Functional metal ions in nucleic acids
Jens
Müller
Westfälische Wilhelms-Universität Münster, Institut für Anorganische und Analytische Chemie, Corrensstr. 28/30, 48149 Münster, Germany. E-mail: mueller.j@uni-muenster.de; Fax: +49 251 833 6007; Tel: +49 251 833 6006
First published on 18th March 2010
Abstract
Metal ions are inevitably involved in almost every aspect of nucleic acid chemistry. Their most prominent role is certainly the maintenance of nucleic acid structural integrity. However, they serve various other roles as well, e.g. as catalytic co-factors in ribozymes, in determining the secondary structure of tetra-stranded helices, during RNA folding, and possibly even in enabling nucleobase-centred acid/base catalysis under physiological conditions and in modulating homogeneous genetic recombination. This tutorial review gives a compact overview of the multifaceted situations in which metal ions exert specific functions in nucleic acids, also including a discussion of structural metal ions in regular DNA and RNA as well as triple helices, guanine quadruplexes, and helical junctions. It has a strong focus on metal ions that are naturally present in living systems and only occasionally includes a description of the interaction of exogenous metal complexes with nucleic acids (or their components), provided that the results of these studies can be transferred into the context of endogenous metal ions.
 Jens Müller | Jens Müller is Professor of Inorganic Chemistry in the Department of Chemistry and Pharmacy at Münster University. He received his PhD working with Bernhard Lippert at TU Dortmund University (1999) and spent two years as a postdoctoral fellow (2000–2002) with Stephen J. Lippard (MIT) and Gerhard Wagner (Harvard Medical School). After returning to Germany, he became Independent Research Group Leader at TU Dortmund University. In 2008, he joined the faculty at Münster University. His research interests comprise the bioinorganic chemistry of nucleic acids, and in particular the characterization and utilization of nucleic acids with metal-mediated base pairs. |
Introduction
Due to their polyanionic nature, the natural nucleic acids DNA and RNA always occur in combination with cations. In living systems, these cations are usually polyamines as well as mono- and divalent metal ions. It is probably the diversity of the possible interactions between nucleic acids and metal ions that has been fascinating scientists for a long time. The unwaning interest in this area of research became apparent in the recent publication of two monographs on nucleic acid interactions with metal ions and metal complexes.1,2It is the goal of this tutorial review to give a brief overview of the various situations in which metal ions carry out certain functions in nucleic acids. In this context, “function” may be the maintenance of structural integrity, the induction of proper folding, the enabling of catalysis, and many more. Prior to any discussion of the effect of metal ions on nucleic acids, it is surely worthwhile to reflect on which metal ions (or, to be more precise, which metal complexes comprising aqua ligands) are typically associated with nucleic acids. Based on availability and solubility under physiological conditions, only Na+, K+, Mg2+, and Ca2+ need to be seriously considered. Hence, other metal ions or metal complexes will be discussed only when they might serve as models for one of these cations. A detailed discussion of, e.g., metal-based drugs or of the toxicity of exogenous metal species is beyond the scope of this article and is covered by other recent reviews.2–5
Metal ions and nucleic acid structural integrity
Nucleic acids are polyanions. Basically, each nucleotide carries one negative charge that needs to be shielded to enable the formation of a stable secondary structure. This shielding can be provided by a variety of cations, typically polyamines (such as spermine and spermidine) and/or mono- and divalent metal ions (mainly Na+). The counterion condensation theory for linear polyions6 can be used to describe the localization of metal ions in the vicinity of a nucleic acid. It states that independent of the total counterion concentration, a certain percentage of metal ions stays in close proximity of the nucleic acid and thereby reduces its effective charge density. A prediction valid for monovalent cations suggests that about 75% of the cations are located within close distance of the DNA surface, with hardly any dependence on the total cation concentration.6 This diffuse binding is highly unspecific. Based on the assumption that non-directed electrostatic forces are the key determinants in nucleic acid metal ion interactions, the possibility of additional sequence-specific cation binding has long been neglected. Moreover, difficulties in the experimental localization of metal ions in nucleic acids structures as a result of their typical high disorder seemed to confirm a mostly statistical distribution of the cations. This view has changed over the past one or two decades. Theoretical calculations have tremendously contributed to our current knowledge of metal-ion binding sites in nucleic acids. As it turned out, a significant percentage of metal ions binds in a sequence-specific manner. Typical metal binding patterns strongly depend on the nucleic acid conformation. They are therefore discussed individually in the following subchapters.B-DNA
B-DNA is the most prominent DNA conformation (Fig. 1b). It adopts the geometry originally proposed by Watson and Crick7 and represents the biologically relevant form of DNA. The best-studied sequence typically adopting B-type geometry is the self-complementary Dickerson-Drew dodecamer, d(CGCGAATTCGCG).8 Based on the calculated Na+ density around the duplex formed by this sequence it was concluded that in addition to the non-specific binding outside the groove regions of the DNA, sequence-specific binding inside the grooves exists as well.9 Most metal ions were observed in the central AATT region, some at the GpC steps but none at the CpG steps. A closer inspection showed that the Na+ ions found at the ApT steps are located in the minor groove, coordinated by carbonyl groups of thymine residues from opposite strands. This localization indicates that probably several of the “water molecules” observed in the so-called spine of hydration along the minor groove are actually Na+ ions, or that the water positions are at least partially occupied by Na+ (Na+ and O2– are isoelectronic, making them virtually indistinguishable by single crystal X-ray diffraction analysis).10 Recent magnetic relaxation dispersion data suggest that the ApT site in the minor groove of the Dickerson-Drew dodecamer has a Na+ occupancy of up to 50%.11 It is therefore obvious that in addition to the non-specific binding based on electrostatic interactions, metal ions bind to nucleic acids in a site-specific manner (as had previously been established for short nucleic acid fragments12,13).14 Accordingly, different sequences exhibit different binding patterns and concomitantly lead to different local structures of the duplex. One of the best-known examples for a recurring structural distortion is the bending of so-called A-tracts (i.e., duplex DNA with sequences of at least three ApA, ApT or TpT steps without any interfering flexible TpA step) that is accompanied by a narrowing minor groove. In good agreement with the above-mentioned computational result that metal ions are preferentially localized at the central AATT region of the Dickerson-Drew dodecamer,9 the sequence-specific nature of the cation binding provides an elegant way of explaining the origin of the A-tract bending. However, at present the question remains regarding the causal relationship: Does the presence of an unusually narrow minor groove result in a higher metal ion affinity, or does the binding of metal ions lead to the formation of an unusually narrow groove?15,16 | ||
| Fig. 1 Schematic representation of various nucleic acid double helix conformations: (a) A-DNA; (b) B-DNA; (c) Z-DNA. | ||
A-DNA
Typical conditions for the formation of A-type duplex DNA (Fig. 1a) include low humidity (e.g. decreasing water content in DNA fibres or less water and concomitantly more alcohol in the crystallizing solution). In addition, it represents the natural conformation of RNA double helices and DNA/RNA chimeric helices. It has been long known that metal ions are to some extent involved in the structural transition from B-DNA to A-DNA, as evidenced by the fact that the Na+ salt of DNA can undergo this type of transition whereas the Li+ salt does not (but rather forms C-DNA).17,18 Unfortunately, structural information on the metal-binding behaviour of nucleic acids adopting an A-type duplex is scarce, especially with respect to endogenous metal ions. It has been suggested that the binding of exogenous metal ions like Ba2+ or metal complexes like [Co(NH3)6]3+ destabilizes the B-form, and that subsequently a competition between the A-form and the Z-form is evoked, the outcome of which strongly depends on the nucleic acid sequence.19Z-DNA
The Z-DNA conformation (Fig. 1c) has attracted a lot of attention as it is the only polymorph based on a left-handed double helix.20 Its repeating unit is a dinucleotide, with CpG being the best sequence for Z-DNA formation. Originally, Z-DNA formation was believed to require an extremely high ionic strength,21 but it is now known that in the presence of cellular cations it can be induced to form even under physiological conditions. In fact, its transient formation in vivo is nowadays accepted, and it was shown that the formation of a continuously stacked double helix with B→Z and Z→B transitions is feasible.22 In Z-DNA, the distance between the phosphate groups of neighbouring nucleotides is significantly smaller than in A- and B-DNA. As a result, higher charged cations (such as Mg2+ or some of the cellular polyamines) are necessary for an effective stabilization of this conformation. In addition, highly charged exogenous complexes such as [Co(NH3)6]3+ and [Ru(NH3)6]3+ are known stabilizers of Z-DNA.20 They bind via outer-sphere interactions, i.e. via hydrogen bonds involving their ammine ligands. The potential hydrogen bonding sites in Z-DNA are identical to those in B-DNA (with the exception of the purine N3 position which is not accessible in Z-DNA). Obviously, these sites are also involved in stabilizing the Z conformation by forming hydrogen bonds to the aqua ligands of solvated Mg2+ and Na+ ions. Current research regarding the metal interactions of Z-DNA also includes the design of metal complexes that are capable of either sensing Z-DNA or promoting the B→Z transition under low-salt conditions.23,24Metal-ion involvement in the formation of higher-order structures
DNA triple helices
It has been reported as early as 1957 that two pyrimidine strands and one purine strand can form a triple helix.25 Since then, various other sequence combinations have been shown to be able to adopt a triplex structure.26 A base triplet within a triplex always consists of a regular Watson–Crick base pair with the third base binding in the major groove of the former double helix. Depending on whether the third strand aligns in a parallel or an antiparallel orientation with respect to the purine-rich strand, different triplex conformations are feasible. At present, no systematic studies focusing on the metal-binding behaviour of triple helices have been reported. However, it is obvious that as a result of the higher charge density divalent cations are necessary to shield effectively the negative charge and enable the formation of this type of secondary structure. Typically, a concentration as low as 5 mM Mg2+ is sufficient. Moreover, theoretical calculations suggest that cation-induced polarization of the base triple significantly contributes to its stabilization.27 Exogenous metal complexes like the trans-[Pt(NH3)2]2+ entity have also been shown to be able to stabilize triple helices.28Guanine quadruplex
Oligonucleotides with stretches of consecutive guanine nucleotides can aggregate by forming a four-stranded helix.29 This quadruplex comprises stacked planar tetrads of guanine bases held together by eight hydrogen bonds (Scheme 1). The structural integrity of the guanine quadruplex is strongly dependent on the presence of monovalent cations, especially K+ and Na+. Guanine quadruplexes are of enormous interest as there is (indirect) evidence that they exist in vivo.30–32 For example, guanine-rich sequences occur as overhangs on the 3′-ends of eukaryotic chromosomes (known as telomeres) with sequences such as GnTn, GnTnGn, or (TTAGGG)n. The human telomeres in somatic cells are typically 8000–10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 bases long, with the terminal 100–200 bases being “single-stranded”. This overhang can fold back and stabilize the telomeres via formation of guanine tetrads.32,33 Such stabilization is interesting in terms of the development of a selective anticancer therapy:34 As a result of the inability of DNA polymerase to fully replicate single strands, the telomeres progressively shorten during successive rounds of replication, eventually leading to cell death. In more than 80% of all tumour cells an enzyme called telomerase is active that catalyzes the synthesis of telomeric DNA repeats onto the single-stranded 3′-end, thereby maintaining telomere length and resulting in immortal cells. Telomerase is not active in healthy human somatic cells. Hence, the stabilization of the telomeric DNA in the form of a guanine quadruplex might enable a selective anticancer therapy. In this context, numerous small organic molecules as well as metal complexes have been devised as potential quadruplex-inducing agents.35–38 The probable biological importance of guanine quadruplexes also stems from various other observations,39 a few of which are briefly summarized in the following: (1) Ligands known to stabilize the guanine quadruplex structure in the promotor region of a gene are able to suppress40 or increase41 transcription of that gene. Combined with the observation that guanine-rich sequences capable of forming quadruplexes are prevalent within the promotor regions of numerous genes this implies a genome-wide regulatory role for guanine quadruplexes.42 (2) Formation of guanine-quadruplex structures has been implied in the molecular pathology of the Fragile-X syndrome.43 (3) A combinatorially selected guanine quadruplex was shown to be a potent inhibitor of HIV cell fusion.44
000 bases long, with the terminal 100–200 bases being “single-stranded”. This overhang can fold back and stabilize the telomeres via formation of guanine tetrads.32,33 Such stabilization is interesting in terms of the development of a selective anticancer therapy:34 As a result of the inability of DNA polymerase to fully replicate single strands, the telomeres progressively shorten during successive rounds of replication, eventually leading to cell death. In more than 80% of all tumour cells an enzyme called telomerase is active that catalyzes the synthesis of telomeric DNA repeats onto the single-stranded 3′-end, thereby maintaining telomere length and resulting in immortal cells. Telomerase is not active in healthy human somatic cells. Hence, the stabilization of the telomeric DNA in the form of a guanine quadruplex might enable a selective anticancer therapy. In this context, numerous small organic molecules as well as metal complexes have been devised as potential quadruplex-inducing agents.35–38 The probable biological importance of guanine quadruplexes also stems from various other observations,39 a few of which are briefly summarized in the following: (1) Ligands known to stabilize the guanine quadruplex structure in the promotor region of a gene are able to suppress40 or increase41 transcription of that gene. Combined with the observation that guanine-rich sequences capable of forming quadruplexes are prevalent within the promotor regions of numerous genes this implies a genome-wide regulatory role for guanine quadruplexes.42 (2) Formation of guanine-quadruplex structures has been implied in the molecular pathology of the Fragile-X syndrome.43 (3) A combinatorially selected guanine quadruplex was shown to be a potent inhibitor of HIV cell fusion.44
 | ||
| Scheme 1 Chemical structure of a guanine tetrad. A total of eight hydrogen bonds are formed within one tetrad. It is additionally stabilized by coordinative bonds to a central metal ion (represented by a grey sphere, not necessarily located within the tetrad plane, see Fig. 2a). | ||
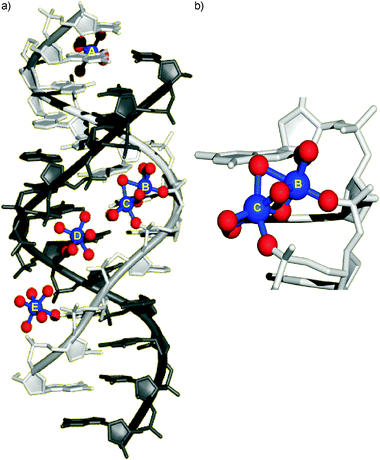
These examples show that guanine quadruplexes represent promising therapeutic targets;45 therefore a plethora of studies exists regarding their precise characterization. Numerous different topologies have been found for telomeric quadruplexes, depending on the flanking sequences, probably the nucleic acid concentration, and particularly the nature of the stabilizing monovalent metal ions.46,47 For example, a total of five different topologies have been reported as yet for the intramolecular quadruplex formed from four repeats of the human telomeric DNA sequence d(TTAGGG)n.48–52 In all guanine quadruplexes, the cations are located along the helical axis and coordinate to the O6 atoms of guanine. While Na+ ions can be found either in plane with a tetrad or in-between two neighbouring tetrads, the larger K+ ions have only been observed in the latter position (Fig. 2). Hence, the typical coordination geometry of a K+ ion is that of a distorted square antiprism. It is generally anticipated that the K+ forms represent the biologically more relevant topologies because K+ is much more abundant in the intracellular environment than Na+.
![Structure of the intramolecular guanine quadruplex from the human telomeric sequence d[AGGG(TTAGGG)3] in the presence of K+ ions as determined by single crystal X-ray diffraction analysis.49 Guanine residues are shown in orange, adenine and thymine residues in grey and green, respectively. The localization of the K+ ions (purple) in-between neighbouring nucleobase tetrads can clearly be discerned. View (a) perpendicular to, and (b) along the internally aligned cations. Coordinates taken from PDB code 1KF1.49](/image/article/2010/MT/c000429d/c000429d-f2.gif) | ||
| Fig. 2 Structure of the intramolecular guanine quadruplex from the human telomeric sequence d[AGGG(TTAGGG)3] in the presence of K+ ions as determined by single crystal X-ray diffraction analysis.49 Guanine residues are shown in orange, adenine and thymine residues in grey and green, respectively. The localization of the K+ ions (purple) in-between neighbouring nucleobase tetrads can clearly be discerned. View (a) perpendicular to, and (b) along the internally aligned cations. Coordinates taken from PDB code 1KF1.49 | ||
As a result of the site-specific localization of the metal ions, they can easily be detected in single crystal X-ray diffraction studies. In addition, several heteronuclear NMR studies helped to provide direct proof for the presence of metal ions along the helical axis in solution53,54 and to elucidate the affinity of different cations for binding to the surface (i.e. to external phosphate groups) versus binding along the helical axis.55 Even EXAFS studies have been performed to identify the metal binding sites in guanine quadruplexes.56 Solid-state NMR data gathered using the guanine tetrad structure formed by guanosine 5′-monophosphate showed that the affinity of monovalent cations for binding along the helical axis follows the order K+ > NH4+ > Rb+ > Na+ > Cs+ > Li+,55 strongly suggesting that size effects influence the binding affinity. It can be concluded from these data that the preference for binding K+ ions is not only based on its higher abundance in the cells but that K+ is also favoured thermodynamically. More precisely, the free energy difference for the competition between K+ and Na+ amounts to −(8.0 ± 1.6) kJ mol−1 at 25 °C (for guanine quartets formed by 5′-GMP).55 Interestingly, when replacing guanine with the non-natural nucleobase isoguanine, formation of pentads instead of tetrads is observed due to the different geometrical arrangement of hydrogen bond donor and acceptor groups.57 As a result of the increased size of the cavity along the helical axis, this artificial pentaplex requires Cs+ for optimal stabilization.
Helical junctions
Helical junctions are typically divided into four-way and three-way junctions. They contain double helical segments that intersect with axial discontinuities. At these intersections, strands are exchanged between different helical segments.58 The best-known four-way DNA junction is the Holliday junction,59 representing the central intermediate in homogeneous genetic recombination. In addition, helical junctions are important architectural elements in RNA structures. Although often drawn as if the helices were held together only by the continuity of the strands, the unconstrained Holliday junction (termed stacked X structure) in practice adopts an arrangement with pairwise antiparallel coaxial stacking of the individual double helical segments (Fig. 3b). Metal ions exert a profound effect on the structure of the Holliday junction. As a result of the increased charge density at the junction site due to the accumulation of phosphate groups from different helical segments, micromolar concentrations of divalent metal ions such as Mg2+ (>80 μM) or of cellular polyamines like spermine (>25 μM) are necessary for proper folding.60 Even very high concentrations of monovalent cations such as Na+ or K+ result in partial folding of the junction only. The unfolded structure is expected to have an approximately square arrangement, providing maximum extension at the junction site and thereby minimal electrostatic repulsion (Fig. 3a). As has also been observed for duplex DNA, stabilization of helical junctions not only occurs via non-directed electrostatic shielding but—at least for the divalent metal ions and metal complexes—also via site-specific interactions.58 An interesting suggestion has been made with respect to an involvement of the hydration spheres of the metal ions: Based on the results of single crystal X-ray diffraction analyses, hydrogen bond formation involving aqua ligands originating from site-specifically bound metal ions result in a stabilising ordering effect of the hydration inside the junction arms.61 | ||
| Fig. 3 Representative structures of DNA four-way junctions in (a) an extended, unstacked conformation and (b) the stacked X conformation. The four individual strands are colour-coded. In the presence of sufficient amounts of divalent cations, the junction adopts the stacked X conformation. The structures shown here contain different oligonucleotide sequences and have been determined by single crystal X-ray diffraction analyses under different conditions.108,109 In particular, the four-way junction in the extended conformation is stabilized by the RuvA protein (protein not shown). The chemical equilibrium is shown only to demonstrate the possibility of a conformational change induced by divalent cations. Coordinates taken from PDB codes 1C7Y and 3IGT, respectively.108,109 | ||
The significant dependence of the structure on the metal ion concentration is reflected in the kinetics of homogenous genetic recombination. During this biological process, the junction can undergo sequential exchange of base pairs, termed branch migration. The time necessary for one step of branch migration strongly depends on the metal ion concentration. It varies over several orders of magnitude and has been reported to amount to 300 μs in the absence of Mg2+ and 300 ms in the presence of 10 mM Mg2+.62 Obviously, the extended, unstacked conformation that is adopted under low-salt conditions is required for fast branch migration (Fig. 3).
The formation of three-way junctions shows a similar dependence on metal ion concentration. From a structural point of view, the perfect three-way junction does not fold by coaxial stacking as a result of its rigid framework. This restraint is released in the preferentially formed bulged three-way junctions, resulting in increased stability. Again, proper folding of a stacked structure requires the presence of suitable metal ions.58 Recently, an exogenous metal complex has been reported to be capable of binding and stabilising a DNA three-way junction,63 thereby opening up a new path towards potential therapeutic agents based on metal complexes that target DNA in a non-covalent manner.64
Effects of metal coordination to nucleobases
Stabilization of rare tautomers
It is obvious that the mutual recognition of complementary nucleobases vitally depends on the tautomeric structures of the two bases (Scheme 2). The presence of a nucleobase in its rare tautomeric form during replication will lead to the formation of a mismatched base pair as a result of the modified hydrogen bond donor and acceptor capabilities of that nucleobase. If not repaired, such a mispair is likely to lead to a mutation event.65 For example, the rare imino form of adenine is in principle capable of forming a base pair with guanine, so that after two rounds of replication an original adenine–thymine base pair is eventually replaced by a cytosine–guanine base pair,66i.e., a transversion mutation takes place (Scheme 3). Fortunately, for each of the four common nucleosides the predominant tautomer is favoured over the second most abundant one by a factor of about 104–105, equivalent to a relative stability in the order of 23–29 kJ mol−1,67 making such a scenario apparently unlikely. However, the coordination of metal ions to nucleobases is known to lead frequently to the stabilization of rare tautomeric forms,67 with numerous examples existing for the various nucleobases.66–70 In these metal-stabilized rare tautomers, the metal occupies a position that is usually occupied by a proton, forcing the proton to move to another position and thereby generating the rare tautomer (Scheme 4). Although the structural information on metal-stabilized rare tautomers is often gathered from single crystal X-ray diffraction analyses of transition metal complexes of model nucleobases, the possibility exists that also the transient formation of a metal-stabilized rare tautomer in vivo might in the end lead to a mutation event, thereby adding this scenario to the possible reasons for metal-induced mutagenicity.65 Interestingly, even the formation of short-lived metal-free rare tautomers seems feasible, provided that the process of tautomerization that starts immediately after the demetalation is somehow impeded, e.g., by the non-aqueous environment provided by the binding pocket of a DNA-binding protein. It should therefore be possible that the transiently binding metal ion responsible for generating the rare tautomer occupies a site normally involved in Watson–Crick base pairing. | ||
| Scheme 2 Chemical structures of base pairs involving guanine. (a) When both nucleobases are present in their predominant tautomeric form, a regular guanine–cytosine base pairs is formed. (b) Guanine in its rare 6H, 9H-enol (trans) tautomeric form can (from a geometrical point of view) form a base pair with thymine. | ||
 | ||
| Scheme 3 The rare imino tautomer of adenine (A*) is in principle capable of forming a base pair with guanine (G) (the latter nucleoside must be present in its syn conformation for proper base pairing), eventually replacing an adenine–thymine base pair with a cytosine–guanine base pair after two successive rounds of replication, leading to a transversion mutation. | ||
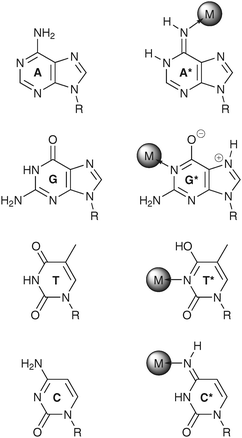 | ||
| Scheme 4 Examples for metal-stabilized rare tautomers of adenine (A*),66 guanine (G*),110 thymine (T*),70 and cytosine (C*).111 For comparison, the respective predominant tautomers are shown as well. | ||
Shifted acidity constants
The complexation of a nucleobase by a metal ion (or metal complex) dramatically changes the electronic situation of the base pair. As the positively charged metal ions withdraw electron density from the aromatic heterocycles, they induce an acidification of the exchangeable protons of the nucleobase and concomitantly a decrease in basicity of the endocyclic nitrogen atoms that carry no protons or of the exocyclic carbonyl groups.71 Depending on the extent of this effect, the pKa values may be perturbed in such a way that formerly neutral nucleosides become deprotonated and hence charged. It is obvious that metal ion coordination is by no means the only contributor to the phenomenon of shifted pKa values, but it needs to be noted that local differences in the microenvironment (e.g., changes in the dielectric constant, electrostatic effects due to closely positioned negative or positive charges, hydrogen bond effects) should not be considered the sole reasons. A highly topical feature closely related to shifted acidity constants is that of acid–base catalysis involving nucleobases, especially in the context of ribozymes.72,73 For example, it has been proposed that the hairpin ribozyme cleaves the phosphodiester bond by using a guanine moiety as the general base and an adenine moiety as the general acid.74 Moreover, it has been reported that the apparent pKa value of the active-site cytosine in the hepatitis delta virus ribozyme is shifted towards neutrality, and more importantly in the context of this review, that it depends on the metal ion concentration.75,76As is the case with the tautomeric shifts induced by metal ions, most available information stems from structural work on inert metal complexes of model nucleobases. Numerous examples exist of altered pKa values of nucleobases, with the metal complexes responsible for these alterations (a) replacing endocyclic imino protons, (b) replacing exocyclic amino protons, (c) coordinating to neutral bases, or (d) occupying several of these positions (Scheme 5).71 When also taking into account a possible stabilization of the deprotonated nucleobase via intramolecular hydrogen bonding, an acidification by almost 10 log units is feasible.77 It therefore appears likely that metal-ion coordination contributes—at least partially—to the shift of “natural” pKa values towards neutrality as observed in several large nucleic acid structures such as ribozymes.
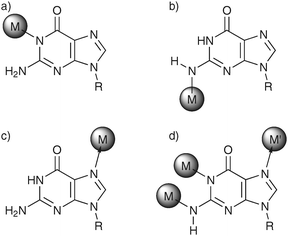 | ||
| Scheme 5 Various nucleobase binding patterns are possible, leading to altered pKa values as a result of metal complexation (here: nucleobase = guanine): The metal complex (a) replaces an endocyclic imino proton,110 (b) replaces an exocyclic amino proton,112 (c) coordinates to a neutral base,113 or (d) occupies several of these positions.114 | ||
Metal ions and RNA
The view of the role of RNA in living systems has changed dramatically over the past decades. It is now evident that this nucleic acid is not merely a passive carrier of genetic information but rather exerts a large variety of functions, including (but not limited to) RNA splicing, protein transport, and gene regulation. The involvement of metal ions in some of these functions as well as structural aspects of metal ion–RNA interactions will be dealt with in the following.Metal-dependent structural motifs
Metal ions most commonly required for the stabilization of functional RNA structures are Mg2+ and K+.78–80 Contrary to DNA with its double helical structure (for exceptions see above) the structural chemistry of RNA is not limited to one type of conformation. Instead, a plethora of different structural motifs—many of then containing non-Watson–Crick base pairs—can be adopted. A variety of these motifs strictly requires the presence of appropriate metal ions. In the following, two representative examples for structured non-Watson–Crick motifs will be presented, namely the loop E motif and an AA platform.The loop E motif is adopted by a dodecamer duplex and comprises seven non-canonical base pairs (Fig. 4a). It forms only in the presence of millimolar Mg2+.81 In addition to a “metal ion zipper” along the major groove, a particularly interesting structural feature is the presence of a site-specific binuclear Mg2+-cluster containing three bridging aqua ligands (Fig. 4b).82 Optical melting studies were able to show that the Mg2+ ions indeed bind to the loop E motif in a specific fashion (rather than by non-specific electrostatic attractions).83 Similarly to what had been observed for A-tracts in DNA (see above), this localization of positive charge density is accompanied by an unusually narrow groove, illustrating again a tight correlation between the presence of metal ions and the formation of (unusual) nucleic acid structures.
 | ||
| Fig. 4 (a) Loop E motif within structured RNA with five site-specifically bound Mg2+ ions (blue) as determined by single crystal X-ray diffraction analysis.82 The oxygen atoms in the coordination sphere of the metal ions are shown in red. Coordinates taken from PDB code 354D.82 Mg2+ ions A and D bind exclusively in an outer-sphere fashion, ions B, C, and E also via inner-sphere interactions. A “metal ion zipper” formed by Mg2+ ions B–E is clearly discernible. (b) A close-up of the binuclear Mg2+ cluster with three bridging aqua ligands indicates the presence of two unusually close phosphate groups. | ||
An AA platform is a structural motif in which two contiguous nucleobases (here: two adenine residues) are located side-by-side instead of stacking on top of one another (Fig. 5). AA platforms can be found in tetraloop receptors, tertiary structural motifs occurring frequently within RNA. Interestingly, the formation of stable tetraloop receptors has been correlated with the site-specific binding of a dehydrated monovalent metal ion.84 Based on single crystal X-ray diffraction analyses and additional biochemical experiments (metal-ion rescue experiments, see below), this monovalent metal ion was shown to be K+. It is bonded to the sugar-phosphate backbone of the platform-forming nucleotides (phosphate-O, 2′-OH) as well as to neighbouring nucleobases (namely guanine-N7, guanine-O6, uracil-O4). These experiments show that not only divalent but also monovalent metal ions can bind in an inner-sphere fashion (i.e., via direct contacts between metal ion and nucleic acid) to RNA and are likely to play a role in the activity of this nucleic acid.
 | ||
| Fig. 5 Experimental structure of an AA platform motif within structured RNA as determined by single crystal X-ray diffraction analysis.84 The inner-sphere binding of the K+ ion (orange) can clearly be discerned. Reprinted with permission from ref. 84 (Nat. Struct. Biol., 1998, 5, 986-992). Copyright 1998, Macmillan Publishers Ltd. | ||
Folding of RNA
As a result of its often highly complex yet compact tertiary structure, the folding of RNA into the native state has attracted much attention. Interestingly, even large ribozymes can fold through direct pathways via discrete conformationally defined intermediates.85 In addition to proteins that play an important role in the folding process, metal ions have been found to be a crucial determinant of the pathway towards the folded state.86–88 Several models exist for the tertiary folding of large RNA molecules, all of which strongly depend on the availability of certain metal ions at a given folding stage.85 While a detailed description of these models is beyond the scope of this tutorial review, it can be summarized that in most cases monovalent metal ions such as K+ are necessary at an early stage to ensure formation of the required secondary structure while the presence of divalent metal ions like Mg2+ is essential for stabilizing the complex tertiary structure at later stages (Fig. 6). From a kinetic point of view, in most models a Mg2+-dependent fast collapse into a misfolded state is followed by a slow folding into the native state.89 Interestingly, several cases are known in which the most prominent state is not the “native” one but rather a “near-native” one. For example, recent single-molecule studies of group II intron ribozymes showed that the catalytically active conformer is adopted only transiently, requires the substrate for stabilization, and is strongly dependent on the presence of a significant concentration of Mg2+ (>15–20 mM).89 Moreover, the presence of additional metal ions such as Ca2+ can induce the formation of distinct subpopulations, leading to an even more complicated picture.90 These examples show the importance of metal ions with respect to the structural (and hence functional) integrity of nucleic acids in general and of catalytic RNA in particular.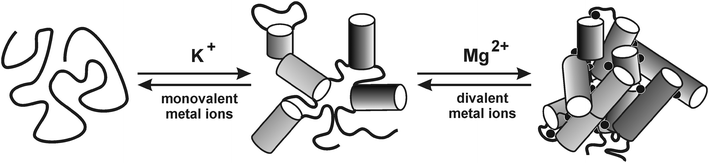 | ||
| Fig. 6 Schematic simplified representation of the folding pathway of a group II intron ribozyme. The folding is strongly dependent on type and concentration of metal ions. Monovalent cations enable formation of the secondary structure by screening of the negative charge. Divalent cations are required for formation of the catalytically active tertiary structure. Reproduced with permission from ref. 92 (R. K. O. Sigel, "Group II Intron Ribozymes and Metal Ions - A Delicate Relationship", Eur. J. Inorg. Chem., 2005, 2281-2292). Copyright 2005, Wiley-VCH Verlag GmbH & Co. KGaA. | ||
Metal ions in ribozymes
The ability of ribozymes to perform catalysis is often strongly correlated with the presence of metal ions.73,79,91–93 While predominantly divalent metal ions are required for catalysis, certain ribozymes can also function in the presence of molar concentrations of monovalent cations.94 Obviously, structures obtained by single crystal X-ray diffraction analysis are able to provide a precise overview of site-specifically bound metal ions.95,96 Moreover, NMR spectroscopy can help to shed light on the metal-ion binding properties of RNA.97–99 However, detailed studies on the actual role of the respective functional metal ions (function = catalysis and maintenance of structural integrity, respectively) are hampered by the fact that the RNAzymes typically depend on spectroscopically silent closed-shell metal ions. Hence, experimental techniques complementary to those used to study metalloenzymes are required for the detection of (catalytically active) metal species in ribozymes, two of which will be described below.Lanthanide ions can be applied as structural probes for metal-ion binding sites.100 In a representative example, Tb3+ was used as a mimic for Mg2+.101 The similar radii of hexacoordinate Tb3+ (92 pm) and Mg2+ (72 pm) in combination with the same preference for coordinating oxygen ligands makes Tb3+ a good structural substitute of Mg2+ in nucleic acids. In fact, Tb3+ coordinates to RNA even more tightly than Mg2+.102 Conveniently, the aqua ligands of solvated Tb3+ display a pKa value close to neutral pH, making them ideal acid/base catalysts for efficient RNA cleavage under physiological conditions. Hence, by exposing an RNA molecule in its native folded state to increasing Tb3+-concentrations and by mapping the lanthanoid-induced backbone cleavage to specific RNA residues, potential metal-binding sites within the nucleic acid can be identified. While this method is not suitable for the precise identification of the respective RNA ligands, it certainly helps to localize areas with a particularly large number of high-affinity metal-binding sites.
Metal-ion rescue experiments are another means of identifying functionally relevant metal ions as well as their precise position within a nucleic acid. Previous anticipation of a metal-binding site is required for this type of experiment, however. In a typical set of experiments, suspected metal-binding groups are systematically chemically mutated (typically by sulfur substitution of the ribose or phosphoryl oxygen atoms) to prevent binding of the natural metal ion and hence to inhibit RNA functionality.103 If the addition of a different, usually non-endogenous metal ion restores functionality, the mutated group most probably was involved in site-specific metal-ion binding. Obviously, the preservation of the local structure after the mutation is a prerequisite for a successful metal-ion rescue experiment. As mentioned above, the binding of K+ to an AA platform motif could be confirmed by a metal-ion rescue experiment: When replacing the guanine residue located directly below the AA platform by 6-thioguanosine 5′-monothiophosphate, the AA platform motif loses its structural integrity (as evidenced by the loss of binding affinity to another RNA structural motif), which in turn can be restored upon the addition of the thiophilic Tl+.84
The actual function of metal ions in a ribozyme-catalyzed phosphoryl transfer reaction becomes evident upon inspection of the nucleic acid active site residues including the three Mg2+ ions required for catalysis in the Tetrahymena thermophila ribozyme (Scheme 6).91 This ribozyme catalyzes a phosphotransesterification between a guanosine and a short oligonucleotide substrate that base pairs with the ribozyme.104 In the catalytic mechanism, the divalent metal ions serve as Lewis acids, stabilizing the transition state by efficiently shielding the negative charge of the attacking 3′-oxygen of the guanosine nucleophile, the departing 3′-oxygen of the oligonucleotide substrate, and the phosphoryl oxygen atoms of the reactive phosphoryl group. Moreover, one of the Mg2+ ions is believed to facilitate the deprotonation of the 3′-OH group under physiological conditions, thereby enabling its activation as the nucleophile.105
 | ||
| Scheme 6 Experimentally verified interactions of three Mg2+ ions (represented by grey spheres) that stabilize the transition state of the Tetrahymena thermophila group I ribozyme. Nucleobase abbreviations are printed in italics (A = adenine, C = cytosine, G = guanine, U = uracil). Figure adapted from ref. 91. | ||
Summary
It is astonishing to see that less than a handful of different metal ions are necessary for the stabilization of a wealth of nucleic acid structures—from regular DNA double helices to non-Watson–Crick RNA motifs, by way of four-way junctions and quadruplexes. While it is undisputed that the nucleic acid sequence plays the predominant role in determining the secondary (and where applicable tertiary) structure, metal ions are unequivocally required for nucleic acid folding—and in some cases such as the guanine quadruplex, they also influence the final topology. In addition to their role as sustainer of structural integrity, metal ions are involved in a plethora of other functions. For example, many ribozymes are catalytically active only in the presence of suitable metal ions. Considering the widespread occurrence of self-cleaving ribozymes,106 functional metal ions in nucleic acid chemistry are widely distributed in nature. Other possible functions such as the modulation of homogeneous genetic recombination by influencing the conformational equilibrium of a Holliday junction, or the enabling of nucleobase-centred acid/base catalysis under physiological conditions by shifting pKa values of nucleobases, are not yet proven to be of importance in vivo, but they certainly are interesting hypotheses. Given the ever increasing number of types of functional RNA, it is almost certain that even more examples of currently unknown interrelationships between metal ions and nucleic acids will be established in the future. And when looking beyond one's own nose, even in the non-biological context of DNA-based nanostructures, the distinct metal-binding properties of nucleic acids have been shown to be useful in regulating the precise outcome of the self-assembled structures.107References
- Nucleic Acid-Metal Ion Interactions, ed. N. V. Hud, RSC Publishing, 2009 Search PubMed.
- Metal Complex-DNA Interactions, ed. N. Hadjiliadis and E. Sletten, John Wiley & Sons, 2009 Search PubMed.
- A. R. Timerbaev, Metallomics, 2009, 1, 193–198 RSC.
- R. C. Todd and S. J. Lippard, Metallomics, 2009, 1, 280–291 RSC.
- A. Arita and M. Costa, Metallomics, 2009, 1, 222–228 RSC.
- G. S. Manning, Q. Rev. Biophys., 1978, 11, 179–246 CrossRef CAS.
- J. D. Watson and F. H. C. Crick, Nature, 1953, 171, 737–738 CAS.
- R. Wing, H. Drew, T. Takano, C. Broka, S. Tanaka, K. Itakura and R. E. Dickerson, Nature, 1980, 287, 755–758 CrossRef CAS.
- M. A. Young, B. Jayaram and D. L. Beveridge, J. Am. Chem. Soc., 1997, 119, 59–69 CrossRef CAS.
- X. Shui, L. McFail-Isom, G. G. Hu and L. D. Williams, Biochemistry, 1998, 37, 8341–8355 CrossRef CAS.
- F. C. Marincola, V. P. Denisov and B. Halle, J. Am. Chem. Soc., 2004, 126, 6739–6750 CrossRef CAS.
- N. C. Seeman, J. M. Rosenberg, F. L. Suddath, J. J. P. Kim and A. Rich, J. Mol. Biol., 1976, 104, 109–144 CrossRef CAS.
- J. M. Rosenberg, N. C. Seeman, R. O. Day and A. Rich, J. Mol. Biol., 1976, 104, 145–167 CrossRef CAS.
- M. Egli, Chem. Biol., 2002, 9, 277–286 CrossRef CAS.
- N. V. Hud and M. Polak, Curr. Opin. Struct. Biol., 2001, 11, 293–301 CrossRef CAS.
- N. V. Hud and A. E. Engelhart, in Nucleic Acid-Metal Ion Interactions, ed. N. V. Hud, RSC Publishing, 2009, pp. 75–117 Search PubMed.
- R. E. Franklin and R. G. Gosling, Nature, 1953, 171, 740–741 CAS.
- D. A. Marvin, M. Spencer, M. H. F. Wilkins and L. D. Hamilton, Nature, 1958, 182, 387–388 CrossRef CAS.
- M. C. Wahl and M. Sundaralingam, in Oxford Handbook of Nucleic Acid Structure, ed. S. Neidle, Oxford University Press, 1999, pp. 117–144 Search PubMed.
- B. Basham, B. F. Eichman and P. S. Ho, in Oxford Handbook of Nucleic Acid Structure, ed. S. Neidle, Oxford University Press, 1999, pp. 199–252 Search PubMed.
- F. M. Pohl and T. M. Jovin, J. Mol. Biol., 1972, 67, 375–396 CAS.
- S. C. Ha, K. Lowenhaupt, A. Rich, Y.-G. Kim and K. K. Kim, Nature, 2005, 437, 1183–1186 CrossRef CAS.
- B. Spingler and C. Da Pieve, Dalton Trans., 2005, 1637–1643 RSC.
- B. Spingler and P. M. Antoni, Chem.–Eur. J., 2007, 13, 6617–6622 CrossRef CAS.
- G. Felsenfeld, D. R. Davies and A. Rich, J. Am. Chem. Soc., 1957, 79, 2023–2024 CrossRef CAS.
- E. Wang and J. Feigon, in Oxford Handbook of Nucleic Acid Structure, ed. S. Neidle, Oxford University Press, 1999, pp. 355–388 Search PubMed.
- J. Muñoz, J. L. Gelpí, M. Soler-López, J. A. Subirana, M. Orozco and F. J. Luque, J. Phys. Chem. B, 2002, 106, 8849–8857 CrossRef CAS.
- J. Müller, M. Drumm, M. Boudvillain, M. Leng, E. Sletten and B. Lippert, J. Biol. Inorg. Chem., 2000, 5, 603–611 CrossRef CAS.
- M. Gellert, M. N. Lipsett and D. R. Davies, Proc. Natl. Acad. Sci. U. S. A., 1962, 48, 2013–2018 CAS.
- K. Paeschke, T. Simonsson, J. Postberg, D. Rhodes and H. J. Lipps, Nat. Struct. Mol. Biol., 2005, 12, 847–854 CrossRef CAS.
- C. Schaffitzel, I. Berger, J. Postberg, J. Hanes, H. J. Lipps and A. Plückthun, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 8572–8577 CrossRef CAS.
- C.-C. Chang, I.-C. Kuo, I.-F. Ling, C.-T. Chen, H.-C. Chen, P.-J. Lou, J.-J. Lin and T.-C. Chang, Anal. Chem., 2004, 76, 4490–4494 CrossRef CAS.
- Y. Xu, T. Ishizuka, K. Kurubayashi and M. Komiyama, Angew. Chem., Int. Ed., 2009, 48, 7833–7836 CrossRef CAS.
- D. R. Corey, Chem. Biol., 2009, 16, 1219–1223 CrossRef CAS.
- J. E. Reed, S. Neidle and R. Vilar, Chem. Commun., 2007, 4366–4368 RSC.
- A. Arola-Arnal, J. Benet-Buchholz, S. Neidle and R. Vilar, Inorg. Chem., 2008, 47, 11910–11919 CrossRef CAS.
- K. Suntharalingam, A. J. P. White and R. Vilar, Inorg. Chem., 2009, 48, 9427–9435 CrossRef CAS.
- P. Wu, D.-L. Ma, C.-H. Leung, S.-C. Yan, N. Zhu, R. Abagyan and C.-M. Che, Chem.–Eur. J., 2009, 15, 13008–13021 CrossRef CAS.
- J. L. Huppert, Philos. Trans. R. Soc. London, Ser. A, 2007, 365, 2969–2984 CrossRef CAS.
- A. Siddiqui-Jain, C. L. Grand, D. J. Bearss and L. H. Hurley, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 11593–11598 CrossRef CAS.
- Z. A. E. Waller, S. A. Sewitz, S.-T. D. Hsu and S. Balasubramanian, J. Am. Chem. Soc., 2009, 131, 12628–12633 CrossRef CAS.
- A. Verma, V. K. Yadav, R. Basundra, A. Kumar and S. Chowdhury, Nucleic Acids Res., 2009, 37, 4194–4204 CrossRef CAS.
- M. Fry and L. A. Loeb, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 4950–4954 CrossRef CAS.
- A. T. Phan, V. Kuryavyi, J.-B. Ma, A. Faure, M.-L. Andréola and D. J. Patel, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 634–639 CrossRef CAS.
- S. Balasubramanian and S. Neidle, Curr. Opin. Chem. Biol., 2009, 13, 345–353 CrossRef CAS.
- S. Burge, G. N. Parkinson, P. Hazel, A. K. Todd and S. Neidle, Nucleic Acids Res., 2006, 34, 5402–5415 CrossRef CAS.
- T. I. Gaynutdinov, P. Brown, R. D. Neumann and I. G. Panyutin, Biochemistry, 2009, 48, 11169–11177 CrossRef CAS.
- Y. Wang and D. J. Patel, Structure, 1993, 1, 263–282 CrossRef CAS.
- G. N. Parkinson, M. P. H. Lee and S. Neidle, Nature, 2002, 417, 876–880 CrossRef CAS.
- K. N. Luu, A. T. Phan, V. Kuryavyi, L. Lacroix and D. J. Patel, J. Am. Chem. Soc., 2006, 128, 9963–9970 CrossRef CAS.
- A. T. Phan, K. N. Luu and D. J. Patel, Nucleic Acids Res., 2006, 34, 5715–5719 CrossRef CAS.
- K. W. Lim, S. Amrane, S. Bouaziz, W. Xu, Y. Mu, D. J. Patel, K. N. Luu and A. T. Phan, J. Am. Chem. Soc., 2009, 131, 4301–4309 CrossRef CAS.
- D. Rovnyak, M. Baldus, G. Wu, N. V. Hud, J. Feigon and R. G. Griffin, J. Am. Chem. Soc., 2000, 122, 11423–11429 CrossRef CAS.
- S. Basu, A. A. Szewczak, M. Cocco and S. A. Strobel, J. Am. Chem. Soc., 2000, 122, 3240–3241 CrossRef.
- A. Wong and G. Wu, J. Am. Chem. Soc., 2003, 125, 13895–13905 CrossRef CAS.
- I. V. Smirnov, F. W. Kotch, I. J. Pickering, J. T. Davis and R. H. Shafer, Biochemistry, 2002, 41, 12133–12139 CrossRef CAS.
- J. C. Chaput and C. Switzer, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 10614–10619 CrossRef CAS.
- D. M. J. Lilley, Q. Rev. Biophys., 2000, 33, 109–159 CrossRef CAS.
- R. Holliday, Genet. Res., 1964, 5, 282–304 CrossRef.
- D. M. J. Lilley and R. M. Clegg, Annu. Rev. Biophys. Biomol. Struct., 1993, 22, 299–328 CrossRef CAS.
- J. H. Thorpe, B. C. Gale, S. C. M. Teixeira and C. J. Cardin, J. Mol. Biol., 2003, 327, 97–109 CrossRef CAS.
- I. G. Panyutin and P. Hsieh, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 2021–2025 CrossRef CAS.
- A. Oleksy, A. G. Blanco, R. Boer, I. Usón, J. Aymamí, A. Rodger, M. J. Hannon and M. Coll, Angew. Chem., Int. Ed., 2006, 45, 1227–1231 CrossRef CAS.
- J. Müller and B. Lippert, Angew. Chem., Int. Ed., 2006, 45, 2503–2505 CrossRef.
- J. Müller, R. K. O. Sigel and B. Lippert, J. Inorg. Biochem., 2000, 79, 261–265 CrossRef CAS.
- F. Zamora, M. Kunsman, M. Sabat and B. Lippert, Inorg. Chem., 1997, 36, 1583–1587 CrossRef CAS.
- B. Lippert and D. Gupta, Dalton Trans., 2009, 4619–4634 RSC.
- B. Lippert, H. Schöllhorn and U. Thewalt, J. Am. Chem. Soc., 1986, 108, 6616–6621 CrossRef CAS.
- H. Schöllhorn, U. Thewalt and B. Lippert, J. Am. Chem. Soc., 1989, 111, 7213–7221 CrossRef.
- O. Renn, B. Lippert and A. Albinati, Inorg. Chim. Acta, 1991, 190, 285–289 CrossRef CAS.
- B. Lippert, Prog. Inorg. Chem., 2005, 54, 385–447 CAS.
- B. Lippert, Chem. Biodiversity, 2008, 5, 1455–1474 CrossRef CAS.
- R. K. O. Sigel and A. M. Pyle, Chem. Rev., 2007, 107, 97–113 CrossRef CAS.
- P. C. Bevilacqua, Biochemistry, 2003, 42, 2259–2265 CrossRef CAS.
- B. Gong, J.-H. Chen, E. Chase, D. M. Chadalavada, R. Yajima, B. L. Golden, P. C. Bevilacqua and P. R. Carey, J. Am. Chem. Soc., 2007, 129, 13335–13342 CrossRef CAS.
- M. D. Smith, R. Mehdizadeh, J. E. Olive and R. A. Collins, RNA, 2008, 14, 1942–1949 CrossRef CAS.
- M. S. Lüth, M. Willermann and B. Lippert, Chem. Commun., 2001, 2058–2059 RSC.
- D. E. Draper, RNA, 2004, 10, 335–343 CrossRef CAS.
- A. M. Pyle, J. Biol. Inorg. Chem., 2002, 7, 679–690 CrossRef CAS.
- E. Freisinger and R. K. O. Sigel, Coord. Chem. Rev., 2007, 251, 1834–1851 CrossRef CAS.
- N. B. Leontis, P. Ghosh and P. B. Moore, Biochemistry, 1986, 25, 7386–7392 CrossRef CAS.
- C. C. Correll, B. Freeborn, P. B. Moore and T. A. Steitz, Cell, 1997, 91, 705–712 CrossRef CAS.
- M. J. Serra, J. D. Baird, T. Dale, B. L. Fey, K. Retatagos and E. Westhof, RNA, 2002, 8, 307–323 CrossRef CAS.
- S. Basu, R. P. Rambo, J. Strauss-Soukup, J. H. Cate, A. R. Ferré-D'Amaré, S. A. Strobel and J. A. Doudna, Nat. Struct. Biol., 1998, 5, 986–992 CrossRef CAS.
- A. M. Pyle, O. Fedorova and C. Waldsich, Trends Biochem. Sci., 2007, 32, 138–145 CrossRef CAS.
- S. A. Woodson, Curr. Opin. Chem. Biol., 2005, 9, 104–109 CrossRef CAS.
- D. P. Giedroc and N. E. Grossoehme, in Nucleic Acid-Metal Ion Interactions, ed. N. V. Hud, RSC Publishing, 2009, pp. 180–220 Search PubMed.
- S. Moghaddam, G. Caliskan, S. Chauhan, C. Hyeon, R. M. Briber, D. Thirumalai and S. A. Woodson, J. Mol. Biol., 2009, 393, 753–764 CrossRef CAS.
- M. Steiner, K. S. Karunatilaka, R. K. O. Sigel and D. Rueda, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 13853–13858 CrossRef CAS.
- M. Steiner, D. Rueda and R. K. O. Sigel, Angew. Chem. Int. Ed., 2009, 48, 9739–9742 CAS.
- J. K. Frederiksen, R. Fong and J. A. Piccirilli, in Nucleic Acid-Metal Ion Interactions, ed. N. V. Hud, RSC Publishing, 2009, pp. 260–306 Search PubMed.
- R. K. O. Sigel, Eur. J. Inorg. Chem., 2005, 2281–2292 CrossRef CAS.
- V. J. DeRose, Curr. Opin. Struct. Biol., 2003, 13, 317–324 CrossRef CAS.
- J. B. Murray, A. A. Seyhan, N. G. Walter, J. M. Burke and W. G. Scott, Chem. Biol., 1998, 5, 587–595 CrossRef CAS.
- N. Ban, P. Nissen, J. Hansen, P. B. Moore and T. A. Steitz, Science, 2000, 289, 905–920 CrossRef CAS.
- D. M. Shechner, R. A. Grant, S. C. Bagby, Y. Koldobskaya, J. A. Piccirilli and D. P. Bartel, Science, 2009, 326, 1271–1275 CrossRef CAS.
- E. M. Osborne, W. L. Ward, M. Z. Ruehle and V. J. DeRose, Biochemistry, 2009, 48, 10654–10664 CrossRef CAS.
- M. C. Erat, O. Zerbe, T. Fox and R. K. O. Sigel, ChemBioChem, 2007, 8, 306–314 CrossRef CAS.
- M. C. Erat and R. K. O. Sigel, Inorg. Chem., 2007, 46, 11224–11234 CrossRef CAS.
- R. K. O. Sigel and A. M. Pyle, Metal Ions Biol. Syst., 2003, 40, 477–512 CAS.
- R. K. O. Sigel, A. Vaidya and A. M. Pyle, Nat. Struct. Biol., 2000, 7, 1111–1116 CrossRef CAS.
- M. S. Kayne and M. Cohn, Biochemistry, 1974, 13, 4159–4165 CrossRef CAS.
- J. L. Hougland, A. V. Kravchuk, D. Herschlag and J. A. Piccirilli, PLoS Biol., 2005, 3, e277 CrossRef.
- D. Herschlag and T. R. Cech, Biochemistry, 1990, 29, 10159–10171 CrossRef CAS.
- S.-o. Shan, A. Yoshida, S. Sun, J. A. Piccirilli and D. Herschlag, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 12299–12304 CrossRef CAS.
- C.-H. T. Webb, N. J. Riccitelli, D. J. Duminski and A. Lupták, Science, 2009, 326, 953 CrossRef CAS.
- Y. He, Y. Tian, Y. Chen, T. Ye and C. Mao, Macromol. Biosci., 2007, 7, 1060–1064 CrossRef CAS.
- P. Khuu and P. S. Ho, Biochemistry, 2009, 48, 7824–7832 CrossRef CAS.
- M. Ariyoshi, T. Nishino, H. Iwasaki, H. Shinagawa and K. Morikawa, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 8257–8262 CrossRef CAS.
- G. Frommer, I. Mutikainen, F. J. Pesch, E. C. Hillgeris, H. Preut and B. Lippert, Inorg. Chem., 1992, 31, 2429–2434 CrossRef CAS.
- J. Müller, E. Zangrando, N. Pahlke, E. Freisinger, L. Randaccio and B. Lippert, Chem.–Eur. J., 1998, 4, 397–405 CrossRef CAS.
- S. E. Taylor, E. Buncel and A. R. Norris, J. Inorg. Biochem., 1981, 15, 131–141 CrossRef.
- G. Schröder, B. Lippert, M. Sabat, C. J. L. Lock, R. Faggiani, B. Song and H. Sigel, J. Chem. Soc., Dalton Trans., 1995, 3767–3775 RSC.
- M. S. Lüth, E. Freisinger, F. Glahé and B. Lippert, Inorg. Chem., 1998, 37, 5044–5045 CrossRef.
| This journal is © The Royal Society of Chemistry 2010 |