster, Neuroscience, 1999, 93(3), 1179–1187 CrossRef CAS.
- F. Hussain, A. Rodriguez-Granillo and P. Wittung-Stafshede, J. Am. Chem. Soc., 2009, 131(45), 16371–16373 CrossRef CAS.
- L. Banci, I. Bertini, F. Cantini, N. Della-Malva, M. Migliardi and A. Rosato, J. Biol. Chem., 2007, 282(32), 23140–23146 CrossRef CAS.
- L. A. Yatsunyk and A. C. Rosenzweig, J. Biol. Chem., 2007, 282(12), 8622–8631 CrossRef CAS.
- M. Ralle, S. Lutsenko and N. J. Blackburn, J. Inorg. Biochem., 2004, 98(5), 765–774 CrossRef CAS.
- T. Miyayama, K. T. Suzuki and Y. Ogra, Toxicol. Appl. Pharmacol., 2009, 237(2), 205–213 CrossRef CAS.
- R. McRae, B. Lai and C. J. Fahrni, JBIC, J. Biol. Inorg. Chem., 2010, 15(1), 99–105 CrossRef CAS.
- I. H. Hung, R. L. Casareno, G. Labesse, F. S. Mathews and J. D. Gitlin, J. Biol. Chem., 1998, 273(3), 1749–1754 CrossRef CAS.
- G. S. Kelner, M. Lee, M. E. Clark, D. Maciejewski, D. McGrath, S. Rabizadeh, T. Lyons, D. Bredesen, P. Jenner and R. A. Maki, J. Biol. Chem., 2000, 275(1), 580–584 CrossRef CAS.
- S. Itoh, H. W. Kim, O. Nakagawa, K. Ozumi, S. M. Lessner, H. Aoki, K. Akram, R. D. McKinney, M. Ushio-Fukai and T. Fukai, J. Biol. Chem., 2008, 283(14), 9157–9167 CrossRef CAS.
- I. Hamza, J. Prohaska and J. D. Gitlin, Proc. Natl. Acad. Sci. U. S. A., 2003, 100(3), 1215–1220 CrossRef CAS.
- S. Itoh, K. Ozumi, H. W. Kim, O. Nakagawa, R. D. McKinney, R. J. Folz, I. N. Zelko, M. Ushio-Fukai and T. Fukai, Free Radical Biol. Med., 2009, 46(1), 95–104 CrossRef CAS.
- I. Hamza, A. Faisst, J. Prohaska, J. Chen, P. Gruss and J. D. Gitlin, Proc. Natl. Acad. Sci. U. S. A., 2001, 98(12), 6848–6852 CrossRef CAS.
- D. M. Glerum, A. Shtanko and A. Tzagoloff, J. Biol. Chem., 1996, 271(24), 14504–14509 CrossRef CAS.
- A. B. Maxfield, D. N. Heaton and D. R. Winge, J. Biol. Chem., 2003, 279(7), 5072–5080 CrossRef.
- Y. Takahashi, K. Kako, S. Kashiwabara, A. Takehara, Y. Inada, H. Arai, K. Nakada, H. Kodama, J. Hayashi, T. Baba and E. Munekata, Mol. Cell. Biol., 2002, 22(21), 7614–7621 CrossRef CAS.
- C. Oswald, U. Krause-Buchholz and G. Rodel, J. Mol. Biol., 2009, 389(3), 470–479 CrossRef CAS.
- L. Banci, I. Bertini, S. Ciofi-Baffoni, A. Janicka, M. Martinelli, H. Kozlowski and P. Palumaa, J. Biol. Chem., 2008, 283(12), 7912–7920 CrossRef CAS.
- L. Banci, I. Bertini, S. Ciofi-Baffoni, T. Hadjiloi, M. Martinelli and P. Palumaa, Proc. Natl. Acad. Sci. U. S. A., 2008, 105(19), 6803–6808 CrossRef CAS.
- L. Hiser, M. Di Valentin, A. G. Hamer and J. P. Hosler, J. Biol. Chem., 2000, 275(1), 619–623 CrossRef CAS.
- L. Banci, I. Bertini, F. Cantini, S. Ciofi-Baffoni, L. Gonnelli and S. Mangani, J. Biol. Chem., 2004, 279(33), 34833–34839 CrossRef CAS.
- B. C. Mobley, G. M. Enns, L. J. Wong and H. Vogel, Clin. Neuropathol., 2009, 28(2), 143–149 Search PubMed.
- L. C. Papadopoulou, C. M. Sue, M. M. Davidson, K. Tanji, I. Nishino, J. E. Sadlock, S. Krishna, W. Walker, J. Selby, D. M. Glerum, R. V. Coster, G. Lyon, E. Scalais, R. Lebel, P. Kaplan, S. Shanske, D. C. De Vivo, E. Bonilla, M. Hirano, S. DiMauro and E. A. Schon, Nat. Genet., 1999, 23(3), 333–337 CrossRef CAS.
- I. Valnot, S. Osmond, N. Gigarel, B. Mehaye, J. Amiel, V. Cormier-Daire, A. Munnich, J. P. Bonnefont, P. Rustin and A. Rotig, Am. J. Hum. Genet., 2000, 67(5), 1104–1109 CAS.
- J. C. Williams, C. Sue, G. S. Banting, H. Yang, D. M. Glerum, W. A. Hendrickson and E. A. Schon, J. Biol. Chem., 2005, 280(15), 15202–15211 CrossRef CAS.
- S. C. Leary, F. Sasarman, T. Nishimura and E. A. Shoubridge, Hum. Mol. Genet., 2009, 18(12), 2230–2240 CrossRef CAS.
- F. Pierrel, P. A. Cobine and D. R. Winge, BioMetals, 2007, 20(3–4), 675–682 Search PubMed.
- M. J. Niciu, X. M. Ma, R. El Meskini, J. S. Pachter, R. E. Mains and B. A. Eipper, Neurobiol. Dis., 2007, 27(3), 278–291 CrossRef CAS.
- N. Barnes, R. Tsivkovskii, N. Tsivkovskaia and S. Lutsenko, J. Biol. Chem., 2004, 280(10), 9640–9645 CrossRef.
- R. Linz and S. Lutsenko, J. Bioenerg. Biomembr., 2007, 39(5–6), 403–407 CrossRef CAS.
- M. J. Niciu, X. M. Ma, R. El Meskini, G. V. Ronnett, R. E. Mains and B. A. Eipper, Neuroscience, 2006, 139(3), 947–964 CrossRef CAS.
- E. Nishihara, T. Furuyama, S. Yamashita and N. Mori, NeuroReport, 1998, 9(14), 3259–3263 CrossRef CAS.
- Y. M. Kuo, J. Gitschier and S. Packman, Hum. Mol. Genet., 1997, 6(7), 1043–1049 CrossRef CAS.
- T. Saito, M. Okabe, T. Hosokawa, M. Kurasaki, A. Hata, F. Endo, K. Nagano, I. Matsuda, K. Urakami and K. Saito, Neurosci. Lett., 1999, 266(1), 13–16 CrossRef CAS.
- S. Lutsenko, N. L. Barnes, M. Y. Bartee and O. Y. Dmitriev, Physiol. Rev., 2007, 87(3), 1011–1046 CrossRef CAS.
- S. Lutsenko, E. S. LeShane and U. Shinde, Arch. Biochem. Biophys., 2007, 463(2), 134–148 CrossRef CAS.
- A. N. Barry, U. Shinde and S. Lutsenko, JBIC, J. Biol. Inorg. Chem., 2010, 15(1), 47–59 CrossRef CAS.
- A. A. Gybina and J. R. Prohaska, Genes Nutr., 2006, 1(1), 51–59 Search PubMed.
- R. El Meskini, L. B. Cline, B. A. Eipper and G. V. Ronnett, Dev. Neurosci., 2005, 27(5), 333–348 Search PubMed.
- A. Bohlken, B. B. Cheung, J. L. Bell, J. Koach, S. Smith, E. Sekyere, W. Thomas, M. Norris, M. Haber, D. B. Lovejoy, D. R. Richardson and G. M. Marshall, Br. J. Cancer, 2009, 100(1), 96–105 CrossRef CAS.
- X. Li, S. Chen, Q. Wang, D. J. Zack, S. H. Snyder and J. Borjigin, Proc. Natl. Acad. Sci. U. S. A., 1998, 95(4), 1876–1881 CrossRef CAS.
- M. Lovicu, M. B. Lepori, S. Incollu, V. Dessi, A. Zappu, R. Iorio, M. D'Ambrosi, M. T. Pellecchia, P. Barone, G. Maggiore, S. De Virgiliis, A. Cao and G. Loudianos, Genetic Testing and Molecular Biomarkers, 2009, 13(2), 185–191 Search PubMed.
- G. Loudianos, M. Lovicu, V. Dessi, M. Tzetis, E. Kanavakis, L. Zancan, L. Zelante, C. Galvez-Galvez and A. Cao, Hum. Mutat., 2002, 20(4), 260–266 CrossRef CAS.
- A. B. Shah, I. Chernov, H. T. Zhang, B. M. Ross, K. Das, S. Lutsenko, E. Parano, L. Pavone, O. Evgrafov, I. A. Ivanova-Smolenskaya, G. Anneren, K. Westermark, F. H. Urrutia, G. K. Penchaszadeh, I. Sternlieb, I. H. Scheinberg, T. C. Gilliam and K. Petrukhin, Am. J. Hum. Genet., 1997, 61(2), 317–328 CrossRef CAS.
- K. Petrukhin, S. Lutsenko, I. Chernov, B. M. Ross, J. H. Kaplan and T. C. Gilliam, Hum. Mol. Genet., 1994, 3(9), 1647–1656 CrossRef CAS.
- M. C. Reddy, S. Majumdar and E. D. Harris, Biochem. J., 2000, 350(3), 855–863 CrossRef CAS.
- L. B. Moller, Z. Tumer, C. Lund, C. Petersen, T. Cole, R. Hanusch, J. Seidel, L. R. Jensen and N. Horn, Am. J. Hum. Genet., 2000, 66(4), 1211–1220 CrossRef CAS.
- M. Qi and P. H. Byers, Hum. Mol. Genet., 1998, 7(3), 465–469 CrossRef CAS.
- S. G. Kaler, L. K. Gallo, V. K. Proud, A. K. Percy, Y. Mark, N. A. Segal, D. S. Goldstein, C. S. Holmes and W. A. Gahl, Nat. Genet., 1994, 8(2), 195–202 CrossRef CAS.
- S. La Fontaine and J. F. Mercer, Arch. Biochem. Biophys., 2007, 463(2), 149–167 CrossRef.
- B. Hardman, A. Michalczyk, M. Greenough, J. Camakaris, J. Mercer and L. Ackland, Cell. Physiol. Biochem., 2007, 20(6), 1073–1084 CrossRef CAS.
- L. Nyasae, R. Bustos, L. Braiterman, B. Eipper and A. Hubbard, Am. J. Physiol.: Gastrointest. Liver Physiol., 2006, 292(4), G1181–1194 CrossRef.
- M. Y. Bartee and S. Lutsenko, BioMetals, 2007, 20(3–4), 627–637 Search PubMed.
- Y. Guo, L. Nyasae, L. T. Braiterman and A. L. Hubbard, Am. J. Physiol.: Gastrointest. Liver Physiol., 2005, 289(5), G904–916 CrossRef CAS.
- H. Roelofsen, H. Wolters, M. J. Van Luyn, N. Miura, F. Kuipers and R. J. Vonk, Gastroenterology, 2000, 119(3), 782–793 CrossRef CAS.
- L. Braiterman, L. Nyasae, Y. Guo, R. Bustos, S. Lutsenko and A. Hubbard, Am. J. Physiol.: Gastrointest. Liver Physiol., 2008, 296(2), G433–444 CrossRef.
- C. Lane, M. J. Petris, A. Benmerah, M. Greenough and J. Camakaris, BioMetals, 2004, 17(1), 87–98 Search PubMed.
- C. Cobbold, J. Coventry, S. Ponnambalam and A. P. Monaco, Hum. Mol. Genet., 2003, 12(13), 1523–1533 CrossRef CAS.
- M. J. Petris and J. F. Mercer, Hum. Mol. Genet., 1999, 8(11), 2107–2115 CrossRef CAS.
- M. J. Francis, E. E. Jones, E. R. Levy, R. L. Martin, S. Ponnambalam and A. P. Monaco, J. Cell. Sci., 1999, 112(Pt 11), 1721–1732 CAS.
- M. J. Petris, J. Camakaris, M. Greenough, S. LaFontaine and J. F. Mercer, Hum. Mol. Genet., 1998, 7(13), 2063–2071 CrossRef CAS.
- C. M. Lim, M. A. Cater, J. F. Mercer and S. La Fontaine, J. Biol. Chem., 2006, 281(20), 14006–14014 CrossRef CAS.
- S. E. Stephenson, D. Dubach, C. M. Lim, J. F. Mercer and S. La Fontaine, J. Biol. Chem., 2005, 280(39), 33270–33279 CrossRef CAS.
- W. C. Singleton, K. T. McInnes, M. A. Cater, W. R. Winnall, R. McKirdy, Y. Yu, P. E. Taylor, B. X. Ke, D. R. Richardson, J. F. Mercer and S. La Fontaine, J. Biol. Chem. Search PubMed.
- A. Ibricevic, S. L. Brody, W. J. Youngs and C. L. Cannon, Toxicol. Appl. Pharmacol., 2010, 243(3), 315–322 CrossRef CAS.
- N. Barnes, M. Y. Bartee, L. Braiterman, A. Gupta, V. Ustiyan, V. Zuzel, J. H. Kaplan, A. L. Hubbard and S. Lutsenko, Traffic, 2009, 10(6), 767–779 CrossRef CAS.
- L. S. Mangala, V. Zuzel, R. Schmandt, E. S. Leshane, J. B. Halder, G. N. Armaiz-Pena, W. A. Spannuth, T. Tanaka, M. M. Shahzad, Y. G. Lin, A. M. Nick, C. G. Danes, J. W. Lee, N. B. Jennings, P. E. Vivas-Mejia, J. K. Wolf, R. L. Coleman, Z. H. Siddik, G. Lopez-Berestein, S. Lutsenko and A. K. Sood, Clin. Cancer Res., 2009, 15(11), 3770–3780 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |