Formation and study of single metal ion–phospholipid complexes in biphasic electrospray ionization mass spectrometry
Michel
Prudent
a,
Manuel A.
Méndez
a,
Daniel F.
Jana
b,
Clémence
Corminboeuf
b and
Hubert H.
Girault
*a
aLaboratoire d’Electrochimie Physique et Analytique, Ecole Polytechnique Fédérale de Lausanne, Station 6, CH-1015 Lausanne, Switzerland. E-mail: hubert.girault@epfl.ch
bLaboratory for Computational Molecular Design, Ecole Polytechnique Fédérale de Lausanne, Batochime, CH-1015 Lausanne, Switzerland
First published on 20th May 2010
Abstract
Single metal ion–phospholipid complexes are observed in biphasic electrospray ionization mass spectrometry (BESI-MS) using a dual-channel microsprayer. Such a microsprayer makes it possible to put into contact two immiscible liquids within the Taylor cone. Thus, L-α-dipalmitoyl phosphatidylcholine (DPPC) dissolved in 1,2-dichloroethane (DCE) reacts with aqueous metal cations (M = Na+, K+, Ca2+, Cu2+, La3+) yielding the formation of [M-DPPCn]z+ complexes. The number of phospholipid molecules ranges from 1 to 4 for monovalent ions, to 8 for divalent and to more than 10 for trivalent ions respectively. The large number of ligands observed involves the formation of solvent free single ion–phospholipid complexes.
Introduction
Lipids, one of the major constituents of cell membranes, serve many purposes such as cellular and sub-cellular partitioning, messenger cell signalling, to maintain electrochemical gradients, etc. Nonetheless, due to its apparent low reactivity, the lipidic component of cell membranes has been commonly regarded as an inert matrix that contains the receptors with which chemical messengers can interact and trigger a response from the interior of the cell. This perspective has been modified over the years, describing the cell membranes not as a passive barrier and/or matrix for receptors but more like an active interface with specific interactions with different types of molecules, ranging from inorganic ions to proteins. This realization is essential as it impacts both the structure, dynamics and stability of membranes, as well as the binding and insertion of proteins to or into membranes. Those processes are responsible for messenger–receptor catalyzed interactions,1 membrane fusion and transport of small molecules across membranes. In particular, the interaction of metal ions with phospholipids has received a great deal of attention. Biological membranes are indeed always surrounded by aqueous solutions containing cations in high concentrations. In addition, increasing evidence suggests that electrostatic interactions between cations and lipid molecules are crucial for structural and dynamical changes in the polar head group region.The interaction of monovalent ions with lipids is generally accepted to be rather weak, although non-negligible. As a matter of fact, alkali ions can induce phase transitions as it was observed over multilamellar vesicles.2 Analogously, divalent metal cations, such as calcium, essential to a large number of life processes, effectively interact with cell membranes, modifying their conformation, structure and/or stability, and playing a key role in membrane fusion processes. The interactions between calcium and lipidic bilayers have been thoroughly investigated over the years.3–5 Experimental evidence obtained by multiple techniques (NMR,6 atomic force microscopy,7 X-ray diffraction,8 scanning electrochemical microscopy,9 fluorescence,10 light scattering measurements,8etc.) strongly suggests the formation of calcium–phospholipid complexes6 whose importance has lead to the introduction of calcium within the minimal fusion machinery.8 Indeed, calcium ion bridges generated by the local dehydration of phospholipid head groups and the calcium ions themselves, ultimately leads to the membrane fusion as it has been corroborated by classical molecular dynamics (MD).11 Additional classical MD simulations also predicted the sequential multistep binding and coordination of Ca2+ cations by three or four lipid carbonyl oxygens, supporting this idea.4 Copper is also known to participate in lipid metabolism and may react at the cell surface.12,13
Other recent theoretical studies (MD) have shown that interfacial charge differences stemming from electrostatic interactions in both cell membrane leaflets can also be responsible in a great extent for the creation of intense electric fields across the lipidic bilayer.3
In the field of electrochemistry at polarized liquid–liquid interfaces, the presence of lipidic monolayers adsorbed at the interface between two immiscible electrolyte solutions (ITIES) facilitates the transfer of ions from the aqueous to the organic phase. It indicates that phospholipids have ionophoric properties forming complexes in the organic phase.14 Analogously, Monzón and Yudi have shown that alkali metal ions like Li+ increase lipid organization inducing the interfacial packing of monolayers.15
In a more general sense, the analysis of the entire lipidic content of a biological system (i.e. the lipidome) has given rise to a new emerging branch of metabolomics denominated lipidomics.16–18 Among the techniques most commonly employed to analyze the vast number of compounds present in a biological sample, mass spectrometry (MS) has proven to be a very versatile and powerful technique. Owing to its soft ionization process, electrospray ionization (ESI) has even allowed the transfer of surfactant micelles into the gas phase conserving their structures.19 Nevertheless, there are only a few examples in the literature reporting the ion–lipid interactions in ESI-MS with an emphasis on the fragmentation process.20–24
The major difficulties in studying lipid–ion interactions stem from the low solubility of lipids in water and the high hydrophilic character of small inorganic ions. Both species are normally found in different and immiscible solvents unless prior formation of vesicles,25 monolayers26 or bilayers.27 Nonetheless, recent developments of ESI sources in MS provide adequate tools for such kind of studies.28 Efficient analysis of complexes occurring at the interface formed between water and 1,2-dichloroethane (DCE) has been recently carried out29 combining dual-channel microsprayers30 with the classic ESI technique. These microsprayers enable the injection of two immiscible liquids placing them in contact only at the tip where the Taylor cone is formed.29 The application of a high external voltage leads to electrocapillary emulsification31 along with intense swirls at the Taylor cone,32 making the analysis by MS of interfacial complexes feasible. In order to obtain valuable insight information of metal ion–phospholipid complexation reactions, we here apply this new approach, called biphasic electrospray ionization (BESI).28,29,33 Reactions between different metal ions dissolved in an aqueous phase and a phospholipid dissolved in DCE will thus be followed by MS.
Experimental
Chemicals
Calcium sulfate dihydrate (CaSO4·2H2O), cupric sulfate pentahydrate (CuSO4·5H2O), sodium chloride (NaCl), potassium chloride (KCl) and 1,2-dichloroethane (DCE) were bought from Fluka (Buchs, Switzerland). Lanthanum chloride (LaCl3·2.21H2O) was provided by the Laboratory of Lanthanides Supramolecular Chemistry (EPFL, Lausanne, Switzerland). The phospholipid (L-α-dipalmitoyl phosphatidylcholine, DPPC, M = 734.0 g mol−1) was from Sigma (St Louis, MO). Deionized water (18.2 MΩ cm−1) was prepared using a Milli-Q system from Millipore (Bedford, MA). All the compounds were used as received.DPPC was diluted to 200 μM in DCE. The salts (NaCl, KCl, CaSO4, CuSO4 and LaCl3) were diluted in water to 200 μM or 1 mM. La3+ solution was titrated with a complexometric method at 0.16 or 0.79 mM.
Biphasic electrospray and MS setup
The biphasic microsprayer, described previously,29 consists of a double microchannel (20 μm × 50 μm × 1 cm) polyimide microchip developed by DiagnoSwiss SA (Monthey, Switzerland).30 The chip was fixed in a holder connected to a syringe pump (KdScientific, Holliston, MA) enabling to hold two syringes (100 μL, Hamilton, Bonaduz, Switzerland) to introduce different solutions. Thus, two immiscible liquids were infused and contacted within the Taylor cone. In order to avoid any contamination, one line is dedicated to the aqueous phase and the other line to the organic phase.Mass spectrometric measurements were carried out in an LCT time of flight (TOF) mass spectrometer (Micromass, Manchester, UK) used in positive ionization mode. The commercial electrospray interface was removed and the BESI source was mounted in front of the MS. The high voltage was connected to the stainless steel needle of the syringe containing the aqueous solution. The pump was switched on (1 μL min−1 for each line, i.e. a total flow rate of 2 μL min−1) and the MS power supply was set at 5.0 kV. The source temperature was fixed at 130 °C. The ion optics parameters were tuned in order to maximize high molecular weights. The mass spectra were averaged during 1 min.
Conversion rates χ of [M-DPPCn]z+ were calculated as follows:
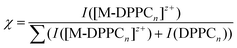 | (1) |
Computational methodology
Geometry optimizations and energy computations were performed on model systems including one metal cation (i.e. K+ or Ca2+) coordinated to one to four truncated phospholipid molecules (the 15 carbon atom aliphatic chain is reduced to a propyl group; structures with up to 233 atoms). Considering the large number of atoms in the model systems, the binding enthalpies were calculated following the reaction at the B3LYP34/3-21G* level (ZPE and thermal corrections included):35| Mz+ + nDPPC → [M-DPPCn]z+ | (2) |
Results
Complexation of potassium, sodium, calcium, copper and lanthanum ions with phospholipids were carried out with the BESI source coupled to a TOF-MS. DPPC concentration was equal to 200 μM allowing the formation of a layer at the water|DCE interface, the critical value being approximately 10 μM.37 In general, singly charged DPPC and 2DPPC were the most abundant species observed, as previously reported.33 Trimer and bigger complexes were not abundant or not even observed. The reaction of DPPCn with one metal ion only ([M-DPPCn]z+) was observed when infusing the two immiscible liquids containing the reactants. However complexes containing several metal ions were not observed. The mono metal ion complex charge corresponds to that of the metal ion itself since DPPC is a zwitterionic species. The salts were used in excess because of the rather poor abundance of clusters at lower concentrations.Fig. 1a shows the mass spectrum obtained for K+. It clearly exhibits a mass shift of 38 Th of one, two and three DPPC, the most abundant being [K-DPPC]+ at m/z = 772.4 Th. A fragment of 2 DPPC was also observed at m/z = 1230.0 Th, which has been characterized with other metal ions.22,23 Na+ ions exhibit the same trend (MS not shown). Interaction with the divalent ion Ca2+ induces a different peak pattern with doubly charged clusters observed up to 8 DPPC (Fig. 1b). [Ca-DPPC4]2+ was the most abundant cluster. The tandem mass spectra (obtained with an ion trap MS) of the dimer and tetramer bound to Ca2+ ion showed the loss of DPPC ligands and fatty acid (palmitate) residues.23 Similar coordination was observed using a Cu2+ salt (Fig. 1c), i.e. copper(II) ions bound to 3–7 DPPC forming doubly charged clusters, and in addition copper(I) ions bound to one and two DPPC. [Cu-DPPC4]2+ was the most abundant cluster, in agreement with MS data reported for diacylglycerophosphocholines.20 The formation of copper(I) complexes from a copper(II) salt is well documented in ESI-MS and stems from the reduction of CuII to CuI during the ESI process.38–42 Moreover, different fragments observed and attributed to copper(II) ions, result from a higher yield of fragmentation induced by transition metal ions.24 Finally, La3+ was tested. The mass spectrum (Fig. 1d) shows high conversion rates despite a rather lower salt concentration used (0.8 mM instead of 1 mM) compared to the other ions tested and higher values of n, from 4 to 10. All the complexes were triply charged and no special fragments were induced by this cation.
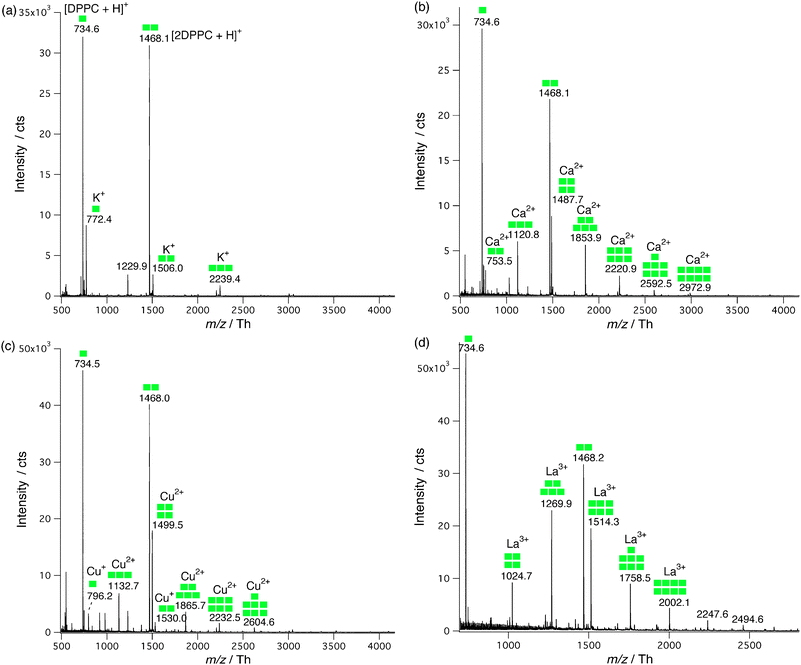 | ||
Fig. 1 Mass spectra of metal ion-DPPC complexes obtained with a BESI source coupled to a TOF-MS. (a) KCl, (b) CaSO4 and (c) CuSO4 at 1 mM each, and (d) LaCl3 at 0.8 mM, in water and DPPC 200 μM in DCE. Mass spectra were summed during 1 min.  stands for one DPPC molecule. stands for one DPPC molecule. | ||
Fig. 2 summarizes the conversion rates of [M-DPPCn]z+ as a function of the number of DPPC molecules. Except for the monovalent ions K+, Na+ and Cu+ where χ decreases monotonically as n increases, the distributions exhibit Gaussian curves and La3+ gives the highest abundance taking into account the sum of all the [La-DPPCn]3+. In particular, the most abundant complexes were of stoichiometry 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 for K+, Na+ and Cu+, 1:4 for Ca2+ and Cu2+ and 1:5 for La3+. The total conversion rate of the [M-DPPCn]z+ complexes decreases according to the following order La3+ > Ca2+ > Cu2+ > K+ > Na+ (Cu+ complexes were the least abundant because they stemmed from the reduction of Cu2+; therefore their concentration was much lower, affecting also the abundance of copper(II)-phospholipid complexes).
:1 for K+, Na+ and Cu+, 1:4 for Ca2+ and Cu2+ and 1:5 for La3+. The total conversion rate of the [M-DPPCn]z+ complexes decreases according to the following order La3+ > Ca2+ > Cu2+ > K+ > Na+ (Cu+ complexes were the least abundant because they stemmed from the reduction of Cu2+; therefore their concentration was much lower, affecting also the abundance of copper(II)-phospholipid complexes).
 | ||
| Fig. 2 Conversion rates of the metal ion–DPPCn complexes as a function of n. The curve shapes are explained by the presence of a first and a second coordination spheres (marked with dashed lines). Colored dashed lines were added between markers for clarity. | ||
Discussion
Metal–DPPC complexes
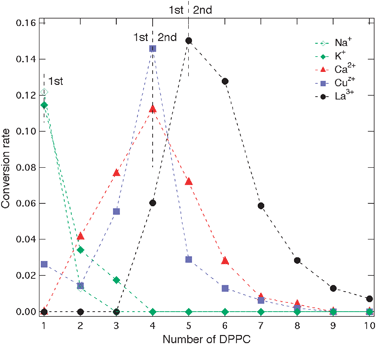
To rationalize the present observations, we computed the binding energy between K+ and the polar head of the truncated phospholipids on the one hand and compared them with K+ and water molecules on the other hand. In the latter case, the six solvent molecules provide an interaction energy that approaches the hydration energy of −360 kJ mol−1.36 As shown in Fig. 3a, the ion-solvent interaction energy increases quasi-linearly with the number of water molecules yielding an average value of around 60 kJ mol−1 indicative of an ion–dipole interaction. In the case of the ion–phospholipid interaction, the increase is also quasi-linear but with an average value of 300 kJ mol−1. Even if these values cannot be compared stricto sensu (the binding energies for the water complexes are computed at a much higher theoretical level, see computational details), it is clear that the interaction between K+ and DPPC is stronger than that with the water molecules, due to the binding mode of the phospholipid (Fig. 4). From a steric and charge density viewpoint, 3 DPPCs are sufficient to complete the first coordination shell and it is not surprising not to observe potassium ions with more than 3 DPPCs forming a single ion complex in the gas phase. | ||
| Fig. 3 Enthalpy of binding for (a) K+ and (b) Ca2+ in the exchange process between free ion and water, and free ion and lipid molecules. (Computational data). Colored dashed lines were added between markers for clarity. | ||
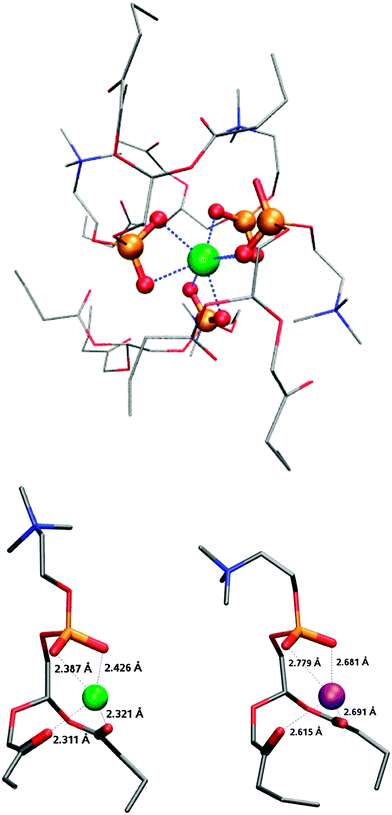 | ||
| Fig. 4 Computational structure of Ca2+-DPPC4 (top), Ca2+-DPPC (left) and K+-DPPC (right). Green: calcium, purple: potassium and red: oxygen from phosphate (thickened). The dashed blue lines represent metal–O bonds. The aliphatic chains were cut for computational reasons. | ||
In the case of calcium–water interactions, Fig. 3b shows that the first six water molecules provide a bit more than half of the total hydration energy (−1180 kJ mol−1)43 also with a monotonic variation of the binding energy. Indeed, Bako et al. have shown that for clusters with 7 and 8 water molecules, those which are formed with six water molecules in the first hydration shell and with one or two in the second shell are more stable than those which are hepta- or octa-coordinated.43 They further suggested that these six water molecules of the first hydration shell in solution are arranged in well-defined octahedral geometry, each of these molecules being linked by hydrogen bonds to three molecules in the secondary shell. Here, the average binding energy for molecules of the first hydration shell is about 140 kJ mol−1 reflecting the higher charge of Ca2+ when compared to K+. With DPPC, computations show that four lipid molecules saturate the space around the cation by seven oxygen ligands coming from phosphate groups in close vicinity as shown in Fig. 4. Again, although the two sets of calculations cannot be compared directly, it is clear that complexation by a phospholipid is more favorable than that by four water molecules. While attempts to coordinate additional DPPC molecules to the calcium cation did not succeed, experimental results clearly show that complexes with 8 DPCCs are formed in the gas phase. From a steric viewpoint, the hexa-coordination of the calcium ion is ensured by 4 DPPC molecules (Fig. 4, top). Additional DPPC ligands could therefore result either from longer range ion–dipole interactions with the polar heads of the DPPC squeezed between the alkyl chains of the primary solvation shell to form a second solvation shell, or from van der Waals interactions thereby forming single ion–phospholipid complexes with the polar heads of the outer phospholipid molecules being located on the outside of the structures.
In the case of lanthanum, the number of water molecules in the inner and second hydration layer is equal to 9 and 16 respectively, as recently reviewed by Dognon et al.44 Here, we can observe up to 10 DPPC molecules. The maximum shown in Fig. 2 suggests that five must be involved in the inner coordination layer. As in the case of calcium, the additional DPPCs can either be aligned with the inner shell or forming a single ion complex in a head-to-tail conformation.
In the case of copper, Fig. 2 shows a mixed curve shape first decreasing before increasing. This stems from the two possible oxidation states of copper, Cu+ behaving as K+ and Cu2+ as Ca2+.
Mechanism of reactions
The formation of these species can be explained by the following general biphasic reaction mechanism:| [M(H2O)x]z+(w) + nDPPC(o) → [M-(DPPC)n]z+(g) + xH2O(g) | (3) |
Regarding the gas phase, and taking into account the equality of the flow rates of both phases, two different situations can be envisaged: formation and ejection of biphasic droplets or ejection of monophasic aqueous and/or DCE droplets (Fig. 5). In the first case, biphasic droplets are likely to comprise an inner organic core surrounded by an outer aqueous shell (o@w, pathway I). One can show that the aqueous phase will surround spontaneously the organic core only if the following condition is fulfilled:
| γDCE-a > γw-a + γw-DCE | (4) |
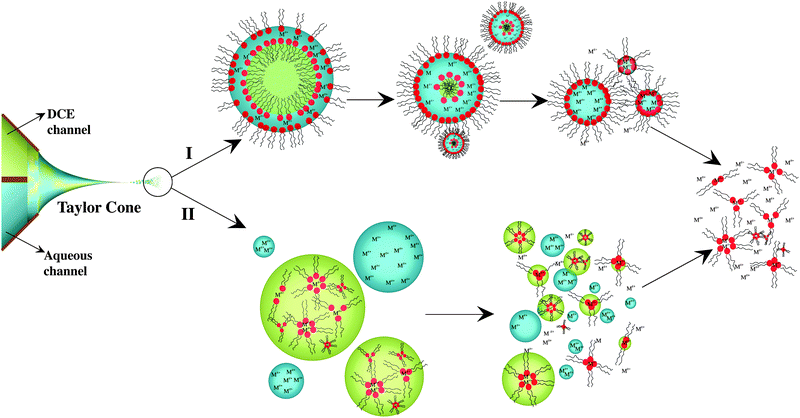 | ||
| Fig. 5 Schematic drawing of the two postulated pathways for the complexation/ionization process taking place after spraying an aqueous and an organic (containing DPPC) solution using the dual-channel microsprayer during BESI experiments. | ||
In the second case, the presence of monophasic aqueous and organic droplets within the fine mist formed during the electrospray process is considered (pathway II). In this case, inverted micelles formed within the Taylor cone are dissolved in the organic phase.
Finally, for both pathways, consecutive desolvation steps lead to stable cation–phospholipid complexes, as observed by MS. The ionization mechanisms would differ for the two pathways. Droplets fission proposed by the charge residue model (CRM)47,48 would be favored when o@w droplets are considered (Pathway I). On the contrary, the ion evaporation model (IEM)49 would predominate for the pathway II due to the prior presence of the complexes in the organic phase and absence of a blocking barrier at the DCE–air interface.
Independent of the ionization mechanism, important implications at the membrane scale can be drawn from the mass spectrometric evidence of these cation–DPPC clusters. First, structuring and packing effects caused by the cations adsorption/binding are known to have major influences on biologic membranes50,51 and can be rationalized in terms of the cation–lipid interactions. Hence, cations without the adequate charge and/or incapable of forming a second coordination sphere (e.g. K+ and Na+) will have a smaller impact on the membrane properties than cations with this ability (e.g. Ca2+, Cu2+ or La3+). In the second group of cations, La3+ will perturb in a greater extent the lipidic membrane. Indeed, it accommodates around it a higher number of lipid molecules (either at the first or at the second coordination sphere), as it has been reported by Lehrmann and Seelig.52 Therefore, intra-micellar interactions, specially taking place in the second coordination shell, should be responsible for the effects experimentally observed of metal cations over lipidic membranes, like packing effect. Analogously, it is shown that such interactions are strong enough to preserve the structure of the complexes in the gas phase.
As a last remark, clusters of [M-DPPCn]z+ can be formed at the liquid–liquid interface within either the Taylor cone or the expelled droplets from it, according to our previous results and discussion on the BESI source.29 The multistep binding of calcium to bilayers has been reported to be sub–microseconds,4 which is compatible with reactions driven under our conditions. These comparisons consolidate the validity of BESI sources for the analysis of in situ formed lipid complexes.
Conclusions
The present liquid–liquid mass spectrometric study shows for phospholipid–metal ion complexes evidences of a first coordination sphere for monovalent ions and of a second coordination sphere for multivalent ions. Single metal-ion-containing clusters are formed by metal ions reacting with multiple DPPC molecules. The charge of the metal ions correlates with the number of DPPC as indicated by the [M-DPPCn]z+ abundance in the mass spectra. In the first coordination sphere, the driving forces for the complexes formation are the phosphate-ion electrostatic interactions (even though carbonyl oxygens are involved in the complexes for low lipid coordination number). Beyond a certain number of DPPC molecules, the formation of a second coordination shell involving London dispersion forces, leads to the formation of larger clusters. These results do not only consolidate the potential of the liquid–liquid interface methodology for MS analysis of lipids, but also suggest that the reaction between metal cations and phospholipids goes beyond the polar head group region. In consequence, the existence of a second coordination sphere would induce perturbations in the membrane at longer distances than those expected if only a single coordination sphere was involved.Acknowledgements
The Fonds National Suisse pour la Recherche Scientique is thanked for financial support through the projects “Analytical tools for fast phosphoproteome analysis” (Grant 200020-113413/1) and “Electrochemical methodology for the study of peptide lipid interaction” (Grant 200020-113428). The authors also appreciate the fruitful discussions with Fernando Cortés Salazar.References
- D. F. Sargent and R. Schwyzer, Proc. Natl. Acad. Sci. U. S. A., 1986, 83, 5774–5778 CrossRef CAS.
- M. Rappolt, G. Pabst, H. Amenitsch and P. Laggner, Colloids Surf., A, 2001, 183–185, 171–181 CrossRef CAS.
- A. A. Gurtovenko and I. Vattulainen, J. Phys. Chem. B, 2008, 112, 4629–4634 CrossRef CAS.
- R. A. Böckmann and H. Grubmüller, Angew. Chem., Int. Ed., 2004, 43, 1021–1024 CrossRef.
- R. A. Böckmann, A. Hac, T. Heimburg and H. Grubmüller, Biophys. J., 2003, 85, 1647–1655 CrossRef.
- C. Altenbach and J. Seelig, Biochemistry, 1984, 23, 3913–3920 CrossRef CAS.
- B. P. Jena, C. Sang-Joon, A. Jeremic, M. H. Stromer and R. Abu-Hamdah, Biophys. J., 2003, 84, 1337–1343 CrossRef CAS.
- A. Jeremic, M. Kelly, J. A. Cho, S. J. Cho, J. K. H. Horber and B. P. Jena, Cell Biol. Int., 2004, 28, 19–31 CrossRef CAS.
- D. P. Burt, J. Cervera, D. Mandler, J. V. Macpherson, J. A. Manzanares and P. R. Unwin, PhysChemChemPhys, 2005, 7, 2955–2964 RSC.
- H. Y. D. Ke, W. L. Anderson, R. M. Moncrief, G. D. Rayson and P. J. Jackson, Environ. Sci. Technol., 1994, 28, 586–591 CrossRef CAS.
- J. J. Potoff, Z. Issa, C. W. Manke Jr. and B. P. Jena, Cell Biol. Int., 2008, 32, 361–366 CrossRef CAS.
- D. C. Kennedy, R. K. Lyn and J. P. Pezacki, J. Am. Chem. Soc., 2009, 131, 2444–2445 CrossRef CAS.
- D. Huster, T. D. Purnat, J. L. Burkhead, M. Ralle, O. Fiehn, F. Stuckert, N. E. Olson, D. Teupser and S. Lutsenko, J. Biol. Chem., 2007, 282, 8343–8355 CrossRef CAS.
- Y. Yoshida, K. Maeda and O. Shirai, J. Electroanal. Chem., 2005, 578, 17–24 CrossRef CAS.
- L. M. A. Monzón and L. M. Yudi, in Electrochimica Acta, editon edn, AA, AA, editon edn, 2006, vol. 51, pp. 4573–4581 Search PubMed.
- A. Carrasco-Pancorbo, N. Navas-Iglesias and L. Cuadros-Rodríguez, TrAC, Trends Anal. Chem., 2009, 28, 263–278 CrossRef CAS.
- N. Navas-Iglesias, A. Carrasco-Pancorbo and L. Cuadros-Rodríguez, TrAC, Trends Anal. Chem., 2009, 28, 393–403 CrossRef CAS.
- M. R. Wenk, Nat. Rev. Drug Discovery, 2005, 4, 594–610 CrossRef CAS.
- G. Siuzdak and B. Bothner, Angew. Chem., Int. Ed. Engl., 1995, 34, 2053–2055 CrossRef CAS.
- P. F. James, M. A. Perugini and R. A. J. O’Hair, Rapid Commun. Mass Spectrom., 2007, 21, 757–763 CrossRef CAS.
- P. F. James, M. A. Perugini and R. A. J. O’Hair, J. Am. Soc. Mass Spectrom., 2008, 19, 978–986 CrossRef CAS.
- P. F. James, M. A. Perugini and R. A. J. O’Hair, J. Am. Soc. Mass Spectrom., 2006, 17, 384–394 CrossRef CAS.
- M. Pulfer and R. C. Murphy, Mass Spectrom. Rev., 2003, 22, 332–364 CrossRef CAS.
- Y. P. Ho, P. C. Huang and K. H. Deng, Rapid Commun. Mass Spectrom., 2003, 17, 114–121 CrossRef CAS.
- L. Hugonin, V. Vukojevic, G. Bakalkin and A. Graslund, Biochim. Biophys. Acta, Biomembr., 2008, 1778, 1267–1273 CrossRef CAS.
- S. O. Hagge, M. U. Hammer, A. Wiese, U. Seydel and T. Gutsmann, BMC Biochem., 2006, 7, 15 CrossRef.
- A. Aroti, E. Leontidis, M. Dubois, T. Zemb and G. Brezesinski, Colloids Surf., A, 2007, 303, 144–158 CrossRef CAS.
- M. Prudent and H. H. Girault, Analyst, 2009, 134, 2189–2203 RSC.
- M. Prudent, M. A. Méndez and H. H. Girault, Anal. Sci., 2008, 24, 1399–1404 CrossRef CAS.
- M. Prudent, J. S. Rossier, N. Lion and H. H. Girault, Anal. Chem., 2008, 80, 2531–2538 CrossRef CAS.
- A. Watanabe, K. Higashitsuji and K. Nishizawa, J. Colloid Interface Sci., 1978, 64, 278–289 CrossRef CAS.
- T. C. Rohner, N. Lion and H. H. Girault, PhysChemChemPhys, 2004, 6, 3056–3068 RSC.
- M. A. Méndez, M. Prudent, B. Su and H. H. Girault, Anal. Chem., 2008, 80, 9499–9507 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. Montgomery, J. A., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. B. Tomasi, V. B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian, Inc., Wallingford, CT, editon edn, 2004.
- J. S. Rao, T. C. Dinadayalane, J. Leszczynski and G. N. Sastry, J. Phys. Chem. A, 2008, 112, 12944–12953 CrossRef CAS.
- Z. Samec, A. Trojanek and P. Krtil, Faraday Discuss., 2005, 129, 301–313 RSC.
- L. Gianelli, V. Amendola, L. Fabbrizzi, P. Pallavicini and G. G. Mellerio, Rapid Commun. Mass Spectrom., 2001, 15, 2347–2353 CrossRef CAS.
- H. Lavanant, H. Virelizier and Y. Hoppilliard, J. Am. Soc. Mass Spectrom., 1998, 9, 1217–1221 CrossRef.
- M. Prudent and H. H. Girault, J. Am. Soc. Mass Spectrom., 2008, 19, 560–568 CrossRef CAS.
- M. Prudent, C. Roussel and H. H. Girault, Electrochem. Commun., 2007, 9, 2067–2074 CrossRef CAS.
- D. Schröder, M. C. Holthausen and H. Schwarz, J. Phys. Chem. B, 2004, 108, 14407–14416 CrossRef.
- I. Bako, J. Hutter and G. Palinkas, J. Chem. Phys., 2002, 117, 9838–9843 CrossRef CAS.
- C. Clavaguera, R. Pollet, J. M. Soudan, V. Brenner and J. P. Dognon, J. Phys. Chem. B, 2005, 109, 7614–7616 CrossRef CAS.
- J. B. Li, R. Miller and H. Mohwald, Colloids Surf., A, 1996, 114, 123–130 CrossRef CAS.
- I. G. Loscertales, A. Barrero, I. Guerrero, R. Cortijo, M. Marquez and A. M. Gañan-Calvo, Science, 2002, 295, 1695–1698 CrossRef CAS.
- M. Dole, L. L. Mack, R. L. Hines, D. O. Chemistry, R. C. Mobley, L. D. Ferguson and M. B. Alice, J. Chem. Phys., 1968, 49, 2240–2249 CrossRef CAS.
- L. L. Mack, P. Kralik, A. Rheude and M. Dole, J. Chem. Phys., 1970, 52, 4977–4986 CrossRef.
- J. V. Iribarne and B. A. Thomson, J. Chem. Phys., 1976, 64, 2287–2294 CrossRef CAS.
- M. Suwalsky, B. Ungerer, L. Quevedo, F. Aguilar and C. P. Sotomayor, J. Inorg. Biochem., 1998, 70, 233–238 CrossRef CAS.
- P. K. Yeap, K. O. Lim, C. S. Chong and T. T. Teng, Chem. Phys. Lipids, 2008, 151, 1–9 CrossRef CAS.
- R. Lehrmann and J. Seelig, Biochim. Biophys. Acta, Biomembr., 1994, 1189, 89–95 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |