Cytosolic zinc buffering and muffling: Their role in intracellular zinc homeostasis
Robert A.
Colvin
ab,
William R.
Holmes
ab,
Charles P.
Fontaine
a and
Wolfgang
Maret
c
aDepartment of Biological Sciences, Ohio University, Athens, OH 45701, USA. E-mail: colvin@ohio.edu; Fax: 740-593-0300; Tel: 740-593-0198
bNeuroscience Program, Ohio University, Athens, OH 45701, USA
cKing's College London, School of Biomedical and Health Sciences, Nutritional Sciences Division, London, United Kingdom SE1 9NH
First published on 16th April 2010
Abstract
Our knowledge of the molecular mechanisms of intracellular homeostatic control of zinc ions is now firmly grounded on experimental findings gleaned from the study of zinc proteomes and metallomes, zinc transporters, and insights from the use of computational approaches. A cell's repertoire of zinc homeostatic molecules includes cytosolic zinc-binding proteins, transporters localized to cytoplasmic and organellar membranes, and sensors of cytoplasmic free zinc ions. Under steady state conditions, a primary function of cytosolic zinc-binding proteins is to buffer the relatively large zinc content found in most cells to a cytosolic zinc(II) ion concentration in the picomolar range. Under non-steady state conditions, zinc-binding proteins and transporters act in concert to modulate transient changes in cytosolic zinc ion concentration in a process that is called zinc muffling. For example, if a cell is challenged by an influx of zinc ions, muffling reactions will dampen the resulting rise in cytosolic zinc ion concentration and eventually restore the cytosolic zinc ion concentration to its original value by shuttling zinc ions into subcellular stores or by removing zinc ions from the cell. In addition, muffling reactions provide a potential means to control changes in cytosolic zinc ion concentrations for purposes of cell signalling in what would otherwise be considered a buffered environment not conducive for signalling. Such intracellular zinc ion signals are known to derive from redox modifications of zinc-thiolate coordination environments, release from subcellular zinc stores, and zinc ion influx via channels. Recently, it has been discovered that metallothionein binds its seven zinc ions with different affinities. This property makes metallothionein particularly well positioned to participate in zinc buffering and muffling reactions. In addition, it is well established that metallothionein is a source of zinc ions under conditions of redox signalling. We suggest that the biological functions of transient changes in cytosolic zinc ion concentrations (presumptive zinc signals) complement those of calcium ions in both spatial and temporal dimensions.
 Robert A. Colvin | Bob Colvin received his PhD in cell physiology from Rutgers University. After postdoctoral studies at the University of Connecticut in cardiology, he accepted a position as assistant professor of pharmacology at Oral Roberts University School of Medicine. In 1990, he joined the Biological Sciences Department, Neuroscience Program at Ohio University and was promoted to full professor in 2006. His work has centered on the mechanism and regulation of transporters for both zinc and calcium and cellular ion homeostasis, particularly as it relates to neurodegenerative diseases such as Alzheimer’s disease and stroke. |
 William R. Holmes | William Holmes received his PhD in Biomathematics from UCLA. Currently he is a Professor in the Neuroscience Program in the Department of Biological Sciences at Ohio University where he is also Director of the Quantitative Biology Institute. His research interests include the development of mathematical and computational models at the levels of the individual neuron, the synapse and biochemical reactions within cells, particularly as they relate to mechanisms of learning and memory. Recent work concerns estimation of parameters governing the electrical properties of neurons, calcium dynamics and reaction-diffusion in dendritic spines, and zinc homeostasis in cortical neurons. |
 Charles P. Fontaine | Charles P. Fontaine received a Bachelor of Science degree in Microbiology from Ohio University in 1998. In 2003 he began a position as a research technician under the guidance of Robert Colvin in the Neuroscience Program in the department of Biological Sciences at Ohio University. During the six years he performed research with Dr Colvin he also pursued and was awarded a Master of Science in Environmental Sciences, at Ohio University. Charles has since moved to Diagnostic Hybrids Inc. where he operates as a Research Associate and Laboratory Manager in the R&D department. |
 Wolfgang Maret | Wolfgang Maret received his diploma in chemistry and his PhD in natural sciences from the Saarland University, Saarbruecken, Germany. After postdoctoral studies at the University of Chicago, he accepted a position as assistant professor at Harvard Medical School. Since 2003, he has been an associate professor in the Division of Human Nutrition (Department of Preventive Medicine and Community Health) at the University of Texas Medical Branch in Galveston, TX. His work focuses on molecular mechanisms of cellular metal homeostasis, sulfur redox chemistry, structure and function of metalloenzymes, as well as the functions of micronutrients in chronic and degenerative diseases. |
Introduction: Metals in biology and a description of buffering and muffling reactions for the control of cytosolic zinc ions
It is thought that the availability of metal ions as organisms evolved determined cellular metal levels and thus how metals are now utilized for biological functions. Prokaryotic organisms evolved in a reductive environment when certain transition metal ions became available.1,2 This condition along with the redox chemical activity of many transition metal ions resulted in the vanishingly small free cytosolic concentrations observed in modern prokaryotic organisms.3 Prokaryotic cells evolved metal-binding proteins to maintain these extremely low cytosolic metal ion concentrations and protect the cell against cytotoxic increases in intracellular metal ions. One sees this primordial state mirrored by the relatively low cytosolic concentrations of most metal ions in modern eukaryotic cells. However, eukaryotic organisms evolved in an environment of increasing oxygen availability, a condition that was conducive to increased availability and abundance of free metal ions, such as zinc. Thus, steady state cytosolic free zinc ion concentrations are higher in eukaryotic cells when compared to prokaryotic cells, but are much lower than cytosolic free calcium ion concentrations.3–8 Zinc and calcium have complementary roles in modern eukaryotic cells. Through its binding to thousands of proteins, zinc ions are critical for cell proliferation, differentiation, signalling, and survival.9 Factors that contributed to zinc's prominence among metal ions include its chemical characteristics as a relatively strong Lewis acid in enzymes, and most importantly, the fact that zinc is the only essential transition metal ion that lacks biological redox activity.10 The lack of redox activity along with zinc's relatively strong affinity to proteins makes it well suited as a structural cofactor. An analysis of metalloproteomes that carry evolutionary footprints of metal geochemistry11 demonstrated that the use of zinc increased whereas the use of iron declined.12 Thus, zinc now performs functions that were once the domain of other metal ions.For all modern eukaryotic cells, steady state concentrations of free intracellular zinc ions are maintained within narrow limits by buffering systems. Steady state free zinc ion concentrations must be under especially tight control, because eukaryotic cells generally contain higher levels of total zinc than other transition metals and each cell needs to distribute zinc to a staggering number of proteins for a myriad of functions. Zinc ions cannot have uncontrolled access to metalloproteins, since metal binding to proteins generally follows the Irving-Williams series where zinc is highly preferred over most other metals except copper.13 Such an uncontrolled access to zinc would render many metalloproteins dysfunctional and likely result in cell death. The converse is true also and zinc deficiency leads to cell death as well.14
One of the key features of eukaryotic evolution was the compartmentalization of the cell, and naturally it included compartmentalization of intracellular zinc. Zinc compartmentalization and sequestration evolved in part to allow cellular zinc utilization to increase significantly, while keeping the cytosolic free zinc ion concentration remarkably low to prevent zinc ions from binding to the wrong proteins. Zinc ion-binding and transport proteins probably first evolved for a protective function. As eukaryotic cells compartmentalized, cellular zinc ion homeostasis necessarily became more elaborate and the ability to modulate non-steady state changes in cytosolic free zinc ion concentrations evolved. The modulation of cytosolic free zinc ion concentrations and intracellular distribution became the task for the concerted action of cytosolic binding proteins, transporters, and sequestration in organelles; such activities have been termed muffling reactions.15 Muffling reactions and the resulting control of cytosolic zinc ion transients provide a basis for zinc ions to take on new intracellular functions, such as the regulation of gene expression, enzymatic activity, and signalling.
A satisfying description of intracellular zinc ion buffering and muffling will include the spatial and temporal distributions and concentrations of both free and bound zinc ions in the cytosol and in each cellular compartment, i.e., the zinc metallome, and how zinc ion levels are maintained and change in response to various physiological and/or pathophysiological perturbations. These zinc ion homeostatic processes interact with and are linked to the metabolic and developmental state of the cell as well.16 Thus, the first step to a better understanding of zinc ion homeostasis is an accurate description of the cellular zinc proteome, the cellular concentrations of zinc-binding proteins, and free and total zinc ions in each cellular compartment under steady state conditions. The next steps are to describe the interactions of zinc-binding proteins, zinc transporters, sequestering organelles, and zinc buffering and muffling reactions in both temporal and spatial dimensions, under non-steady state conditions. Solving these problems will be challenging, but important inroads have been made as we will describe.
The cellular zinc metallome: Fundamental considerations
The cellular zinc metallome is defined as the distribution of free and bound zinc ions in every cellular compartment.17 Since transition metals bind to proteins with generally high affinity that follows the Irving-Williams series, changes in protein expression necessarily result in altered metallomes. However, direct experimental evidence of such interactions remains scarce.Quantitative, functional, and structural metallomics approaches are being undertaken to understand the biological significance of metals. Quantitative metallomics endeavors to determine the free and total concentrations of metals in each cellular compartment including the cytosol and various organelles. Functional metallomics attempts to describe how metals affect cellular attributes and activities in cellular time and space and how metal levels are regulated. Structural metallomics considers the different coordination environments of metals in biology. In order to decipher the complex interplay between proteomics and metallomics, computational approaches of systems biology are used. Computational models of cellular metal homeostasis based on experimental data provide the best promise of eventually understanding the complex interactions of metals, proteins, and compartments in a single cell and will allow predictions on how nutrients, drugs, and toxicants interfere with control of cellular zinc homeostasis.
Zinc ions are known to be accumulated in cells because measured total cellular concentrations are much higher than physiological extracellular concentrations.18 We expect the zinc metallome to change throughout the life cycle of a cell reflecting the cell's developmental stage, level of activity and function, as is indeed borne out by recent experiments.19 Thus, phenotypically distinct cells might be expected to have similarly distinct zinc metallomes and may vary considerably in their steady state cytosolic free zinc ion concentrations as well.
The cellular zinc proteome: A large number of zinc-binding proteins with diverse functions
What is so remarkable about zinc biology is the large number of zinc-binding proteins.20 A major change in utilization of zinc during the evolution of eukarya occurred when a large number of proteins adopted zinc as a structural factor and for interactions with other proteins, nucleic acids and lipids.21 Since the discovery of carbonic anhydrase as a zinc protein in 1939,22 estimates of the number of zinc-binding proteins have been constantly revised upwards. Zinc-dependent enzymes were found in all classes of enzymes, i.e., oxidoreductases, transferases, hydrolases, lyases, isomerases, and ligases.23 When zinc was found in the Xenopus laevis transcription factor IIIA (TFIIIA),24,25 new approaches to discovering structural zinc sites became available. It became possible to predict zinc-binding sites based on homology searches of characteristic ligands and their spacing (signature) in sequence databases, accelerating the pace of discovery and allowing the number of known zinc-binding proteins to almost double in a relatively short period of time. These findings showed that structural zinc is important for many critical interactions between biomolecules. In TFIIIA, nine repetitive sequences of cysteine (C) and histidine (H) residues bind zinc and are referred to as zinc fingers. Subsequently, coordination motifs with four cysteine ligands (C4), with three cysteine ligands and one histidine ligand (C3H), and with cysteine, histidine and aspartate ligands were found in a number of proteins that is at least on par with the number of zinc-requiring enzymes.26With the advent of sequencing entire genomes, databases could be mined, and using bioinformatics approaches, predictions were made that at least 10% (about 2800 gene products) of the human genome encodes for zinc-binding proteins.27 The zinc-binding proteins include 397 hydrolases; 302 ligases; 167 transferases; 43 oxidoreductases and 24 lyases/isomerases; 957 transcription factors; 221 signaling proteins; 141 transport/storage proteins; 53 proteins with structural metal sites; 19 proteins involved in DNA repair, replication, and translation; 427 zinc finger proteins of unknown function; and 456 proteins of unknown function. While all of this work identified proteins with enzymatic or structural functions based on homology to known templates, there are an increasing number of signatures that do not fall in either one of these categories.20,28 Thus, the actual number of zinc-binding proteins might be even larger.29 Zinc is used as a structural element between proteins to establish higher order structures and it binds transiently to some proteins, presumably for regulation.10
A limitation of this bioinformatics approach for predicting the number of zinc-binding proteins is that it does not provide information about the location of the proteins or their concentrations. Thus, additional analytical data are needed to determine the relationship between protein-bound zinc and free zinc ions in the cytosol and various cellular compartments. It seems inevitable to conclude that total and free zinc ion concentrations will be different in each cellular compartment. For example, the frequent association of zinc with thiol ligands of cysteines and different thiol redox states of subcellular compartments30 together suggest that total and free zinc ion concentrations in subcellular compartments will differ significantly.
One of the corollaries of such widespread use of zinc in proteins—and of its strong interaction with proteins in general—is that the availability of free zinc ions must be controlled tightly and that zinc ions must be provided to the proteins that require them for their functions at the right time and in the right location. However, given the large number of and structurally different zinc proteins, it is unlikely that specific metallochaperones are involved in recognizing zinc-requiring proteins, like that seen with copper-requiring proteins.31 It has become clear that there are a considerable number of zinc-binding proteins that function in the control of steady state and non-steady state cytosolic free zinc ion concentration. These include zinc sensors, zinc transporters, and cytosolic zinc-binding proteins.
Experimental approaches to estimating steady state cytosolic free zinc ion concentrations
As stated above the primary goal of zinc homeostatic processes is to control the steady state concentration of cytosolic free zinc ions within narrow limits. However, it has been difficult experimentally to determine with certainty what these concentration limits are. Estimates of intracellular free zinc ion concentrations were first made almost 30 years ago and were based on the strong in vitro zinc inhibition of rabbit muscle phosphoglucomutase, a magnesium-dependent enzyme. A ratio of 1 to 10−6 between the Mg2+ and Zn2+ forms of the enzyme in muscle liver extracts was estimated.32 Since the protein is essentially not zinc-inhibited in vivo, one can estimate, using a free magnesium concentration of 0.13 mM in muscle, a free zinc ion concentration of about 100 pM. With a corrected value of 4 × 10−7 for the ratio,33 the calculated value for zinc ions is 32.5 pM. Subsequently, the same investigators employed the response of this enzyme to metal ions as an indicator for the free zinc ion concentration in equine blood plasma.33 They determined a value of 210 pM for free zinc, and noted that with such low zinc ion concentrations, many of the zinc/protein interactions reported to occur at much higher zinc concentrations cannot be physiologically significant, a conclusion that is not widely appreciated due to a lack of understanding the large difference between total intracellular zinc and free zinc ion concentrations. The free zinc ion concentration in blood plasma was also calculated to be 20 pM by considering the affinities and chelating capacities of different biomolecules.34 Based on original estimates of the affinity of metallothionein (MT) for zinc, it was estimated that cytosolic zinc ion concentrations should be below 100 pM.35 The underlying assumption was that a protein that binds zinc ions as strongly as MT would severely limit the availability of free zinc ions.In the last two decades, the synthesis of chelating agents that are coupled to fluorophores that fluoresce strongly when zinc ions are bound made it possible to measure cytosolic free zinc ion concentrations directly by loading these compounds into living cells. In as much as these agents proved to be powerful tools for relative measurements, their application for absolute determinations needs consideration of their physical chemistry as well as their chemical biology in cells. Initially, their application in cellular studies generated more confusion than clarification. It was discovered that most calcium indicators actually have higher affinity for the zinc ion than the calcium ion.36 This finding led to attempts to use such indicators for the measurement of zinc ions. Mag fura-5 is one compound in this category. It was used quite successfully in neuronal cultures to measure large pathological increases in cytosolic free zinc ions. Its zinc ion affinity is about 30 nM whereas its calcium and magnesium affinities are 20 μM and 2.6 mM, respectively.37,38 Although there was the possibility that magnesium and calcium could interfere with zinc ion measurements, control experiments showed that this was not the case. Resting zinc ion concentrations were near the detection limit of Mag fura-5 for zinc ions in cultured neurons, so based on its affinity for the zinc ion, resting cytosolic free zinc ions could be no higher than low nanomolar.
Zinc-selective compounds for live cell imaging were first developed based on 6-methoxy-(8-p-toluenesulfonamido)quinoline (TSQ), such as Zinquin.39,40 While Zinquin has the selectivity and high affinity required for zinc ion measurements, its utility suffers from the requirement for excitation with damaging ultraviolet light. Furthermore, in most cells Zinquin is highly compartmentalized, making it difficult to determine cytosolic free zinc ion concentrations.41 A later generation of compounds—FluoZin-3 and ZnAF-2—are highly selective for zinc ions, have the prerequisite high affinity for zinc ions, have a large dynamic range or response, and are readily loaded into living cells.36,42–45 Studies using these compounds have consistently estimated the cytosolic free zinc ion concentration to be picomolar to no higher than a few nanomolar in various cell types.7,8 Again, such values are in agreement with estimates of <1 nM free cytosolic zinc ions in other cell lines when using a zinc ion chelator and 19F nuclear magnetic resonance.46,47
Recently, biosensors with high zinc ion affinity and selectivity based on carbonic anhydrase have been engineered.48 Employing these cytosolic sensors, cytosolic free zinc ion concentrations in the 5–10 pM range in PC12 (rat pheochromocytoma) cells kept in serum-free medium were measured. Finally, genetically encoded fluorescence resonance energy transfer (FRET) sensors with zinc ion-selective binding can be targeted to specific cellular locations within cells, such as the cytosol, plasma membrane, mitochondrion, and insulin-storing granules.49,50 Such sensors have been used to estimate free zinc ion concentrations in the cytosol and in subcellular organelles. The CALWY sensors developed and used by Merkx and colleagues50 appear particularly promising and have yielded estimates of 0.4 nM free zinc ions in the cytosol of INS-1 (832/11) pancreatic beta-cells.
One of the more important caveats to the introduction of a chelating agent into a cell for measuring cytosolic free zinc ions is the fact that the chelating agent, which may be at a far greater intracellular than the extracellular concentration, will add an additional cytosolic buffer for zinc ions thereby perturbing the steady state cytosolic free zinc ion concentration. Experimental studies have shown that the intracellular concentration of a chelating fluorophore can be quite high and has a demonstrable effect on the estimation of steady state cytosolic free zinc ion concentration.8,49,51 It has been proposed that corrections for this effect can be made by taking measurements at different intracellular chelator concentrations and extrapolating to a zero concentration.8 The extrapolation function depends on the intrinsic buffering properties of the cells, which can vary under physiological conditions. A linear extrapolation may be permissible only under conditions where zinc is relatively well buffered with regard to the concentrations of the fluorophore. Thus, the chelating fluorophore should be kept to a minimum intracellular concentration to obtain the best estimates of cytosolic free zinc ion concentrations. In cultured primary cortical neurons, computational models of zinc homeostasis predicted that an intracellular concentration of 5 μM ZnAF-2F minimally affected estimates of steady state cytosolic free zinc ion concentrations because steady state zinc buffering in cultured neurons was apparently large enough to overcome the perturbing effect of this concentration of intracellular ZnAF-2F.7 Other issues that need to be addressed are whether or not the fluorescent probe localizes exclusively to the cytosol52 (although this is apparently not a problem for the genetically encoded FRET sensors), whether the binding of other metal ions interfere,42,43 or, whether or not the fluorescence enhancement is due to the probe forming ternary complexes with zinc-binding proteins.
To summarize, a variety of approaches unequivocally come to the same conclusion that steady state cytosolic free zinc ion concentrations are only picomolar to single digit nanomolar in eukaryotic cells. Yet, given the strong interactions of zinc with so many intracellular proteins, the steady state cytosolic free concentration and non-steady state fluctuations have significant implications for the physiology and pathophysiology of the cell.53–57 Thus, it is of much more than just academic interest to determine accurately the steady state concentrations of cytosolic free zinc ions and the size and duration of non-steady state changes in cytosolic free zinc ion concentration.
Control of steady state cytosolic free zinc ion concentration—intracellular zinc buffering
Cytosolic free zinc ion concentrations must be controlled within a narrow range that is optimal for supplying enough zinc ions to maintain proper zinc protein functions but avoids passing a higher threshold that would result in unspecific and potentially detrimental interactions with other metalloproteins. In contrast to the low concentrations of cytosolic free zinc ions, total cellular zinc concentrations are in the hundreds of micromolar.7,8,58 This means that cells normally must maintain micromolar concentrations of high affinity zinc-binding sites that function to buffer intracellular zinc and determine the set point for steady state cytosolic free zinc ions. Cells apparently achieve the required level of steady state cytosolic zinc buffering by using resident cytosolic zinc-binding proteins. If such buffering by cytosolic zinc-binding proteins is the primary source of steady state zinc ion control, then the binding constants of the zinc binding-proteins are critical in determining the free zinc ion concentration. This relationship is expressed with the following equation that is analogous to the familiar Henderson-Hasselbalch equation (here pKZn and pZn (−log [Zn2+]) are equivalent to the pKa value and its relationship to pH, and L and ML are the concentrations of free and zinc-bound buffering ligand, respectively).| pZn = pKZn + log (L/ML) |
For most eukaryotic cells, we are just beginning to understand the factors that either sense or regulate the steady state levels of cytosolic free zinc ions.59 The only type of zinc ion sensor so far described in eukaryotic cells is a transcription factor that binds zinc ions in the cytosol and then translocates to the nucleus to direct the expression of proteins that lower the increased zinc ion concentrations to their steady-state levels. One might expect that the affinity of cytosolic zinc ion sensors defines the set point or upper limit for steady state cytosolic free zinc ion levels. For example, the affinity of the ZAP1 zinc sensor in yeast is nanomolar,60 thus one might predict that resting cytosolic free zinc ion levels in yeast should be no higher than low nanomolar. Only one zinc sensor (metal-response element-binding transcription factor-1: MTF-1) has been characterized in multicellular organisms,61 and its set point is also in the low nanomolar range.62 MTF-1 coordinates the expression of two important zinc homeostatic proteins: MT61 and zinc transporter-1 (ZnT1).63
Modulation of non-steady state cytosolic free zinc ion concentration—intracellular zinc muffling
The cellular processes involved in controlling cytosolic free zinc ions include not only steady state levels (see above) but also more complex time-dependent processes modulating non-steady state changes in cytosolic free zinc ions that involve zinc ion transporters and sequestration into cytoplasmic organelles. The term “muffling” has been coined15 to include all of the processes that act to modulate non-steady state changes in cytosolic free ion concentration, including, but not limited to, extrusion from the cell, sequestration in organelles and cytosolic buffering. Importantly, muffling is not a poorly defined phenomenon and can be quantified in physicochemical terms when adding time-dependent fluxes to the thermodynamic concept of buffering.64,65 One key difference between buffering and muffling that has been noted is that ion sequestration and extrusion are slow compared with thermodynamic buffering processes.66 Muffling has been recognized in the study of acid/base balance,67 Ca2+ homeostasis68 and Mg2+ homeostasis,66 but has not been discussed in relation to intracellular zinc ion homeostasis until recently.7 Muffling allows cells to modulate cytosolic free zinc ion transients, even when faced with zinc ion loads or intracellular zinc ion release that far exceed the apparent available zinc-binding sites of cytosolic proteins. In addition, muffling includes binding of zinc ions to macromolecules that transport zinc ions through the cytosol. Thus, intracellular zinc muffling reactions are largely responsible for the characteristic temporal and spatial features of cytosolic zinc ion transients in any given cell.Computational approaches to understanding intracellular zinc buffering and muffling
One approach to better understanding the complex processes that buffer steady state cytosolic free zinc ion concentrations and restore cytosolic zinc ion transients to steady state free zinc ion concentrations is to construct computational models based on experimental data that can be used to test hypotheses about intracellular zinc homeostatic processes. Discrepancies between experimental data and models then help in directing new experimentation. To develop a computational model describing the control of cytosolic free zinc ion concentrations we focused on cultured neurons because of the many functions for zinc ions in the nervous system.69 Models were constructed based on published data and new experimentation7 to match steady state and experimentally induced transients in cytosolic free zinc ion concentrations. The details of model development along with a listing of all relevant experimental constraints are given in our recent publication.7Based on the above discussion of zinc buffering and muffling, it was not surprising that a steady state buffer model based on only cytosolic zinc-binding proteins was unable to match the time-dependent intracellular changes in cytosolic free zinc ion concentrations when zinc influx was increased. A more complex model that included a muffling reaction was required to match the measured time-dependent changes in cytosolic free zinc ion concentrations. The model is illustrated in Fig. 1A and B. The muffling model as presented contains the minimal number of components that could match the experimental data. The model includes a single intracellular store and cytosolic zinc ion-binding proteins capable of shuttling newly arrived cytosolic zinc ions into a cellular storage compartment. A high affinity cytosolic zinc-binding protein is required for the muffling reaction because cytosolic free zinc ion concentrations are maintained at low levels under all but the most extreme pathophysiological conditions. In addition, a high affinity transport mechanism into a store without cytosolic binding proteins would need to have unacceptably fast uptake kinetics to match the experimental data and likely would exist as nearly fully loaded with zinc ions even under steady state conditions. A cadre of high affinity cytosolic zinc-binding proteins, which can be nearly fully occupied with zinc ion under steady state conditions, (as discussed for MT in the muffler mechanism below) capable of transporting newly arrived zinc ions to the store solves this conundrum. The minimal muffling model satisfied all of the experimental constraints and provided good fits to time-dependent changes observed in cytosolic free zinc ion concentrations.7 However, it is appreciated that our minimal muffling model is just that—a simplification of a much more complex intracellular process and—it represents the aggregate properties of many individual processes cooperating in muffling cytosolic free zinc ion concentrations. We still have a limited understanding of these complex processes, but the next few paragraphs present a critical review of what is currently understood.
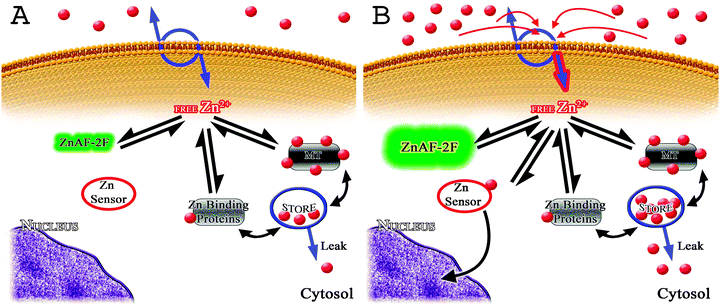 | ||
| Fig. 1 Model schematic of the key elements of intracellular zinc ion homeostasis. The key elements that control the steady state cytosolic free zinc ion concentration and modulate zinc ion transients in cultured rat cortical neurons are illustrated. Red dots represent zinc ions. (A) Steady state cytosolic free zinc ion concentrations are maintained at picomolar to single digit nanomolar concentrations primarily by the buffering action of cytosolic zinc ion-binding proteins that include metallothionein (MT). MT is illustrated with 5 of 7 zinc ion binding sites occupied at steady state. The store is illustrated at steady state with a small amount (relative to its total capacity) of zinc inside and a slow leak. Also illustrated is a zinc ion sensor protein that is unoccupied at steady state but which will bind zinc ions when the cytosolic free concentration increases sufficiently (see B). ZnAF-2F is a fluorophore that allows the experimenter to measure and image both the steady state and spatial and temporal characteristics of zinc ion transients using microfluorometry. ZnAF-2F will increase fluorescence output when bound to zinc ions (see B). (B) Net zinc ion influx is increased as a way to illustrate the functioning of muffling reactions and zinc ion sensing under non-steady state conditions. With an increase in fluorescence ZnAF-2F reports that cytosolic free zinc ion levels are changing as a result of zinc ion influx. Free zinc ions are shuttled to the intracellular store by muffling reactions, which require that zinc ions are first bound to MT or other zinc-binding proteins and then transferred to the store. The store begins to fill with zinc ions. The muffling reactions dampen the zinc ion transient and are responsible for its temporal and spatial characteristics. If cytosolic free zinc ion concentrations reach a sufficiently high level, zinc ions will bind to zinc ion sensors in the cytoplasm. Zinc ion sensors discovered to date are transcription factors that upon binding zinc ions translocate to the nucleus and effect changes in the expression of genes encoding zinc homeostatic proteins. | ||
Cytosolic zinc-binding proteins—focus on metallothionein (MT)
As soluble cytosolic zinc-binding proteins are thought to be key mediators of steady state cytosolic zinc ion buffering and muffling reactions, knowledge of their macroscopic and microscopic binding constants for zinc ions is critical for beginning to understand their functions. It seems that the zinc-binding constants for known intracellular zinc proteins are all within a narrow range.10,70 In fact, we propose that they need to be in such a range in order to keep steady state cytosolic free zinc ions within narrow limits while not disrupting the functions of other metal ions. How narrow is this range? Zinc-binding studies with peptides demonstrate that regardless of the coordination environment provided by the four amino acid ligands of histidine (N) and/or cysteines (S) (N2S2, NS3 or S4), similar binding constants pertain at physiological pH,71 albeit the binding constants are highly pH-dependent. Information about the binding constants for zinc enzymes is limited, but they also seem to be in the single digit picomolar range regardless of the coordination environment.72 From these considerations one would expect cytosolic free zinc ion concentration to be in the picomolar range. Actually, a measured cytosolic free zinc ion concentration in the range of hundreds of picomolar seems to be optimal, because cytosolic free zinc ions should be 1–2 orders of magnitude higher than the affinity of zinc for proteins in order to keep them saturated with zinc.Aside from the zinc ion transporters and the zinc ion sensor MTF-1, metallothioneins are the only proteins for which a participation in controlling zinc ion availability has been discussed. Mice null for the MT-1,2 genes are sensitive to the adverse effects of additional zinc in the diet and they are more susceptible to developing zinc deficiency, demonstrating a role of MTs in cellular zinc metabolism.73 However, the MT-k.o. mice are viable and reproduce. Thus, compensatory mechanisms may exist and/or MTs are only one of several zinc-binding proteins that participate in the control of cytosolic free zinc ion concentrations. There are at least two additional major isoforms, MT-3 and MT-4, in the mouse. These genes have not been ablated in the MT-1,2 k.o. mice. The human metallothionein family has at least a dozen proteins.74 With the exception of human MT-1B, MTs have twenty conserved cysteines, but they differ in their amino acid composition. They are expressed in a tissue-specific and protein-specific way and their expression is controlled by a large number of trans-acting factors. In particular, the expression of MTs, with the exception of MT-3, is controlled by zinc binding to MTF-1.
Changed expression patterns of MTs under many physiological, pathophysiological, and toxicological conditions (studied as altered transcript levels or total expressed protein) do not provide critical information about functional protein structure, such as how many of the twenty cysteines are in the reduced state and whether all or only a fraction of the twenty binding sites are occupied with zinc. The structural model of the protein shows twenty cysteines binding seven divalent metal ions in two metal/thiolate clusters.75,76 The biological significance of this structural model of MT is limited because interactions of the sulfur donors of the cysteines with metals and their redox activity are dynamic and are a basis for a biochemical mechanism of action as a redox-sensitive metal ion donor or acceptor.77 Interpretations of the functions of MTs need to consider the linkage between metal binding and the reactivity of the thiol ligands with a variety of agents, including its thiol/disulfide (redox) buffering capacity.78–80 In addition to changing its concentration as a way of altering its contribution to zinc buffering, oxidizing its thiol ligands or reducing its disulfides also changes its capacity to buffer zinc ions. In this way, redox changes affect steady state cytosolic free zinc ion concentrations.57
Experimental data on zinc binding to human MT-2 were interpreted with reference to the structural model with seven zinc ions binding with strong and virtually the same affinity in the two domains of the protein (Kd = 1.4 × 10−13 M).81 At a first glance, this interpretation seems credible, because, formally, all seven zinc ions are in tetrathiolate coordination environments in the two zinc/thiolate clusters. However, the coordination environments differ with regard to the number of bridging thiolates. The three zinc ions in the Zn3S9 cluster and two of the four zinc ions in the Zn4S11 cluster have two terminal sulfurs and two bridging sulfurs. The remaining two zinc ions in the Zn4S11 cluster have three bridging sulfurs, however. Examination of the zinc-binding properties of human MT-2 with fluorescent chelating agents demonstrated considerable differences in affinities. One zinc ion binds with only nanomolar affinity (log K = 7.7), two zinc ions bind with higher affinity (log K ≈ 10), and four zinc ions bind with picomolar affinity (log K = 11.8).82 Thus, the cluster structure decreases the affinities for some of the zinc ions and provides zinc ion affinities that vary over four orders of magnitude. As a consequence of these properties, metallothionein exists as multiple species: Zn7T, Zn6T, Zn5T, and Zn4T. The distribution of these species depends on the total concentrations of both the protein, i.e., thionein (T) and zinc ions. The low affinity for the seventh zinc ion and cellular zinc ion concentrations in only the picomolar range suggest that Zn7T, the form for which the 3D structure was determined, does not exist under normal physiological conditions. Indeed, the cellular protein binds additional zinc when zinc ions are added to a liver homogenate.72 The conclusion from these studies is that the protein can control zinc ion concentrations over at least three orders of magnitude and well into the nanomolar range, thus in a much wider range than originally thought based on a single average dissociation constant for all seven zinc ions. These properties demonstrate that MT is not a thermodynamic sink for zinc as was implicated by the model based on average tight binding. They also suggest that zinc is not buffered at a single pZn value throughout different cellular states. Changes in the distribution of MT species allow fluctuations of pZn that may be used for controlling biological functions. Further insights into the function of MT in zinc ion buffering and muffling have come from computational studies.
One of the first complexities added to the minimal muffler model was to test specific hypotheses about how cytosolic MT might participate in zinc ion buffering and muffling reactions. Recently obtained data on the binding constants of all seven zinc binding sites on MT-2 provide the necessary data to begin such an analysis.82 However, on and off rate constants for each of the binding sites and data on additional MT proteins, which are not available at present, would allow a much more sophisticated mathematical analysis of MTs' roles and considerably reduce the number of assumptions required to perform the analysis.
When MT was incorporated into the muffler model, using recently obtained data on the binding site affinities for its seven sites,82 and asked to function as the muffler, the model returned interesting insights into MT function. If MT were to be exclusively responsible for sequestration of zinc ions into the store, the required MT concentration would be 2 μM or higher. Remarkably, in these simulations the low affinity zinc binding site, site 7 (Kd of 20 nM), was capable of shuttling zinc ions to the intracellular store at least as effectively as site 6 (Kd of 100 pM) and more effectively than site 5 (Kd of 40 pM), despite having much less zinc ions bound at steady state (illustrated in Fig. 1B). This interpretation was dependent on the assumption that interaction of protein-bound zinc ions with the intracellular store was driven solely by the binding site affinity for zinc ions. This does not have to be the case if additional mechanisms control the transfer. For example, the zinc-binding proteins may release their bound zinc ions preferentially to different components of the intracellular store.
It is important to note that the zinc buffering and muffling functions of MT in the model could be obtained only with different affinities for zinc ions and not with a single global affinity for all seven zinc ions, emphasizing the significance of having discrete affinities for zinc ions for the function of MT. This interpretation contrasts with older notions of cellular MT with a single, average affinity for all seven zinc ions and existing as either fully metal-loaded or entirely metal-free. Thus, the zinc ion binding sites of cellular MT are likely to be partially loaded with zinc ions, as was borne out by cellular studies,83–85 and the lower affinity sites 5–7, in particular, function quite efficiently in a cellular muffling reaction. The observation of MT moving within the cell, e.g., the mitochondrial intermembrane space, further supports the interpretation that MT serves as a muffler. In cultured cortical neurons, MT is likely one component of a cadre of zinc-binding proteins with high affinity for zinc ions and participating in the muffling reaction in response to a zinc ion load. In resting cultured neurons, MT is poised to have a more prominent role in inducible changes in steady state zinc ion buffering. Increases in MT protein expression would force MT to become a more dominant cytosolic zinc ion-binding protein in the muffling reactions as well, which could change the spatial and temporal characteristics of subsequent cytosolic zinc ion transients. In other words, adaptations in intracellular zinc-binding capacity affect the way cells respond to zinc ions in time. Many physical and chemical treatments increase cellular MT levels several-fold.86 Importantly, many induction pathways include physiological signals, suggesting considerable regulation. There is direct experimental evidence of increased steady state cytosolic zinc buffering when MT expression increases in cultured cells.8,87
Zinc transporters and sequestration in organelles
Cellular muffling mechanisms require zinc ions to be sequestered in cellular organelles. There is much experimental evidence for endogenous stores of zinc ions and transport between the cytosol and mitochondria, the nucleus, the Golgi, endosomes/vesicles and lysosomes; and for the uptake and release of zinc ions from these compartments.Zinc transporter families
Two families of zinc transporters, ZnTs (SLC30) and ZIPs (SLC39), have been established based on homology searches in sequence databases using cloned family members and functional expression studies.63,88 Generally, ZnTs are involved in zinc ion efflux from the cytosol whereas ZIPs effect zinc ion influx. Both family members are localized to the plasma membrane and various subcellular organelles. Recent reviews89,90 are available and we direct the reader's attention there for a comprehensive discussion. The following paragraphs are more narrowly focused on specific experimental findings that relate to zinc ion muffling. However, we note that recent 3D structures of a zinc ion export protein show that it likely senses the cytosolic zinc ion concentration and does not function in isolation.91 The exporter is allosterically regulated with highly specialized functions, in which zinc sensing and zinc transport are linked through conformational changes.92Mitochondria
Recent studies93 show that RhodZin-3, a Zn2+-selective fluorophore that accumulates in mitochondria, yields TPEN-sensitive punctate staining in neurons. RhodZin-3 fluorescence colocalized with Mitotracker Green fluorescence confirming the mitochondrial identity of the compartmentalized zinc ions. In addition, when neuronal cytosolic free zinc ion concentrations rise significantly, zinc ions are taken up by mitochondria with derangements of mitochondrial structure and function.93–99 Mitochondrial zinc uptake under physiological conditions was observed previously. When zinc(II) solutions are administered orally to rats, the zinc content of liver mitochondria increases.100 None of the known zinc transporters localizes to the mitochondria. However, when MT is imported into the intramembrane space of mitochondria, it releases zinc ions, which in turn inhibit mitochondrial respiration.101 In addition, zinc ions can enter the matrix of isolated brain mitochondria by uniporter-dependent and -independent mechanisms.99,102 Using FluoZin-3,93 release of endogenous mitochondrial zinc ions to the cytosolic compartment is detected and the released zinc ions are transferred to a cytosolic protein-bound pool that is redox-sensitive and presumably identical with MT. Genetically encoded zinc ion sensors targeted to mitochondria of cultured hippocampal neurons demonstrate endogenous zinc ion stores that increase upon glutamate/zinc exposure and can release zinc ions to the cytosol.49Nucleus
Known zinc transporters are not localized to the nuclear membrane either. When rats received zinc(II) solutions by intraperitoneal injection, the nuclei of rat hepatocytes showed the highest uptake.103 Given the low cytosolic free zinc ion concentrations, diffusion of zinc ions would not supply the nucleus with zinc in a timely manner. Nuclear translocation of MT, which is stimulated by the oxidation of a cytosolic factor, inhibited by depletion of ATP, and is dependent on the small GTPase Ran, could be the main route of zinc traffic between the nucleus and the cytosol.104–107Golgi apparatus
The two different zinc ion transporter gene families thus far identified ZnT (SLC39) and ZIP (SLC30) have members that are localized to the Golgi apparatus. Although most ZIP family transporters are localized to the plasma membrane and are involved in cellular zinc ion import, ZIP7 (SLC39a7) and ZIP9 (SLC39a9)108,109 family members are also localized to the Golgi apparatus and are capable of transporting zinc ions from the lumen of the Golgi to the cytosolic compartment. In living neurons, the zinc-selective fluorophore Zinpyr-3 localizes to a subcellular compartment within living neurons that is suggestive of the Golgi apparatus. Fluorescence was TPEN-sensitive, indicating the presence of endogenous stores of zinc ions.110 Several recent studies demonstrate the localization of members of the ZnT (SLC30a5-7) family of proteins to the secretory pathway, in particular the Golgi apparatus and vesicular compartments, and their importance in zinc ion homeostatic mechanisms.111–114 For example, SLC30a5 and -7 are required for the activation of alkaline phosphatases.115,116 ZnT family members are up-regulated by endoplasmic reticulum stress and may be part of the cell's response to such stress.117 SLC30a5/6 proteins form heteromeric complexes in the secretory pathway,111,118 whereas the SLC30a7 protein appears to be homomeric.112,116Vesicles/endosomes/lysosomes
ZnT3 (SLC30a3), which is abundant only in brain and testis,119 is thought to be responsible for the sequestration of zinc ions into many glutamatergic synaptic vesicles. Using mouse brain, the protein is found in the same regions (hippocampus and cerebral cortex) that show the highest levels of histochemically reactive zinc ions. Electron microscopy shows localization of the SLC30a3 protein to synaptic vesicles that stain positively for zinc ions.120 In addition, sequestration of zinc ions into synaptic vesicles requires SLC30a3 protein in the vesicle membrane, as disruption of this gene in mice eliminated histochemically reactive zinc ions in the synaptic vesicles of these animals.121 SLC30a3 is not the only transporter that influences zinc levels in synaptic vesicles; vglut1 (vesicular glutamate transporter) is also involved.122 Also, there is a role of MT-3 in loading zinc into vesicles, in a pathway that also involves ZnT3,123 and this function appears to involve an interaction of MT-3 and the small GTPase Rab3b.124 Why different ZnT proteins are used for storage of vesicular zinc ions in different cells is not clear.A zinc ion-containing endosomal/vesicular compartment in cultured neurons and many other cell types can be visualized using the zinc-selective fluorophore Zinquin.125–128 Zinc ions contained in vesicles are in a coordination environment that includes sulfur and nitrogen ligands.129 The vesicular/endosomal compartment is distinct from the Golgi apparatus and mitochondrial compartments in cultured neurons and may be involved in cellular trafficking of zinc ions.41 A transporter mechanism has yet to be shown to be associated with this zinc ion-containing compartment, sometimes referred to as zincosomes.129 Zinc ions accumulate in lysosomes, especially under oxidative stress, in cultured neurons.52 The transporter protein ZIP8 (SLC39a8) is localized to the lysosomal membrane in T cells,130 and ZnT2 (SLC30a2) to the lysosomal membrane in fibroblasts.131 Finally, ZnT8 (SLC30a8) is a zinc transporter of pancreatic islets required for zinc insulin crystallization within secretory vesicles, and it affects insulin secretion.132
Zinc and calcium signalling: Complementary roles
First, let us consider the potential complementary nature between zinc and calcium ion signalling. The evolution of calcium and zinc homeostatic mechanisms was driven by the same overarching need, namely to prevent toxic increases in cytosolic free ion concentrations. Eukaryotic cells developed sophisticated mechanisms involving protein binding, transport, and sequestration in order to accomplish this goal for both ions. Consequently, there are many calcium-binding proteins, transporters, and sequestration sites, such that cytosolic calcium ions are buffered and muffled similar to the situation for cytosolic zinc ions. In fact, the buffering for zinc and calcium ions in several cells where it has been measured is on par. The cytosolic zinc ion buffering by cultured neurons is estimated to be 396 μM/pZn unit (at 1–10 nM intracellular range) or about .025 nM change per μM zinc load.7 In comparison, cytosolic calcium ion buffering is generally 10 to 100 fold less, i.e., for a given load of each ion, the cytosolic free calcium ion concentration changes 10 to 100 times more than zinc will.133–136 However, at steady state, cytosolic free calcium ion concentrations are 100 to 1,000 fold higher than those of zinc ions, so that on a molar basis the intracellular buffering for zinc ions is no higher (probably lower) than that for calcium ions. What differs between these two ions is the much greater concentration of free calcium ions outside of cells (millimolar for free calcium ions; nanomolar for free zinc ions137). The large concentration gradient in free calcium ions between the outside and the cytosol (or the gradient in calcium ions between the endoplasmic reticulum (ER) and the cytosol) is critical for mechanisms used to initiate transient cytosolic spikes in calcium ion concentrations. The ER and plasma membrane contain calcium-specific channels that are capable of creating rapid but short lived microdomains of extremely high calcium ions within the cell by temporarily swamping cytosolic buffering mechanisms in such microdomains. In addition, the EF-hand proteins became the dominant calcium-binding proteins. Their conformational change upon calcium binding was critical in making them signal transducers and calcium sensors instead of simply buffers. Finally, active transport mechanisms exist to rapidly reverse cytosolic calcium ion transients.Since extracellular concentrations of free zinc ions are much lower than those of calcium ions, zinc-specific channels would be ineffective as a mechanism of zinc entry; hence zinc-specific channels apparently do not exist (as is the case for other transition metals as well). However, it should be noted that a large body of experimental evidence shows that zinc ion entry through calcium and glutamate-operated channels occurs in cultured neurons when extracellular zinc ion concentrations are micromolar.96,138–143 As high concentrations of extracellular zinc ions are generally restricted to the nervous system, we believe that the primary source of cytosolic zinc ion signals in most cells stems from intracellular release mechanisms. What is the experimental evidence for the existence of such zinc ion signals? There are at least two biological mechanisms for the release of free zinc ions as putative signals. One is the release of zinc ions from cellular vesicles. It can involve the release of zinc ions to the extracellular space, such as in specialized neurons and the actions of zinc on the postsynaptic membrane,69 the release together with insulin from β-cells, where zinc ions may have a paracrine function on α-cells,144 receptor-stimulated release of zinc ions from an internal store in macrophages,145 or release from an intracellular store through the Zip7 transporter.146 A second mechanism is the function of zinc/thiolate coordination environments in proteins as redox transducers of redox signals into zinc “signals” through the particular chemistry of the cysteine ligands of zinc.79 Though both calcium and zinc are redox-inert metal ions, their mechanisms and functions in signalling are entirely different. The preference of calcium for oxygen ligands and the inclusion of nitrogen and sulfur ligands in the preferences of zinc mean that calcium and zinc select different coordination environments in proteins and thus target different proteins. Taken together, zinc and calcium ions signal over a wide range of cytosolic concentrations with different spatial and temporal characteristics and in this way zinc complements the capabilities of calcium.54 Key cellular processes responsible for such differences are the various muffling reactions specific for cytosolic calcium and zinc ions.
Though functional correlates have been described for each case of experimentally induced localized zinc “signals”, the specific molecular targets of zinc ions are not always well defined. Pleiotropic effects of zinc ions on various cellular signalling pathways have been documented.147 However, most of the experiments reported in the literature have not determined the changes in cytosolic free zinc ions associated with zinc's actions. Clearly, without such knowledge and without ascertaining that intracellular buffering and muffling is intact, a great number of mostly non-physiological effects may have been observed and reported owing to the strong tendency for zinc ions to bind to proteins. Thus we still do not know in most cases whether experiments purported to demonstrate that zinc ions affect signalling pathways reflect nonspecific actions at non-physiological concentrations or if genuine targets of zinc signalling have actually been identified. What we do know, however, is that physiological targets of zinc ion signals will need to be in the picomolar to low nanomolar range of concentrations. Global “signals” may also occur, because changes of steady state cytosolic free zinc ion concentrations have been observed in processes such as proliferation, differentiation, and apoptosis.8,80 Thus, conceptually, in the context of what we do know about cytosolic zinc ion buffering and muffling, there are two possible scenarios: first, release of zinc ions from an operated store (protein or vesicular) that functions rapidly followed by a slower re-adjustment to the previous steady state (muffling reactions), and second, longer lasting changes in steady state cytosolic free zinc ion concentration, either an increase or a decrease, as a result of adjustments in the concentrations of cytosolic zinc-binding proteins.
Conclusions
The use of zinc in thousands of proteins requires tight spatiotemporal control. Over three dozen proteins (ZnTs, ZIPs, MTs, MTF-1) combine to control intracellular zinc ion homeostasis and thus the steady state cytosolic and organellar free zinc ion concentrations. Perhaps the most important aspect of homeostatic control is the tight binding of zinc to proteins and the ensuing very low cytosolic free zinc ion concentrations. Buffering and muffling reactions complement each other and make it possible to make sufficient zinc available in the cell without having to resort to very high cytosolic buffer concentrations, the chelating capacities of which would likely interfere with the homeostatic control of other transition metal ions. Muffling reactions allow transients in cytosolic zinc ion concentration and eventual restoration of steady state cytosolic concentrations. The mechanisms employed seem to be akin to those for calcium but unlike those for other transition metal ions. The various cellular zinc-binding proteins are the key players in these mechanisms. In particular, metallothionein, owing to its zinc-binding and transport properties and as a source of zinc ions for signals under conditions of redox signalling, has a critical role in intracellular zinc homeostasis. Investigations of intracellular zinc homeostasis are an example where a holistic “omics” and quantitative approach is necessary to understand the inner workings of a system. Simply putting together a tools list of the molecules involved does not provide any insights into biologically realistic control of intracellular zinc ions.Acknowledgements
The work in RAC's laboratory was supported by Ohio University. The work in WM's laboratory was supported in part by Grant GM 065388 from the National Institutes of Health, the John Sealy Memorial Endowment Fund, and a sponsored research agreement with Neurobiotex Inc, Galveston, TX.References
- R. J. P. Williams, BioMetals, 2007, 20, 107–112 Search PubMed.
- R. J. P. Williams and J. J. R. Frausto da Silva, in The Chemistry of Evolution: Development of our Ecosystem, Elsevier, Amsterdam, 2006 Search PubMed.
- C. E. Outten and T. V. O'Halloran, Science, 2001, 292, 2488–2492 CrossRef CAS.
- E. Marban, T. J. Rink, R. W. Tsien and R. Y. Tsien, Nature, 1980, 286, 845–850 CrossRef CAS.
- E. Marban, M. Kitakaze, H. Kusuoka, J. K. Porterfield, D. T. Yue and V. P. Chacko, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 6005–6009 CrossRef CAS.
- A. Ghosh and M. E. Greenberg, Science, 1995, 268, 239–247 CrossRef CAS.
- R. A. Colvin, A. I. Bush, I. Volitakis, C. P. Fontaine, D. Thomas, K. Kikuchi and W. R. Holmes, Am. J. Physiol. Cell Physiol., 2008, 294, C726–C742 CrossRef CAS.
- A. Krezel and W. Maret, J. Biol. Inorg. Chem., 2006, 11, 1049–1062 CrossRef CAS.
- B. L. Vallee and K. H. Falchuk, Physiol. Rev., 1993, 73, 79–118 CAS.
- W. Maret and Y. Li, Chem. Rev., 2009, 109, 4682–4707 CrossRef CAS.
- C. L. Dupont, S. Yang, B. Palenik and P. E. Bourne, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 17822–17827 CrossRef CAS.
- C. Andreini, L. Banci, I. Bertini and A. Rosato, J. Proteome Res., 2006, 5, 3173–3178 CrossRef CAS.
- I. Irving and R. J. P. Williams, Nature, 1948, 162, 746–747 CrossRef CAS.
- A. Q. Truong-Tran, J. Carter, R. E. Ruffin and P. D. Zalewski, BioMetals, 2001, 14, 315–330 Search PubMed.
- R. C. Thomas, J. A. Coles and J. W. Deitmer, Nature, 1991, 350, 564 CrossRef CAS.
- H. Tamano, F. Kan, M. Kawamura, N. Oku and A. Takeda, Neurochem. Int., 2009, 55, 536–541 CrossRef CAS.
- J. J. R. Frausto da Silva and R. J. P. Williams, in The Biological Chemistry of the Elements: The Inorganic Chemistry of Life, Oxford University Press, Oxford, 2nd edn, 2001 Search PubMed.
- H. Haase and W. Maret, in Cellular and Molecular Biology of Metals, ed. R. Zalups and J. Koropatnick, Taylor and Francis, London, 2010, pp. 179–210 Search PubMed.
- Y. Li and W. Maret, Exp. Cell Res., 2009, 315, 2463–2470 CrossRef CAS.
- W. Maret, J. Anal. At. Spectrom., 2004, 19, 15–19 RSC.
- Y. Shi and J. M. Berg, Science, 1995, 268, 282–284 CrossRef CAS.
- D. Keilin and T. Mann, Nature, 1939, 144, 442–443 CrossRef CAS.
- B. L. Vallee and A. Galdes, Adv. Enzymol. Relat. Areas Mol. Biol., 1984, 56, 283–430 Search PubMed.
- J. S. Hanas, D. J. Hazuda, D. F. Bogenhagen, F. Y. Wu and C. W. Wu, J. Biol. Chem., 1983, 258, 14120–14125 CAS.
- J. Miller, A. D. McLachlan and A. Klug, EMBO J., 1985, 4, 1609–1614 CAS.
- J. M. Berg and Y. Shi, Science, 1996, 271, 1081–1085 CrossRef CAS.
- C. Andreini, L. Banci, I. Bertini and A. Rosato, J. Proteome Res., 2006, 5, 196–201 CrossRef CAS.
- W. Maret, Pure Appl. Chem., 2008, 80, 2679–2687 CrossRef CAS.
- W. Maret, Metallomics, 2010, 2, 117–125 RSC.
- Y. M. Go and D. P. Jones, Biochim. Biophys. Acta., 2008, 1780, 1273–1290 CrossRef CAS.
- D. H. Nies, Science, 2007, 317, 1695–1696 CrossRef CAS.
- E. J. Peck and W. J. Ray, J. Biol. Chem., 1971, 246, 1160–1167.
- G. R. Magneson, J. M. Puvathingal and W. J. Ray Jr, J. Biol. Chem., 1987, 262, 11140–11148 CAS.
- P. M. May and D. R. Williams, Proc. R. Soc. Med., 1977, 70(Suppl 3), 19–23 Search PubMed.
- R. J. Williams, Endeavour, 1984, 8, 65–70 CrossRef CAS.
- K. R. Gee, Z. L. Zhou, D. Ton-That, S. L. Sensi and J. H. Weiss, Cell Calcium, 2002, 31, 245–251 CrossRef CAS.
- L. M. Canzoniero, S. L. Sensi and D. W. Choi, Neurobiol. Dis., 1997, 4, 275–279 CrossRef CAS.
- S. L. Sensi, L. M. T. Canzoniero, S. P. Yu, H. S. Ying, J.-Y. Koh, G. A. Kerchner and D. W. Choi, J. Neurosci., 1997, 17, 9554–9564 CAS.
- P. Coyle, P. D. Zalewski, J. C. Philcox, I. J. Forbes, A. D. Ward, S. F. Lincoln, I. Mahadevan and A. M. Rofe, Biochem. J., 1994, 303(Pt 3), 781–786 CAS.
- M. S. Nasir, C. J. Fahrni, D. A. Suhy, K. J. Kolodsick, C. P. Singer and T. V. O'Halloran, J. Biol. Inorg. Chem., 1999, 4, 775–783 CrossRef CAS.
- R. A. Colvin, M. Laskowski and C. P. Fontaine, Brain Res., 2006, 1085, 1–10 CrossRef CAS.
- J. Zhao, B. A. Bertoglio, K. R. Gee and A. R. Kay, Cell Calcium, 2008, 44, 422–426 CrossRef CAS.
- J. Zhao, B. A. Bertoglio, M. J. Devinney Jr, K. E. Dineley and A. R. Kay, Anal. Biochem., 2009, 384, 34–41 CrossRef CAS.
- R. B. Thompson, D. Peterson, W. Mahoney, M. Cramer, B. P. Maliwal, S. W. Suh, C. Frederickson, C. Fierke and P. Herman, J. Neurosci. Methods, 2002, 118, 63–75 CrossRef CAS.
- T. Hirano, K. Kikuchi, Y. Urano and T. Nagano, J. Am. Chem. Soc., 2002, 124, 6555–6562 CrossRef CAS.
- F. Adebodun and J. F. Post, J. Cell. Physiol., 1995, 163, 80–86 CrossRef CAS.
- J. Benters, U. Flogel, T. Schafer, D. Leibfritz, S. Hechtenberg and D. Beyersmann, Biochem. J., 1997, 322, 793–799 CAS.
- R. A. Bozym, R. B. Thompson, A. K. Stoddard and C. A. Fierke, ACS Chem. Biol., 2006, 1, 103–111 CrossRef CAS.
- P. J. Dittmer, J. G. Miranda, J. A. Gorski and A. E. Palmer, J. Biol. Chem., 2009, 284, 16289–16297 CrossRef CAS.
- J. L. Vinkenborg, T. J. Nicolson, E. A. Bellomo, M. S. Koay, G. A. Rutter and M. Merkx, Nat. Methods, 2009, 6, 737–740 CrossRef CAS.
- K. E. Dineley, L. M. Malaiyandi and I. J. Reynolds, Mol. Pharmacol., 2002, 62, 618–627 CrossRef CAS.
- J. J. Hwang, S. J. Lee, T. Y. Kim, J. H. Cho and J. Y. Koh, J. Neurosci., 2008, 28, 3114–3122 CrossRef CAS.
- W. Maret, C. Jacob, B. L. Vallee and E. H. Fischer, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 1936–1940 CrossRef CAS.
- W. Maret, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 12325–12327 CrossRef CAS.
- H. Haase and W. Maret, Exp. Cell Res., 2003, 291, 289–298 CrossRef CAS.
- H. Haase and W. Maret, J. Trace Elem. Med. Biol., 2005, 19, 37–42 CrossRef CAS.
- W. Maret, BioMetals, 2009, 22, 149–157 Search PubMed.
- R. D. Palmiter and S. D. Findley, EMBO J., 1995, 14, 639–649 CAS.
- K. J. Waldron, J. C. Rutherford, D. Ford and N. J. Robinson, Nature, 2009, 460, 823–830 CrossRef CAS.
- W. Qiao, M. Mooney, A. J. Bird, D. R. Winge and D. J. Eide, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 8674–8679 CrossRef CAS.
- G. K. Andrews, BioMetals, 2001, 14, 223–237 Search PubMed.
- J. H. Laity and G. K. Andrews, Arch. Biochem. Biophys., 2007, 463, 201–210 CrossRef CAS.
- R. D. Palmiter and L. Huang, Pfluegers Arch., 2004, 447, 744–751 CrossRef CAS.
- B. M. Schmitt, Theor. Biol. Med. Model., 2005, 2, 9 Search PubMed.
- B. M. Schmitt, Theor. Biol. Med. Model., 2005, 2, 8 Search PubMed.
- D. Gunzel, F. Zimmermann, S. Durry and W. R. Schlue, Biophys. J., 2001, 80, 1298–1310 CrossRef CAS.
- B. K. Siesjo and S. C. Sorensen, in Ion Homeostasis of the Brain, ed. B. K. Siesjo and S. C. Sorensen, Academic Press, New York, vol. 71, pp. 457–464 Search PubMed.
- C. J. Schwiening and R. C. Thomas, J. Physiol., 1996, 491(Pt 3), 621–633 CAS.
- C. J. Frederickson, J.-Y. Koh and A. I. Bush, Nat. Rev. Neurosci., 2005, 6, 449–462 CrossRef CAS.
- W. Maret, Biochemistry, 2004, 43, 3301–3309 CrossRef CAS.
- A. R. Reddi and B. R. Gibney, Biochemistry, 2007, 46, 3745–3758 CrossRef CAS.
- A. Krezel and W. Maret, J. Biol. Inorg. Chem., 2008, 13, 401–409 CrossRef CAS.
- E. J. Kelly, C. J. Quaife, G. J. Froelick and R. D. Palmiter, J. Nutr., 1996, 126, 1782–1790 CAS.
- Y. Li and W. Maret, J. Anal. At. Spectrom., 2008, 23, 1055–1062 RSC.
- A. Arseniev, P. Schultze, E. Worgotter, W. Braun, G. Wagner, M. Vasak, J. H. Kagi and K. Wuthrich, J. Mol. Biol., 1988, 201, 637–657 CAS.
- A. H. Robbins, D. E. McRee, M. Williamson, S. A. Collett, N. H. Xuong, W. F. Furey, B. C. Wang and C. D. Stout, J. Mol. Biol., 1991, 221, 1269–1293 CAS.
- W. Maret, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2009, 877, 3378–3383 CrossRef CAS.
- W. Maret and B. L. Vallee, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 3478–3482 CrossRef CAS.
- W. Maret, Antioxid. Redox Signal., 2006, 8, 1419–1441 CrossRef CAS.
- W. Maret and A. Krezel, Mol. Med., 2007, 13, 371–375 CAS.
- J. H. R. Kägi, in Metallothionein III, ed. K. T. Suzuki, N. Imura and M. Kimura, Birkhäuser, Basel, 1993, pp. 29–56 Search PubMed.
- A. Krezel and W. Maret, J. Am. Chem. Soc., 2007, 129, 10911–10921 CrossRef CAS.
- A. Pattanaik, C. F. Shaw 3rd, D. H. Petering, J. Garvey and A. J. Kraker, J. Inorg. Biochem., 1994, 54, 91–105 CrossRef CAS.
- Y. Yang, W. Maret and B. L. Vallee, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 5556–5559 CrossRef CAS.
- A. Krezel and W. Maret, Biochem. J., 2007, 402, 551–558 CrossRef CAS.
- A. K. West, J. Hidalgo, D. Eddins, E. D. Levin and M. Aschner, NeuroToxicology, 2008, 29, 489–503 CrossRef CAS.
- L. M. Malaiyandi, K. E. Dineley and I. J. Reynolds, Glia, 2004, 45, 346–353 CrossRef.
- D. J. Eide, Pfluegers Arch., 2004, 447, 796–800 CrossRef CAS.
- S. L. Sensi, P. Paoletti, A. I. Bush and I. Sekler, Nat. Rev. Neurosci., 2009, 10, 780–791 CrossRef CAS.
- L. A. Lichten and R. J. Cousins, Annu. Rev. Nutr., 2009, 29, 153–176 CrossRef.
- M. Lu and D. Fu, Science, 2007, 317, 1746–1748 CrossRef CAS.
- M. Lu, J. Chai and D. Fu, Nat. Struct. Mol. Biol., 2009, 16, 1063–1067 CrossRef CAS.
- S. L. Sensi, D. Ton-That, P. G. Sullivan, E. A. Jonas, K. R. Gee, L. K. Kaczmarek and J. H. Weiss, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 6157–6162 CrossRef CAS.
- J. Wudarczyk, G. Debska and E. Lenartowicz, Arch. Biochem. Biophys., 1999, 363, 1–8 CrossRef CAS.
- S. L. Sensi, H. Z. Yin, S. G. Carriedo, S. S. Rao and J. H. Weiss, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 2414–2419 CrossRef CAS.
- S. L. Sensi, H. Z. Yin and J. H. Weiss, Eur. J. Neurosci., 2000, 12, 3813–3818 CrossRef CAS.
- D. Jiang, P. G. Sullivan, S. L. Sensi, O. Steward and J. H. Weiss, J. Biol. Chem., 2001, 276, 47524–47529 CrossRef CAS.
- K. E. Dineley, T. V. Votyakova and I. J. Reynolds, J. Neurochem., 2003, 85, 563–570 CAS.
- I. G. Gazaryan, I. P. Krasinskaya, B. S. Kristal and A. M. Brown, J. Biol. Chem., 2007, 282, 24373–24380 CrossRef CAS.
- M. Yamaguchi, M. Kura and S. Okada, Chem. Pharm. Bull (Tokyo), 1981, 29, 2370–2374 CAS.
- B. Ye, W. Maret and B. L. Vallee, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 2317–2322 CrossRef CAS.
- L. M. Malaiyandi, O. Vergun, K. E. Dineley and I. J. Reynolds, J. Neurochem., 2005, 93, 1242–1250 CrossRef CAS.
- U. Weser and E. Bischoff, Eur. J. Biochem., 1970, 12, 571–575 CrossRef CAS.
- A. Krezel, Q. Hao and W. Maret, Arch. Biochem. Biophys., 2007, 463, 188–200 CrossRef CAS.
- D. U. Spahl, D. Berendji-Grun, C. V. Suschek, V. Kolb-Bachofen and K. D. Kroncke, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 13952–13957 CrossRef CAS.
- T. Nagano, N. Itoh, C. Ebisutani, T. Takatani, T. Miyoshi, T. Nakanishi and K. Tanaka, J. Cell. Physiol., 2000, 185, 440–446 CrossRef CAS.
- Y. Takahashi, Y. Ogra and K. T. Suzuki, J. Cell. Physiol., 2005, 202, 563–569 CrossRef CAS.
- L. Huang, C. P. Kirschke, Y. Zhang and Y. Y. Yu, J. Biol. Chem., 2005, 280, 15456–15463 CrossRef CAS.
- W. Matsuura, T. Yamazaki, Y. Yamaguchi-Iwai, S. Masuda, M. Nagao, G. K. Andrews and T. Kambe, Biosci. Biotechnol. Biochem., 2009, 73, 1142–1148 CrossRef CAS.
- C. J. Chang, E. M. Nolan, J. Jaworski, S. C. Burdette, M. Sheng and S. J. Lippard, Chem. Biol., 2004, 11, 203–210 CrossRef CAS.
- L. Huang, C. P. Kirschke and J. Gitschier, J. Biol. Chem., 2002, 277, 26389–26395 CrossRef CAS.
- C. P. Kirschke and L. Huang, J. Biol. Chem., 2003, 278, 4096–4102 CrossRef CAS.
- E. Ohana, E. Hoch, C. Keasar, T. Kambe, O. Yifrach, M. Hershfinkel and I. Sekler, J. Biol. Chem., 2009, 284, 17677–17686 CrossRef CAS.
- Z. H. Chi, X. Wang, Z. Y. Wang, H. L. Gao, A. Dahlstrom and L. Huang, NeuroReport, 2006, 17, 1807–1811 CrossRef CAS.
- T. Suzuki, K. Ishihara, H. Migaki, K. Ishihara, M. Nagao, Y. Yamaguchi-Iwai and T. Kambe, J. Biol. Chem., 2005, 280, 30956–30962 CrossRef CAS.
- T. Suzuki, K. Ishihara, H. Migaki, W. Matsuura, A. Kohda, K. Okumura, M. Nagao, Y. Yamaguchi-Iwai and T. Kambe, J. Biol. Chem., 2005, 280, 637–643 CAS.
- C. D. Ellis, F. Wang, C. W. MacDiarmid, S. Clark, T. Lyons and D. J. Eide, J. Cell Biol., 2004, 166, 325–335 CrossRef CAS.
- C. D. Ellis, C. W. MacDiarmid and D. J. Eide, J. Biol. Chem., 2005, 280, 28811–28818 CrossRef CAS.
- R. D. Palmiter, T. B. Cole, C. J. Quaife and S. D. Findley, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 14934–14939 CrossRef CAS.
- H. J. Wenzel, T. B. Cole, D. E. Born, P. A. Schwartzkroin and R. D. Palmiter, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 12676–12681 CrossRef CAS.
- T. B. Cole, H. J. Wenzel, K. E. Kafer, P. A. Schwartzkroin and R. D. Palmiter, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 1716–1721 CrossRef CAS.
- G. Salazar, B. Craige, R. Love, D. Kalman and V. Faundez, J. Cell Sci., 2005, 118, 1911–1921 CrossRef CAS.
- T. B. Cole, C. A. Robbins, H. J. Wenzel, P. A. Schwartzkroin and R. D. Palmiter, Epilepsy Res., 2000, 39, 153–169 CrossRef CAS.
- M. Knipp, G. Meloni, B. Roschitzki and M. Vasak, Biochemistry, 2005, 44, 3159–3165 CrossRef CAS.
- H. Haase and D. Beyersmann, BioMetals, 1999, 12, 247–254 Search PubMed.
- R. A. Colvin, Am. J. Physiol. Cell Physiol., 2002, 282, C317–C329 CAS.
- G. Ranaldi, G. Perozzi, A. Truong-Tran, P. Zalewski and C. Murgia, Am. J. Physiol. Renal Physiol., 2002, 283, F1365–F1375 CAS.
- A. Q. Truong-Tran, R. E. Ruffin and P. D. Zalewski, Am. J. Physiol. Lung Cell Mol. Physiol., 2000, 279, L1172–L1183 CAS.
- G. Wellenreuther, M. Cianci, R. Tucoulou, W. Meyer-Klaucke and H. Haase, Biochem. Biophys. Res. Commun., 2009, 380, 198–203 CrossRef CAS.
- T. B. Aydemir, J. P. Liuzzi, S. McClellan and R. J. Cousins, J. Leukocyte Biol., 2009, 86, 337–348 Search PubMed.
- J. M. Falcon-Perez and E. C. Dell'Angelica, Exp. Cell Res., 2007, 313, 1473–1483 CrossRef CAS.
- F. Chimienti, S. Devergnas, F. Pattou, F. Schuit, R. Garcia-Cuenca, B. Vandewalle, J. Kerr-Conte, L. Van Lommel, D. Grunwald, A. Favier and M. Seve, J. Cell Sci., 2006, 119, 4199–4206 CrossRef CAS.
- F. J. Brinley Jr., T. Tiffert, A. Scarpa and L. J. Mullins, J. Gen. Physiol., 1977, 70, 355–384 CrossRef CAS.
- J. R. Berlin, J. W. Bassani and D. M. Bers, Biophys. J., 1994, 67, 1775–1787 CrossRef CAS.
- H. Mogami, J. Gardner, O. V. Gerasimenko, P. Camello, O. H. Petersen and A. V. Tepikin, J. Physiol., 1999, 518(2), 463–467 CAS.
- A. Fleet, G. Ellis-Davies and S. Bolsover, Biochem. Biophys. Res. Commun., 1998, 250, 786–790 CrossRef CAS.
- C. J. Frederickson, L. J. Giblin, A. Krezel, D. J. McAdoo, R. N. Muelle, Y. Zeng, R. V. Balaji, R. Masalha, R. B. Thompson, C. A. Fierke, J. M. Sarvey, M. de Valdenebro, D. S. Prough and M. H. Zornow, Exp. Neurol., 2006, 198, 285–293 CrossRef CAS.
- Y. Jia, J. M. Jeng, S. L. Sensi and J. H. Weiss, J. Physiol., 2002, 543, 35–48 CAS.
- H. Z. Yin, S. L. Sensi, F. Ogoshi and J. H. Weiss, J. Neurosci., 2002, 22, 1273–1279 CAS.
- C. T. Sheline, H. S. Ying, C. S. Ling, L. M. T. Canzoniero and D. W. Choi, Neurobiol. Dis., 2002, 10, 41–53 CrossRef CAS.
- A. H. Kim, C. T. Sheline, M. Tian, T. Higashi, R. J. McMahon, R. J. Cousins and D. W. Choi, Brain Res., 2000, 886, 99–107 CrossRef CAS.
- G. Kerchner, L. Canzoniero, S. Yu, C. Ling and D. Choi, J. Physiol., 2000, 528, 39–52 CAS.
- S. L. Sensi, H. Z. Yin and J. H. Weiss, NeuroReport, 1999, 10, 1723–1727 CAS.
- H. Ishihara, P. Maechler, A. Gjinovci, P. L. Herrera and C. B. Wollheim, Nat. Cell Biol., 2003, 5, 330–335 CrossRef CAS.
- S. Yamasaki, K. Sakata-Sogawa, A. Hasegawa, T. Suzuki, K. Kabu, E. Sato, T. Kurosaki, S. Yamashita, M. Tokunaga, K. Nishida and T. Hirano, J. Cell Biol., 2007, 177, 637–645 CrossRef CAS.
- C. Hogstrand, P. Kille, R. I. Nicholson and K. M. Taylor, Trends Mol. Med., 2009, 15, 101–111 CrossRef CAS.
- D. Beyersmann and H. Haase, BioMetals, 2001, 14, 331–341 Search PubMed.
| This journal is © The Royal Society of Chemistry 2010 |