Metalloproteomics and metal toxicology of α-synuclein
Aaron Santner
a and
Vladimir N. Uversky
*bc
aMolecular Kinetics, Inc., 6201 La Pas Trail, Suite 160, Indianapolis, Indiana 46268, USA
bInstitute for Intrinsically Disordered Protein Research, Center for Computational Biology and Bioinformatics, Department of Biochemistry and Molecular Biology, Indiana University School of Medicine, 410 W. 10th Street, HS 5009, Indianapolis, Indiana 46202, USA. E-mail: vuversky@iupui.edu; Fax: +1 317-278-9217; Tel: +1 317-27806448
cInstitute for Biological Instrumentation, Russian Academy of Sciences, 142290 Pushchino, Moscow Region, Russia
First published on 13th May 2010
Abstract
Significant evidence has been accumulated linking exposure to heavy metals and/or distortion of metal homeostasis with the development of various neurodegenerative diseases. α-Synuclein is known to be involved in pathogenesis of a subset of neurodegenerative diseases collectively known as synucleinopathies. Therefore the interplay between metals, α-synuclein and neurodegeneration has attracted significant attention of researchers. This review discusses some of the aspects of the α-synuclein metalloproteomics and represents the peculiarities and consequences of α-synuclein interaction with various metal ions. Both non-specific and specific binding of this protein to metals is considered together with the analysis of the effects of such interactions on α-synuclein structure and aggregation propensity.
 Aaron Santner | Dr Santner earned MS and PhD degrees in Biology from Purdue University, Indianapolis in 2001 and 2006, respectively. Following graduate school, he was a postdoctoral fellow in the Department of Biology at Indiana University, Bloomington. His postdoctoral research utilized techniques from molecular biology, biochemistry and molecular genetics to address the roles of regulated protein turnover in plant hormone signaling cascades. Currently, Dr Santner is using bioinformatics methods to understand how disordered regions of proteins mediate protein–protein interactions. Furthermore, he is developing laboratory tools to reduce expressed protein aggregation that are based on intrinsically disordered protein properties. |
 Vladimir N. Uversky | Dr Uversky received broad training, with an MS in Physics, a PhD and a DSc in Biophysics, and with pre- and postdoctoral research in Structural Biology, Biochemistry and Biophysics. He is using various biophysical methods to study protein folding, misfolding, and non-folding, and to characterize intrinsically disordered proteins. Currently, Dr Uversky is also focused on the development and use of bioinformatics methods for the study of intrinsically disordered proteins. Dr Uversky has authored over 330 scientific publications and edited several books and book series on protein structure, function, folding and misfolding. |
Introduction
α-Synuclein is a 140-amino acid protein, which is encoded by a single gene consisting of seven exons located in chromosome 4.1 This protein was first described by Maroteaux et al. in 1988 as a neuron-specific protein localized in the nucleus and presynaptic nerve terminals.2 It is located in close vicinity to, and loosely associated with, presynaptic vesicles and thus may play a role in synaptic release of neurotransmitters.3,4 α-Synuclein has been estimated to account for as much as 1% of the total protein in soluble cytosolic brain fractions.4 Various observations have implicated α-synuclein in the pathogenesis of several neurodegenerative disorders, collectively known as synucleinopathies.5–7 There are several important observations linking α-synuclein and various neuropathologies.8 Some of these observations are:(a) Missense mutations in the α-synuclein gene cause familial Parkinson's disease (PD);
(b) In addition to PD, antibodies to α-synuclein detect Lewy bodies (LBs) and dystrophic Lewy neurites (LNs), the hallmark lesions of PD, in human diseases such as dementia with LBs, Lewy body variant of Alzheimer's Disease (LBVAD), familial and sporadic Alzheimer's Disease (AD), and in elderly Down's syndrome brains;
(c) α-Synuclein is a building block of glial cytoplasmic inclusions (GCIs) in neurodegeneration with brain iron accumulation type 1, and multiple system atrophy (MSA);
(d) Cortical LBs detected with antibodies to α-synuclein correlate with dementia in PD, DLB and LBVAD;
(e) Insoluble α-synuclein filaments are recovered from DLB brains and purified LBs;
(f) Recombinant α-synuclein assembles into LB-like filaments;
(g) α-Synuclein transgenic mice and flies develop a PD-like phenotype;
(h) Cells transfected with α-synuclein treated with nitric oxide generators develop LB-like α-synuclein inclusions;
(i) Biogenic mice over-expressing mutant human amyloid precursor protein and α-synuclein show an augmentation in α-synuclein inclusions.
Overall, many neurodegenerative diseases are characterized by α-synuclein pathologies, where deposition of α-synuclein lesions may be caused by structural alterations in this typical intrinsically disordered protein (IDP), which normally possesses little or no ordered structure under the physiological conditions in vitro.6,7,9–13
As a link between α-synuclein aggregation and PD is one of the most frequently mentioned in literature, the majority of this review will be dedicated to this connection. Clinically, PD is manifested as a movement disorder characterized by tremor, rigidity and bradykinesia. These symptoms are attributed to the progressive loss of dopaminergic neurons from the substantia nigra region of brain. Some surviving nigral dopaminergic neurons contain cytosolic filamentous inclusions known as LBs and LNs. The etiology of PD is unknown, but recent work has shown that, except in extremely rare cases, there is no direct genetic basis for this disease.14 A positive correlation between the prevalence of PD and industrialization has been recognized15 and this disorder is now considered likely to be an “environmental” disease.
The possible involvement of heavy metals in the etiology of PD follows from the results of epidemiological studies16–21 and from the postmortem analysis of the brain tissues of PD patients.22–24 For example, the analysis of the PD mortality rates in Michigan (1986–1988) with respect to potential heavy metal exposure revealed that counties with an industry in the paper, chemical, iron, or copper related-industrial categories had significantly higher PD death rates than counties without these industries.20 An epidemiological study (1987–1989) of Valleyfield, in southern Quebec (Canada) established that an increased risk for PD is associated with occupational exposure to the three metals manganese, iron and aluminium, especially when the duration of exposure is longer than 30 years.19 A population-based case-control study in the Henry Ford Health System (Detroit) suggested that chronic occupational exposure to manganese or copper, individually, or to dual combinations of lead, iron and copper, is associated with PD.16,21 Postmortem analysis of brain tissues from patients with PD gives further confirmation for the involvement of heavy metals in this disorder, in that a considerable increase in total iron, zinc and aluminium content of the parkinsonian substantia nigra was observed when compared to control tissues.22–25 Moreover, analysis of Lewy bodies in the parkinsonian substantia nigra revealed high levels of iron and the presence of aluminium.22,24
Recently, a detailed analysis of the long-term effects of metal ion exposure on α-synuclein aggregation in fish central nervous system tissues was reported.26 To this end, white sucker fish (Catostomus commersoni; aged 5–8 years) were sampled from two sites within the Red Lake area of Northwestern Ontario, a region highly contaminated by metal ions due to mining activity and a site with low metal contamination. The analysis revealed that the central nervous system tissues of fish exposed to elevated metal ion environments had increased α-synuclein immunoreactive aggregates, reflecting that metal ion exposure can lead to α-synuclein deposition, and clearly suggesting that fish can be used as a new model for the analysis of the effect of environmental factors (such as metal exposure) on α-synuclein aggregation.26
These connections of the PD (and other synucleinopathies) pathology with α-synuclein aggregation and exposure to metal have recently attracted significant attention of researchers. More than 1200 papers have been published to analyze association of metals with PD pathogenesis, and more than 100 papers report on various aspects of metal involvement in the modulation of α-synuclein behavior. Several possible mechanisms for metal-stimulated fibrillation of α-synuclein can be envisaged. The simplest would involve direct interactions between α-synuclein and the metal, leading to structural changes in α-synuclein, and resulting in the enhanced propensity to aggregate. These direct interactions of α-synuclein with metals can be specific or have a non-specific character (e.g., non-specific electrostatic interactions). More complex models involve oxidative stress, which is considered to be one of the possible reasons for the neuronal degeneration in PD, resulting from the enhanced level of redox-active metal ions (copper or iron) within the substantia nigra.27–30 In fact, metal-induced oxidant stress can damage critical biological molecules and initiate a cascade of events including mitochondrial dysfunction, excitotoxicity, and a rise in cytosolic free calcium, leading to cell death. It is known that oxidative stress is manifested by increased metal accumulation, which promotes free radical formation, decreased antioxidant levels, which leads to decreased protection against free radical formation, and oxidative damage—factors characteristic of the affected brain regions in PD.27
This paper reviews the current knowledge in the fields of α-synuclein metalloproteomics and metal toxicology. It is started with the analysis of the consequences of the non-specific interactions of metals with this protein on α-synuclein structure and aggregation propensity. Subsequent sections are dedicated to the various aspects related to the interaction of individual metal ions with α-synuclein.
(Non)-specific interactions of α-synuclein with metal ions
There is increasing evidence that altered metal homeostasis may be involved in the progression of neurodegenerative diseases. The potential link between metal exposure, α-synuclein aggregation and PD pathogenesis was proven by in vitro experiments, which showed that α-synuclein aggregation is facilitated in the presence of Cu2+,31 and that Al3+ may induce structural perturbations in this protein.32 These studies will be considered in more details in the corresponding sections of this review.A systematic analysis revealed that a number of mono-, di-, and trivalent metal ions such as Li+, K+, Na+, Cs+, Ca2+, Co2+, Cd2+, Cu2+, Fe2+, Mg2+, Mn2+, Zn2+, Co3+, Al3+ and Fe3+ can cause significant acceleration of the α-synuclein fibril formation in vitro.33 Among 15 cations analyzed, Al3+ was shown to be the most effective stimulator of protein partial folding and subsequent fibrillation, along with Cu2+, Fe2+, Co3+, and Mn2+.33 Earlier studies revealed that a formation of a partially folded intermediate represented a critical early stage in the fibrillation pathway of the natively unfolded α-synuclein. This highly amyloidogenic partially folded intermediate with properties of the pre-molten globule was shown to be stabilized in α-synuclein by low pH or high temperature.10Fig. 1 shows that in the presence of various metal cations natively unfolded α-synuclein adopted a partially folded conformation even at neutral pH. This partially folded conformation was characterized by an increased amount of ordered secondary structure (Fig. 1A), changed local environment of tyrosine residues (Fig. 1B), increased protection of tyrosine residues against acrylamide quenching (Fig. 1C), and by the appearance of solvent-accessible hydrophobic clusters as detected by the increased affinity of the protein to hydrophobic fluorescent probe ANS (Fig. 1D).33 Interestingly, the efficiency of a given cation to induce structural changes in α-synuclein was correlated with the cation's charge density (defined as a ratio of charge to ionic volume). Consequently, small polyvalent cations were more effective in inducing the structural changes than the large monovalent cations (see Fig. 2).33 The induction of partial folding in the intrinsically disordered α-synuclein at neutral pH by cations was explained as follows. The “natively unfolded” status of α-synuclein is mainly determined by strong electrostatic repulsion between the net negative charges. To some extent this resembles the situation occurring for many proteins at low or high pH. Unfolded proteins under conditions of extreme pHs can be transformed into more compact, structured conformations when the net electrostatic repulsion is reduced by binding of counter-ions.34–38 Thus, the metal ion-stimulated α-synuclein conformational change reflects the effective neutralization of the Coulombic charge-charge repulsion. For polyvalent cations, an additional important factor, namely the potential for cross-linking or bridging between two or more carboxylates, has been proposed.33
 | ||
| Fig. 1 Metal ion-induced conformational changes in α-synuclein. (A) Far-UV circular dichroism spectra of 35 μM α-synuclein measured in the absence or presence of 2 mM of the indicated metals. (B) Comparison of the effect of metal ions on the intrinsic α-synuclein fluorescence. Titration curves measured for Al3+ (squares), Zn2+ (diamonds), Mn2+ (inverse triangles), Fe3+ (circles), Fe2+ (triangles), Cu2+ (hexagons), Co3+ (dotted circles) and Co2+ (dotted squares). (C) Stern–Volmer plots for α-synuclein fluorescence quenching by Cu2+ (circles), Fe3+ (inverse triangles) and Fe2+ (squares). (D) ANS fluorescence spectra measured for free dye (solid line) and in the presence of 7 μM α-synuclein and 10–50 mM of the chloride salts of mono-, di- and trivalent cations. Modified from ref. 33. This research was originally published in J. Biol. Chem.: V. N. Uversky, J. Li and A. L. Fink, Metal-triggered structural transformations, aggregation and fibril formation of human α-synuclein. A possible molecular link between Parkinson's disease and heavy metal exposure. J. Biol. Chem., 2001, 276, 44284–44296. © the American Society for Biochemistry and Molecular Biology. | ||
![The dependence of the folding efficiency of the metals on their ionic charge density (A) or ionic radius (B). The parameters used are: half-transition values for the changes induced in the intrinsic fluorescence, [Ion]1/2 (circles); position of maximal ANS fluorescence (squares). Gray symbols correspond to the monovalent cations, whereas open symbols represent di- and trivalent cations. Reproduced from ref. 33. This research was originally published in J. Biol. Chem.: V. N. Uversky, J. Li and A. L. Fink, Metal-triggered structural transformations, aggregation and fibril formation of human α-synuclein. A possible molecular link between Parkinson's disease and heavy metal exposure, J. Biol. Chem., 2001, 276, 44284–44296. © the American Society for Biochemistry and Molecular Biology.](/image/article/2010/MT/b926659c/b926659c-f2.gif) | ||
| Fig. 2 The dependence of the folding efficiency of the metals on their ionic charge density (A) or ionic radius (B). The parameters used are: half-transition values for the changes induced in the intrinsic fluorescence, [Ion]1/2 (circles); position of maximal ANS fluorescence (squares). Gray symbols correspond to the monovalent cations, whereas open symbols represent di- and trivalent cations. Reproduced from ref. 33. This research was originally published in J. Biol. Chem.: V. N. Uversky, J. Li and A. L. Fink, Metal-triggered structural transformations, aggregation and fibril formation of human α-synuclein. A possible molecular link between Parkinson's disease and heavy metal exposure, J. Biol. Chem., 2001, 276, 44284–44296. © the American Society for Biochemistry and Molecular Biology. | ||
A similar trend was also observed in the ability of metal cations to accelerate α-synuclein aggregation (see Fig. 3A). Fig. 3B shows that in the presence of some cations α-synuclein formed typical amyloid-like fibrils. However, in many cases both amorphous aggregates and amyloid-like fibrils were found at the end of the aggregation process. The most likely explanation for this observation is the existence of at least two competing aggregation processes, fibrillation versus amorphous and/or soluble aggregates formation. Therefore, the observation of both fibrillar and amorphous aggregates for α-synuclein in the presence of metals is consistent with a minimal underlying kinetic scheme of the sort shown below:33
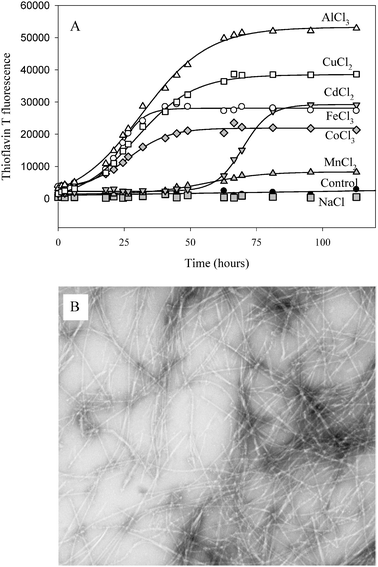 | ||
| Fig. 3 (A) Kinetics of α-synuclein fibril formation in the presence of metal ions. α-Synuclein (35 μM) was incubated with stirring at 37 °C in phosphate buffer, pH 7.5 in the presence of the indicated metals (2 mM), as their chloride salts. Fibril formation was monitored by the increase in ThT fluorescence at 485 nm. Reproduced from ref. 33. This research was originally published in J. Biol. Chem.: V. N. Uversky, J. Li and A. L. Fink, Metal-triggered structural transformations, aggregation and fibril formation of human α-synuclein. A possible molecular link between Parkinson's disease and heavy metal exposure, J. Biol. Chem., 2001, 276, 44284–44296. © the American Society for Biochemistry and Molecular Biology. (B) Negative stained transmission electron micrographs of α-synuclein fibrils prepared In the presence of 2 mM CoCl3. | ||
The variable ratios of fibrils to amorphous aggregates detected in the presence of different cations indicated that there was an underlying branched pathway, and that different cations affected the relative rates of fibril formation or non-fibrillar aggregation differently. The correlation between the amount of fibrils and the rate of fibril formation, suggested that the cations with the high efficiency to accelerate α-synuclein fibrillation produced more fibrils. In other words, these cations interacted with α-synuclein in such a way as to increase the rate of the pathway leading to fibrils.33 Summarizing, this study revealed that the effectiveness of metal cations to induce fibrillation was correlated with the increasing ion charge density and with their ability to induce amyloidogenic partially folded species.33
Subsequent studies revealed that different cations not only accelerated the α-synuclein fibrillation to a different degree, but also resulted in the formation of fibrils with different morphologies.39 For example, the diameter and general structure of fibrils were not affected by the presence of Cu2+ and Zn2+. However, fibrils grown under these conditions were noticeably shorter than fibrils produced in the absence of metals. Furthermore, these metal-enriched fibrils were efficient catalysts of the redox cycling of added Fe2+. On the contrary, fibrils formed in the presence of Al3+ possessed a novel amyloid morphology which consisted of twisted fibrils with a periodicity of about 100 nm and were poorer catalysts of the redox cycling of added Fe2+.39 This study clearly illustrated that the fibril morphology depends significantly on the nature of the metal ion added.
Recently, the peculiarities of the divalent metal ions Fe2+, Mn2+, Co2+, and Ni2+ binding to α-synuclein and their effects on protein aggregation were analyzed by exploiting the different paramagnetic properties of these metal ions.40 NMR spectroscopy revealed that these ions bind preferentially and with low affinity (millimolar) to the C-terminus of α-synuclein. The primary binding site was shown to be the 119DPDNEA124 motif, in which Asp121 acts as the main anchoring residue. Based on these observations and on the results of the backbone residual dipolar coupling measurements it has been concluded that metal binding was not driven exclusively by electrostatic interactions but was mostly determined by the residual structure of the C-terminus of α-synuclein.40
The stoichiometry of Cu2+ and Fe3+ binding to α-synuclein (wild type protein and A30P, A53T, E46K mutant forms associated with the familial PD) was studied using isothermal titration calorimetry (ITC).41 Two Cu2+-binding sites in α-synuclein monomer (wild and mutant forms) were found, with apparent KB of 105 M and 104 M, respectively. However, only one Fe3+-binding site with an apparent KB of 105 M was found in this protein.41
The role of phosphorylation in the interaction of α-synuclein with bivalent and trivalent metal ions was analyzed using set model peptides and phosphopeptides corresponding to the residues 119-132 of α-synuclein (119DPDNEAYEMPSEEG132).42 This study revealed that the 119DPDNEA(pY)EMPSEEG132 phosphopeptide, where tyrosine was replaced with phosphotyrosine, possessed a marked selectivity for trivalent metal ions Tb3+, Fe3+, and Al3+ in comparison with the non-modified peptide or the 119DPDNEAYEMP(pS)EEG132 peptide, where serine was replaced with phosphoserine. Based on the detailed analysis it has been concluded that the phosphoester group on tyrosine provided a metal-binding anchor that was supplemented by carboxylic acid groups at positions 119, 121, and 126 to establish a multidentate ligand, whereas two glutamic acid residues at positions 130 and 131 contributed to binding additional trivalent ions. Furthermore, circular dichroism analysis showed that Fe3+ induced a partially folded structure in 119DPDNEA(pY)EMPSEEG132, whereas no change was observed for 119DPDNEAYEMP(pS)EEG132 or for the unphosphorylated analog.42 Based on these observations it has been concluded that the type and location of a phosphorylated amino acid can affect a metal-binding specificity and affinity of α-synuclein as well as its overall conformation.42
The simultaneous presence of metals and pesticides/herbicides (another group of environmental factors linked to the PD pathogenesis) accelerated the α-synuclein fibrillation synergistically.43Fig. 4 shows that in experiments in which α-synuclein was incubated in the presence of both diethyldithiocarbamate (DDC) and AlCl3, fibril formation was more rapid than that expected from the sum of the effects of the two agents alone, indicating a synergistic effect between the pesticide and the metal ion.43 Furthermore, the simultaneous presence of DDC and AlCl3 led to enhanced conformational change, as detected by circular dichroism, over each additive individually.43 In agreement with these observations, it has been recently established that herbicides preferentially bind to a partially folded intermediate conformation of α-synuclein induced by Mn2+, Al3+, Cd2+, Cu2+ and Zn2+.44 Similarly, some metals were shown to cause a significant acceleration of α-synuclein fibrillation in the presence of high concentrations of various macromolecules mostly through decreasing the fibrillation lag-time. The faster fibrillation in crowded environments in the presence of heavy metals suggested a simple molecular basis for the observed elevated risk of PD due to exposure to metals.45 Intriguingly, the addition of certain metals (Ti3+, Zn2+, Al3+, and Pb2+) was shown to overcome the methionine oxidation-induced inhibition of the α-synuclein fibril formation.46 This is illustrated by Fig. 5 which represents transmission electron micrographs of α-synuclein fibrils induced in non-oxidized and oxidized protein by different metals. Based on observations that methionine can react with essentially all of the known oxidants found in normal and pathological tissues; that α-synuclein is a very abundant brain protein; that the concentration of α-synuclein could be increased significantly as a result of the neuronal response to toxic insult; and that methionine sulfoxide residues in proteins can be cycled back to their native methionines by methionine sulfoxide reductase it has been proposed that the methionine residues in α-synuclein may be used by the cells as a natural scavenger of reactive oxygen species. Here, α-synuclein can serve as the protective antioxidant since the methionine-oxidized α-synuclein does not fibrillate and the addition of the oxidized protein inhibits fibrillation of the non-oxidized α-synuclein. On the other hand, a combination of oxidative stress and environmental metal pollution could play an important role in triggering the fibrillation of α-synuclein and thus possibly PD development.46
 | ||
| Fig. 4 Synergistic interactions between pesticide and metal in the stimulation of α-synuclein fibril formation. α-Synuclein (28 μM) alone (black squares); with 100 μM DDC (open circles); with 4 mM AlCl3 (black circles); with DDC (100 μM) and AlCl3 (4 mM) added in that order (black triangles); and with AlCl3 (4 mM) and DDC (100 μM) added in that order (open triangles). | ||
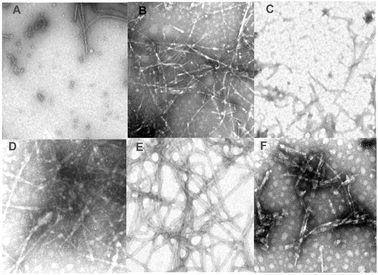 | ||
| Fig. 5 Negatively stained transmission electron micrographs of different α-synuclein fibrils induced in non-oxidized and oxidized protein by different metals: Met oxidized α-synuclein control (A); α-Synuclein control (B). Oxidized α-synuclein in the presence of calcium (C). Oxidized α-synuclein in the presence of aluminium (D). Oxidized α-synuclein in the presence of lead (E). Oxidized α-synuclein in the presence of zinc (F). Reproduced from ref. 46. This research was originally published in J. Biol. Chem.: G. Yamin, C. Glaser, V. N. Uversky and A. L. Fink, Certain metals trigger fibrillation of the methionine-oxidazed α-synuclein. J. Biol. Chem., 2003, 278, 27630–27635. © the American Society for Biochemistry and Molecular Biology. | ||
Data presented in this section support the notions that many metal ions interact with α-synuclein non-specifically, and that binding of certain cations (e.g., Cu2+ and Fe3+) is relatively specific. Interaction with metals can modify α-synuclein structure, modulate its fibrillation rates and change fibril/aggregate morphology. Subsequent sections are dedicated to the analysis of the effects of specific metal ions.
Aluminium
Aluminium is one of the metal ions to which exposure was linked to the PD pathology via the epidemiological studies and the postmortem analysis of the brain tissues of PD patients.19,22–25 As soon as a connection between α-synuclein and PD pathogenesis was established (in 1997, when Polymeropoulos et al. identified a mutation in the α-synuclein gene in familial cases of early-onset PD47 and Spillantini et al. demonstrated the presence of α-synuclein in Lewy bodies,48), the effect of Al3+ on α-synuclein structure was analyzed.32 This first study revealed that the protein structure can be altered by certain environmental factors, including aluminium. This aluminium-altered conformation structure became resistant to proteases such as trypsin, α-chymotrypsin, and calpain.32To gain more information on the structural consequences of cation binding, the effect of increasing concentrations of Al+3 on the spectral properties of α-synuclein has been analyzed.33 The shapes and intensities of the far-UV CD, UV absorbance, intrinsic fluorescence and ANS fluorescence spectra changed significantly with the increase in the Al+3 concentration. This indicated that the structure of natively unfolded α-synuclein was affected by interaction with the cation. Furthermore, the spectral changes induced in α-synuclein by Al+3 occurred simultaneously in a rather co-operative manner, were completely reversible and independent of protein concentration (at least in the range of 0.7–35 μM). These observations were consistent with the assumption that the Al+3-induced structure formation in α-synuclein represented an intramolecular process and not self-association.33 Kinetics analysis revealed that the addition of Al+3 induced a rapid formation of the secondary structure, within the dead time of the manual mixing experiment (∼5 s).33
Structurally, this Al+3-stabilized partially folded intermediate of α-synuclein possessed a strong resemblance to the amyloidogenic pre-molten globule form originally found at acidic pH or elevated temperatures. In agreement with this structural similarity, the rate of α-synuclein fibril formation at neutral pH was dramatically accelerated by the AlCl3 addition to the α-synuclein solution. In the presence of 2.5 mM of AlCl3, the lag-time was ∼3-times shorter, and the apparent rate of fibril formation was ∼1.5-fold higher compared to protein alone. Furthermore, it has been demonstrated that Al3+ was incorporated into the mature fibrils formed in the presence of Al3+.33
Finally, it has been shown that less aluminium was required to induce a comparable acceleration of fibril formation at higher protein concentrations. In agreement with this finding it was established that aluminium had much larger structure-forming effects at high α-synuclein concentrations. In fact, in the range of protein concentration from ∼0.7 to ∼50 μM the Al3+-induced changes in the spectral properties of α-synuclein were completely reversible and independent of protein content. However, at high protein concentration the Al3+-induced folding of α-synuclein was shown to be accompanied by association of the partially folded intermediates.33 Interestingly, these self-associated forms of partially folded α-synuclein were shown to more readily form fibrils, and possessed a larger amount of ordered secondary structure.33
The conclusion on the Al3+-induced partial folding of α-synuclein was recently confirmed using the selective non-covalent adduct protein probing mass spectrometry (SNAPP-MS), which utilized specific, non-covalent interactions between 18-crown-6 ether (18C6) and lysine to probe protein structure in the presence and absence of metal ions.49 It has been shown that the 18C6 SNAPP distributions for α-synuclein changed dramatically in the presence of 3 mM Al3+, suggesting that Al3+ binding caused a significant change in the conformational dynamics of the monomeric form of this disordered protein.49
Calcium
Calcium is known to have a great number of unique and highly diverse functions in biology. For example, neuronal Ca2+ homeostasis and Ca2+ signaling regulate multiple neuronal functions, including synaptic transmission, plasticity, and cell survival. Being properly controlled, Ca2+ fluxes across the plasma membrane and between intracellular compartments play critical roles in fundamental functions of neurons, including the regulation of neurite outgrowth and synaptogenesis, synaptic transmission and plasticity, and cell survival.50 Therefore disturbances in Ca2+ homeostasis can affect the well-being of the neuron in different ways and to various degrees.51 It has been hypothesized that excitotoxicity and disturbance in Ca2+ homeostasis are among the potential mechanisms leading to neurodegeneration.50 Normal substantia nigra neuron activity seems to depend on unusually high calcium entry,52 and α-synuclein was shown to induce Ca2+ influx in rat synaptoneurosomes through N-type voltage-dependent Ca2+ channels.53 Another consequence of disturbed calcium homeostasis is the activation of calpains, a family of cysteine proteases. These are elevated in the mesencephalon of patients with PD but not in other neurodegenerative disorders involving the mesencephalon.52 Perturbed neuronal Ca2+ homeostasis is also implicated in age-related cognitive impairment and AD.54 Overall, neurodegenerative diseases related to aging, such as AD, PD, or Huntington's disease, are characterized by the positive feedback between Ca2+ dyshomeostasis and the aggregation of disease-related proteins such as Aβ, α-synuclein, or huntingtin.51 Therefore high levels of Ca2+ and other metal ions may play a role in pathogenesis of these diseases. It has been proposed that a better understanding of the cellular and molecular mechanisms promoting or preventing disturbances in cellular Ca2+ homeostasis during aging may lead to novel approaches for therapeutic intervention in neurological disorders such as AD and PD.50Using a microdialysis technique it was shown that α-synuclein binds Ca2+ with an IC50 of about 2–300 μM and that this reaction can be uninhibited by a 50-fold excess of Mg2+. The Ca2+-binding site was assigned to a C-terminally localized acidic 32-amino acid domain of this protein.55 The functional properties of α-synuclein (e.g. the ability of this protein to interact with microtubule-associated protein 1A) and the propensity of α-synuclein to aggregate were affected by Ca2+ binding, suggesting that Ca2+ ions may participate in normal α-synuclein functions in the nerve terminal and also may contribute to some pathological processes associated with α-synuclein aggregation and LB formation.55 In agreement with this hypothesis, a recent study revealed that α-synuclein regulates Ca2+ entry pathways and showed that abnormal α-synuclein levels may promote neuronal damage through dysregulation of Ca2+ homeostasis.56 In another study, a link between Ca2+ homeostasis, α-synuclein and cytosolic dopamine was established suggesting that interplay between these three molecules can be responsible for the selective death of the dopaminergic neurons in the substantia nigra.57
The effect of Ca2+ and other metals on the morphology of α-synuclein aggregates has been analyzed by AFM.58 Three classes of effect were observed with different groups of metal ions. First class included Cu2+, Fe3+, and Ni2+ which yielded 0.8–4 nm spherical particles, similar to α-synuclein incubated without metal ions. Second class was contained Mg2+, Cd2+ and Zn2+ which induced larger, 5–8 nm spherical oligomers. Finally, third class included Co2+ and Ca2+ which gave frequent annular oligomers, 70–90 nm in diameter with Ca2+ and 22–30 nm in diameter with Co2+.58 Each annular particle induced by Ca2+ appeared to be composed of a ring of six spherical particles with occasional open-ring structures composed of a chain of five 4 nm-height spherical particles. In agreement with earlier study, which revealed that α-synuclein calcium binding is mediated by the acidic C terminus,55 no annular oligomers were found when truncated α-synuclein (1–125), lacking the C-terminal 15 amino acids, was co-incubated with Ca2+.58 These observations suggested that the Ca2+-binding domain located within the C-terminal half of α-synuclein was involved in the formation of such annular oligomers. Importantly, soluble 30–50 nm-sized annular α-synuclein oligomers were isolated by mild detergent treatment from glial cytoplasmic inclusions (GCIs) purified from multiple system atrophy brain tissue.59 As annular aggregates were specifically induced by Ca2+ binding, it has been hypothesized that the formation of such aggregated species inside the neurons can be influenced by the intracytoplasmic Ca2+ concentration.60
Finally, Ca2+ was shown to modulate the interaction of α-synucelin with membranes.61 In fact, it has been shown that in the absence of Ca2+, the protein was anchored to the lipid surface via the N-terminal domain. The addition of Ca2+ promoted effective interaction between the C-terminal domain of α-synucelin and lipid membrane. Based on these observations it has been suggested that initial lipid interaction of α-synuclein occurs via its N-terminal domain, followed by a Ca2+-triggered membrane association of the acidic tail, potentially leading to α-synuclein aggregation. This model suggests that cellular Ca2+ dysregulation can be one of the critical factors determining the α-synuclein aggregation in PD.61
Cobalt
Similar to Ca2+, Co2+ was shown to induce the fast formation of annular aggregates of α-synuclein.58 These Co2+-induced doughnut-like oligomers had a mean maximum height of 2 ± 0.8 nm and diameter ranging 22–30 nm, being a bit smaller than those formed in the presence of Ca2+, which were characterized by a diameter range of 70–95 nm, and a mean maximum height of 3.9 ± 1.2 nm.58 Based on the detailed morphological analysis of metal-induced oligomers it has been proposed that the aggregation of α-synuclein can be described as a sequential chain of events starting from monomer to ∼4 nm-height spherical oligomer, to ∼4-nm height annular oligomer, then to ∼8 nm-height annular oligomer, from which ∼8–10 nm-height linear filaments then extend. It has been also proposed that the wide range of diameters observed for annular oligomers indicated that these oligomeric species were likely to not be functionally defined quaternary structures, but formed as a result of the competing self-association processes of either linear extension or ring closure.58Copper
It has long been believed that the concentration of free metal ions in the brain is too low to be physiologically significant and that, as a consequence, involvement of metal ions in neurological disorders may only arise from toxicological exposure to Cu, Fe, Zn, or Mn.62 However, in terms of total concentrations, the brain has more than enough of the mentioned metal ions in its tissue to damage or dysregulate numerous proteins and metabolic systems.30,63,64 Copper is very redox active. Therefore it cannot exist in an unbound form in the cell without causing oxidative damage, as free or incorrectly bound Cu2+ may act as a catalyst for the generation of the most damaging radicals, such as the hydroxyl radical. Therefore, distortions in the tight regulation of Cu2+ homeostasis are expected to contribute to several neurodegenerative conditions.62The human brain is estimated to produce more than 1011 free radicals per day,65 and imbalance in pro-oxidant versus antioxidant homeostasis results in “oxidative stress” with the generation of several potentially toxic reactive oxygen species (ROS), including both the radical (such as hydroperoxyl radical, superoxide radical, hydroxyl radical), and nonradical species (such as hydrogen peroxide) that participate in the initiation and/or propagation of radical chain reactions.62 Inappropriate compartmentalization or elevation of Cu2+ in the cell and inappropriate binding of Cu2+ to cellular proteins are currently being explored as sources of pathological oxidative stress in several neurodegenerative disorders.
A first systematic analysis of the effect of Cu2+ on α-synuclein structure and aggregation revealed that this metal ion was able to induce an effect on α-synuclein oligomerization in the presence of coupling reagents such as dicyclohexylcarbodi-imide or N-(ethoxycarbonyl)-2-ethoxy-1,2-dihydroquinoline.31 This study also revealed that the full-length α-synuclein is able to bind 10.4 Cu2+ ions. The Cu2+-induced oligomerization was effectively suppressed by the truncation of the acidic C-terminus of α-synuclein by treatment with endoproteinase Asp-N, suggesting that the Cu2+-induced oligomerization was dependent on the acidic C-terminal region.31
Reactive oxygen species may initiate the covalent modifications of amino acid residues in proteins, formation of protein–protein cross-linkages, and oxidation of the protein backbone resulting in protein fragmentation. Using the high performance liquid chromatography (HPLC) and matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) methods it has been shown that the major targets of the Cu2+/hydrogen peroxide oxidative modification of α-synuclein are methionines, which were shown to be oxidized to methionine sulfoxides and sulfones.66,67
Since oxidatively modified proteins are known to possess an increased propensity to aggregate, the effect of metal-catalyzed oxidation of α-synuclein by Cu2+ and hydrogen peroxide on protein aggregation was examined.68 The oxidized protein was shown to be self-oligomerized into an SDS-resistant ladder as detected by SDS-PAGE. It has been, suggested that abnormalities in Cu2+ and hydrogen peroxide homeostasis could play critical roles in the metal-catalyzed oxidative oligomerization of α-synuclein, which may lead to possible protein aggregation and neurodegenerations.68
Cu2+ was shown to be one of the most potent modulators of α-synuclein structure and a very effective accelerator of α-synuclein fibrillation in vitro.33 Using atomic force microscopy-based single-molecule mechanical unfolding methodology it has been shown that the presence of Cu2+ significantly enhanced the relative abundance of the “beta-like” structure in the conformationally heterogeneous monomeric α-synuclein, which happened to contain three main classes of conformations.69 Even at physiologically relevant concentrations, Cu2+ ions were effective in acceleration of α-synuclein aggregation without altering the resultant fibrillar structures.70 Using a set of spectroscopic techniques such as absorption, CD, EPR, and NMR, the primary Cu2+-binding site was shown to be localized in the N-terminus of α-synuclein. This binding site involved His50 as the anchoring residue and other nitrogen/oxygen donor atoms in a square planar or distorted tetragonal geometry.70 The acidic C-terminus of the protein, originally thought to drive copper binding, was shown to coordinate a second Cu2+ equivalent with a 300-fold reduced affinity in comparison with the N-terminal Cu2+-binding site.70 Using a set of Trp α-synuclein mutants, it has been confirmed that Cu2+ interacted with α-synuclein, and that Cu2+ binds tightly (KD 100 nM) near the α-synuclein's N-terminus.71 Further confirmation of the N-terminal localization of a strong Cu2+-binding site was obtained using electron paramagnetic resonance (EPR) spectroscopy.72 Subsequent comparative analysis of α- and β-synuclein interaction with Cu2+ by combined application of nuclear magnetic resonance, electron paramagnetic resonance, UV-vis, circular dichroism spectroscopy, and matrix-assisted laser desorption ionization mass spectrometry (MALDI MS) revealed the existence of two independent, non-interacting Cu2+-binding sites with significantly different affinities for the metal ion in the N-terminal regions of both proteins.73 The higher-affinity site was assigned to the N-terminal amino group of Met1 and the lower-affinity site was ascribed to the imidazol ring of the sole His residue. This study also confirmed that Cu2+ binding in the C-terminal region of synucleins represents a nonspecific, very low affinity process.73 In agreement with these studies, the minimal high efficiency Cu2+-binding site was identified within the first four residues of α-synuclein, MDVF.74 In addition, the analysis of various mutant peptides clearly showed that neither Lys6 nor Lys10 are necessary for Cu2+ binding. Furthermore, the two quenching modes measured for the Trp4 excited-state decay kinetics suggested the existence of two distinct Cu2+-polypeptide structures.74
When NMR spectroscopy was used to identify a number of independent Cu2+-binding sites in α-synuclein, as many as 16 different sites capable of binding Cu2+ were found.75 Most of the sites involved negatively charged amino acid side chains, but binding was also detected to the sole histidine residue located at position 50 and to the N-terminal amino group. Importantly, the N-terminal and the histidine sites, as well as the sites in the C-terminal tail were shown to bind Cu2+ in the more highly structured conformation adopted by α-synuclein upon binding to detergent micelles or lipid vesicles.75
In the search for the location of the Cu2+-binding sites, six peptide fragments spanning the full length of α-synuclein were synthesized and analyzed for metal binding and toxicity to SH-SY5Y cells.76 Based on this analysis it has been concluded that both the C-terminus of the protein and a region around histidine 50 play a role in Cu2+ binding. Furthermore, it was concluded that the true binding domain is a nonlinear site composed of both areas acting together to bind Cu2+. Three different Cu2+ binding modes utilizing these separated sites were proposed for α-synuclein. In the first mode, single α-synuclein molecule binds Cu2+ by folding which brings the N- and C-terminal sites together. In the second mode, two α-synuclein molecules are orientated head to tail creating two sites at either end of the molecule. In the third mode, Cu2+ binding results in oligomerization where the C-terminus of the first α-synuclein molecule interacts with the N-terminus of the second molecule to form a Cu2+-binding site, whereas the second site results from the interaction of one of those two α-synuclein molecules with a third α-synuclein molecule.76 Therefore, this Cu2+-mediated interaction between the N- and C-termini could facilitate intermolecular interactions that could promote oligomerization and conversion of α-synuclein to a neurotoxic form via inter-protein interactions.76 In agreement with this hypothesis, it was shown that aggregated α-synuclein is neurotoxic and that this neurotoxicity requires the presence Cu2+.77 These cytotoxic oligomeric species of α-synuclein were isolated and shown to be Cu2+-loaded and possess a unique stellate morphology by EM analysis. This work provided a clear link between the Cu2+-induced oligomerization of α-synuclein, cytotoxicity and neurodegeneration.77
Iron
Interconnection between problematic iron homeostasis, α-synuclein aggregation and various neurodegenerative diseases is very strong. As it was already pointed out, numerous epidemiological studies16–21 and the postmortem analysis of the brain tissues22–24 have linked the heavy metal exposure and metal accumulation in the brain with the PD pathogenesis. Postmortem analysis of brain tissues from patients with PD revealed that the substantia nigra of the PD brain is characterized by a shift in the Fe2+/Fe3+ ratio in favor of Fe3+ and a significant increase in the Fe3+-binding protein, ferritin. In parallel, significantly lower glutathione content was shown to be present.23 It was shown that unilateral injection of FeCl3 into the substantia nigra of adult rats resulted in a substantial selective decrease of striatal dopamine (95%). As a result, dopamine-related behavioral responses were significantly impaired in the iron-treated rats. This supported the assumption that iron initiates dopaminergic neurodegeneration in Parkinson's disease.78In fact, an important role of iron in the PD pathogenesis due to its increase in substantia nigra pars compacta dopaminergic neurons and reactive microglia and its capacity to enhance production of toxic reactive oxygen radicals, has been discussed for many years.79 This hypothesis was strongly supported by findings that iron is able to induce aggregation and toxicity of α-synuclein, further suggesting that the disturbances of iron metabolism in PD including iron uptake, storage, intracellular metabolism, release and post-transcriptional control, as well as interaction with specific proteins, such as α-synuclein, can contribute to the process of neurodegeneration and pathogenesis of nigrostriatal injury.79 Overall, it is established that iron levels increase with the severity of neuropathological changes in PD. The potential mechanism of this increase in the iron levels is the increased transport of this metal ion through the blood-brain barrier in late stages of PD. Iron overload may induce progressive degeneration of nigrostriatal neurons by facilitating the formation of reactive biological intermediates, including reactive oxygen species, and the formation of cytotoxic protein aggregates.80 Intriguingly, it has been established that the intracellular iron is crucial for the 1-methyl-4-phenylpyridinium (MPP+)-induced apoptotic cell death that causes PD provoked by exposure to this neurotoxin. More specifically, MPP+-induced iron signaling was shown to be responsible for intracellular oxidant generation, α-synuclein expression, proteasomal dysfunction, and apoptosis.81
Besides PD, an interplay between iron, α-synuclein aggregation and neurodegeneration is obvious in neurodegeneration with brain iron accumulation, type 1 (NBIA 1), or Hallervorden-Spatz syndrome, which is a rare neurodegenerative disorder characterized clinically by Parkinsonism, cognitive impairment, pseudobulbar features, as well as cerebellar ataxia, and neuropathologically by neuronal loss, gliosis, and iron deposition in the globus pallidus, red nucleus, and substantia nigra. In 1998, it was shown that Lewy bodies found in NBIA 1 were positively stained with antibodies against α-synuclein, suggesting that this protein is commonly associated with the formation of LBs in other sporadic and familial neurodegenerative diseases apart from PD.82 This finding was later confirmed by showing that α-synuclein is accumulated in the brains of NBIA 1 patients in the form of LB-like inclusions, glial inclusions, and spheroids.83,84 Generally, neuropathological studies show that synucleinopathies are generally associated with iron accumulation, which is consistent with a pathological link between iron and α-synuclein.85
As a transition metal closely associated with inducing the formation of reactive oxygen species and oxidative stress, iron has been suspected to contribute to PD because of the Fe2+ ability to promote oxidative damage. Hydrogen peroxide (H2O2) is a reactive oxygen species, which in the presence of redox-active metal ions (such as Fe2+) can produce hydroxyl radicals via Fenton chemistry. Since these radicals are only able to diffuse short nanometre distances, the changes observed at protein sites may identify sites of both hydroxyl radical generation and metal ion interaction. Recent studies have added another player to the scene, α-synuclein, and an additional pathological mechanism, by which iron might contribute to PD is by inducing aggregation of the α-synuclein, which then accumulates in LBs and LNs in PD.86 For example, an analysis of human BE-M17 neuroblastoma cells overexpressing wild-type, A53T, or A30P α-synuclein, revealed that iron and free radical generators, such as dopamine or hydrogen peroxide, were able to stimulate the production of intracellular α-synuclein-containing aggregates.87 The efficiency of protein aggregation was dependent on the amount and the type of α-synuclein expressed following a rank order of A53T > A30P > wild-type > untransfected. Furthermore, overexpression of α-synuclein clearly induced toxicity, and α-synuclein overexpressing BE-M17 neuroblastoma were much more vulnerable to toxicity induced by iron.87
In addition to iron-promoted α-synuclein aggregation it has been found that α-synuclein was able produce hydrogen peroxide and hydroxyl radicals upon incubation in vitro in the presence of small Fe2+ amounts.88 These observations suggested that one of the molecular mechanisms in the PD pathogenesis could be the direct production of hydrogen peroxide and hydroxyl radicals by α-synuclein, in a metal-dependent manner, before, during and after the abnormal protein aggregate formation.88
Although all metal ions studies so far were proved to promote α-synuclein aggregation and fibrillation,33 the morphology of the resultant aggregates was shown to be strongly dependent on the nature of the metal ion, as revealed, for example, by the electron microscopy analysis of the effect of Cu2+ and Fe3+ on α-synuclein (wild-type, A30P, A53T, and E46K) fibril formation and morphology.89 Cu2+ and Fe3+ were shown to selectively and differentially induce the formation of distinctive fibrillar species. Cu2+ induced thin long network-like fibrils with the wild-type of α-synuclein, whereas the mutant forms of the protein showed amorphous aggregates with no fibrillar species. Fe3+ induced short and thick fibrils with both wild type and mutant proteins.89 It has been proposed that such metal-specific fibril morphology might have relevance for better understanding the molecular mechanisms of neurodegeneration and the role of metals in this process.
As small oligomers are widely considered as major toxic aggregate species, the formation of α-synuclein at the single particle level was recently monitored using confocal single-molecule fluorescence techniques, scanning for intensely fluorescent targets (SIFT), fluorescence intensity distribution analysis (FIDA), and atomic force microscopy.90,91 It has been shown that both organic solvents and Fe3+ at low micromolar concentrations were effective inducers of α-synuclein oligomerization. The morphologies of the resulting oligomers were different, with organic solvents inducing small oligomers (“intermediate I”) and iron inducing formation of larger oligomers (“intermediate II”). Importantly, Fe3+ only caused an effect on α-synuclein aggregation when added in the presence of intermediate concentrations of ethanol (∼5%), suggesting that the effect of Fe3+ was depend on the presence of the intermediate I species, therefore proposing a synergistic effect of Fe3+ and ethanol on α-synuclein aggregation.
Although both oligomers were on-pathway to amyloid fibrils and could effectively seed amyloid formation, Fe3+-induced oligomers were SDS-resistant and could form ion-permeable pores in a planar lipid bilayer.91 These observations provide a strong support to an important role of ferric iron in the formation of toxic α-synuclein oligomer species providing a potential disease mechanism.91 Recently, the bioluminescent protein-fragment complementation assay (BPCA) was implemented to directly analyze the formation of toxic α-synuclein oligomers in cell culture and to study the effect of iron and potential drug-like compounds in living cells.92 This analysis revealed a converse modulation of toxic α-synuclein oligomers by N′-benzylidene–benzohydrazide (NBB) derivates and Fe3+, where Fe3+ promoted aggregate formation in living cells whereas in-cell oligomerization and cytotoxicity was inhibited by certain NBB compounds.92
As both, protein aggregation and oxidative stress, being pathological hallmarks of several neurodegenerative diseases, are intimately associated with metals (especially iron and copper), chelation-based therapy could provide a valuable therapeutic approach to such disease states.93–95 The important features of a therapeutic iron-chelating agents were proposed to be the ability to scavenge the free redox-active iron (or copper) present in excess in the brain and to form a non-toxic iron complex, which is then excreted. A second mechanism of beneficial chelation would be to cap the iron at its labile binding site, preventing any mediated toxic action (Fenton activity and/or aggregation). Here, stable metal-chelator complex should favor the state in which the metal is not redox active and therefore not toxic. Furthermore, in order to prevent protein aggregation, additional interactions between the drug and the target protein are highly desirable.93
Lead
Contrarily to the metals considered so far, whose levels are typically elevated in PD brains, the content of lead is reduced in brains of patients with PD. This conclusion follows, for example, from the detailed analysis of the elemental concentration in the cerebrospinal fluid of 42 patients with PD and 20 age-matched controls performed by inductively coupled plasma atomic emission spectrometry (ICP-AES) and sector field inductively coupled plasma mass spectrometry (SF-ICP-MS).96 The elemental profiling where the abundance of Al, Ba, Be, Bi, Ca, Cd, Co, Cr, Cu, Fe, Hg, Li, Mg, Mn, Mo, Ni, Pb, Sb, Si, Sn, Sr, Tl, V, W, Zn, and Zr was analyzed revealed up to 50% depletion of Cr2+ and Pb2+ in Parkinsonian brains, suggesting that these two ions can be considered as the most suitable elements to distinguish between normality and PD.96 Exposure to lead (Pb2+) is known to produce aggresome-like inclusion bodies in target cells as a toxic response.97 This process was shown to be controlled by both metallothionine and α-synuclein. In fact, Pb2+ exposure rapidly increased α-synuclein expression in cells stably expressing metallothionine and then decreased over 48 h as Pb2+-induced aggresome-like inclusion bodies were formed. Metallothionine and α-synuclein were colocolized and Pb2+ exposure caused time-dependent increased colocalization of these two proteins in the aggresome-like inclusion bodies. Based on these observations it has been concluded that both metallothionine and α-synuclein are components of Pb-induced inclusion bodies and may play a role in the formation of these deposits.97 In an in vitro study, Pb2+ was one of a few metal ions shown to overcome the methionine oxidation-induced inhibition of the α-synuclein fibril formation.46Magnesium
Similar to lead, the content of magnesium is reduced in PD brains. For example, analysis of the regional metal concentrations by atomic absorption and atomic emission spectroscopy in four brain regions (frontal cortex, caudate nucleus, substantia nigra, and cerebellum) revealed lower concentrations of magnesium in the caudate nucleus in parkinsonian brains (PD and parkinsonism secondary to neurofibrillary tangle disease) in comparison with controls.98 This finding was later confirmed by inductively coupled plasma emission spectrometry analysis of 26 regions of PD and control brains, which revealed that Mg2+ concentration was lower in cortex, white matter, basal ganglia and brain stem of PD brains compared to control brains.99 Magnesium concentration in cerebrospinal fluid decreased with the duration and severity of the disease.100 Another interesting contradiction between Mg2+ and other metal ions is a potential link between low Mg2+ intake over generations and the pathogenesis of substantia nigra degeneration in humans, which was originally proposed as one of the potential mechanisms of the pathogenesis of the Parkinsonism-dementia complex and amyotrophic lateral sclerosis of Guam and later demonstrated for rats that were exposed to low Mg2+ intake over two generations.101When α-synuclein was incubated in the presence of high Mg2+ concentrations (>10 mM), large aggregates composed of densely packed short fibrillar elements were rapidly formed.102 However, the situation was different at lower Mg2+ concentrations. For example, to understand how changes in Fe2+ and Mg2+ levels might affect the PD pathophysiology, the binding of these ions to α-synuclein and their effect on protein aggregation have been investigated.103 This analysis revealed that Mg2+ affected the α-synuclein conformation differently to Fe2+. Furthermore, at low concentrations, Mg2+ inhibited α-synuclein aggregation induced either spontaneously or by incubation with iron, and no SDS-resistant dimers were formed in the presence of Mg2+.103 Later it was also shown that Mg2+ was able to modulate the interaction between α-synuclein and several herbicides (e.g., lindane, 2,4-D-isopropylester, atrazine, chlordimeform, and paraquat) and was able to successfully inhibit the herbicide-induced aggregation and fibrillation of α-synuclein.44 In this essence, Mg2+ was very different from other metals, such as Al3+, Cd2+, Mn2+, Cu2+ and Zn2+, all of which dramatically enhanced the herbicide interaction with α-synuclein and strongly promoted the herbicide-induced α-synuclein aggregation. Furthermore, when the effect of Mg2+ on the herbicide-α-synuclein binding was examined in the presence of 4 μM of other metals (Al3+, Cd2+, Mn2+, Cu2+ and Zn2+), a significant reduction in the apparent association constant K of herbicide-α-synuclein binding was detected with the increasing Mg2+ concentration. This clearly suggested that the apparent affinity of Mg2+ and magnesium for α-synuclein is strong enough to inhibit the conformation change induced by metals, and that the interaction of α-synuclein with different ligands (including metal ions) produced various (non-identical) conformational changes in this protein.44 In line with these observations, a study was recently conducted to clarify the effects of Mg2+ administration in a rat MPP+ model of PD.104 It was shown that Mg2+ might protect dopaminergic neurons in the substantia nigra from degeneration, significantly inhibiting the toxicity of MPP+ at concentration of Mg2+ to 1.2 mM, and completely eliminating the decrease in the number of dopaminergic neurons at a Mg2+ concentration of 4 mM.104 These observations suggest that the interaction of Mg2+ with α-synuclein might play a neuroprotective role.
Manganese
Manganese is an abundant trace element. It is ubiquitous in the environment, and it occurs in water, soil, air, and food. Manganese intoxication was first described almost 175 years ago in brownstone millers and in workers involved in mining and processing manganese ores, who inhaled toxic amounts of manganese dust.105 Chronic manganism produces an irreversible syndrome which bears a striking resemblance to PD, including fixed gaze, bradykinesia, postural difficulties, rigidity, and tremor.106 Parkinsonism due to the chronic manganese intoxication can be separated from PD by presence of dystonia and mental status changes.107 Manganism is also characterized by a particular propensity to fall backwards, failure to achieve a sustained therapeutic response to levodopa, and failure to detect a reduction in fluorodopa uptake by positron emission tomography.108 Furthermore, LBs have never the been observed in manganese-induced parkinsonism, and the major effects of manganese toxicity were found in the cells of the striatum and globus pallidus which are not dopaminergic.109Although manganism is clearly different from PD, and although a link between Mn2+ exposure and PD is still controversial, some facts suggested that α-synuclein can be involved in pathogenesis. This follows from the intriguing suggestion that nigral dopaminergic neurons overexpressing α-synuclein (or its mutations) may need an additional noxious factor (‘second hit’) to trigger nigral death.110 In fact, co-incubation of α-synuclein with Mn2+ induced partial folding of the protein and its effective fibrillation.33 Mn2+ was shown to be one of the most effective promoters of α-synuclein aggregation in vitro along with Al3+, Fe3+, Cu2+ and Co2+. Furthermore, the presence of Mn3+ induced immediate di-tyrosine formation suggesting that this cation is responsible for the metal-induced oxidation of the protein.33 Interestingly, when SK–N-MC neuroblastoma cells stably expressing the human dopamine transporter were transfected with human α-synuclein and cells were exposed to 30–300 μM MnCl2, the viability of cells overexpressing α-synuclein after 72 h of exposure to Mn2+ was dramatically reduced, suggesting that Mn2+ may co-operate with α-synuclein in triggering neuronal cell death such as seen in manganese parkinsonism.111
Terbium
Tb3+ was used a luminescent probe to analyze the peculiarities of metal binding to a peptide derived from α-synuclein.112,113 α-Synuclein fragment 119–132 (119DPDNEAYEMPSEEG132) was chosen for this analysis because its arrangement of carboxylate groups is similar to Ca2+-binding loops and because it contains two identified phosphorylation sites, Tyr125 and Ser129.112 A series of peptides were synthesized, including the non-modified fragment 119DPDNEAYEMPSEEG132, the 119DPDNEAYEMP(pS)EEG132 phosphopeptide (pS129), the 119DPDNEA(pY)EMPSEEG132 phosphopeptide and several truncated forms of the tyrosine phophsophopeptide. It has been shown that 119DPDNEAYEMPSEEG132 and 119DPDNEAYEMP(pS)EEG132 peptides possessed only a weak Tb3+ binding, whereas 119DPDNEA(pY)EMPSEEG132 showed tight 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding together with 2:1 and 3:1 Tb:peptide adducts.112 These data clearly showed that the phosphorylated amino acid must be appropriately positioned among additional ligating residues to establish this phosphorylation-dependent metal binding.112
:1 binding together with 2:1 and 3:1 Tb:peptide adducts.112 These data clearly showed that the phosphorylated amino acid must be appropriately positioned among additional ligating residues to establish this phosphorylation-dependent metal binding.112
Zinc
Zinc is one of the potential environmental factors to which exposure might favor PD.16,20,21,63 The analysis of the parkinsonian substantia nigra revealed the enhanced level of zinc in comparison with the control tissues.23–25 Zinc was shown to be an effective promoter of α-synuclein aggregation in vitro.33,114 Furthermore, the presence of Zn2+ among several other metals (Ti3+, Al3+, and Pb2+) was shown to effectively overcome the α-synuclein fibrillation induced by the methionines oxidation.46 It was proposed that this metal ion-induced acceleration of fibrillation may be due to the relatively strong coordination of the zinc (and other metal) ion between (at least) two methionine sulfoxides. This bridging was assumed to aid the protein in adopting a necessary conformation for fibrillation, or possibly to involve intermolecular cross-bridging, which could facilitate association and subsequent fibrillation.46When the fibrillation of the oxidized form of wild type α-synuclein and its Met-minus mutants, M5L, M116L, M127L, M116L/M127L, and M5L/M116L/M127L, were studied, the presence of Zn2+ was shown to induce a dramatic acceleration of fibril formation for all of the oxidized proteins studied and in a mutation-dependent manner.115 In fact, all of the oxidized mutants fibrillate faster than non-modified wild-type protein. Furthermore, the efficiency of mutation-induced acceleration of fibrillation seems to be proportional to the amount of substituted methionines, since the rates of fibrillation for the oxidized α-synucleins in the presence of Zn2+ can be arranged in the following order (from faster to slower rates of fibrillation): M5L/M116L/M127 > M116L/M127L > M5L ≈ M116L ≈ M127L.115 This suggested that methionine sulfoxides of the oxidized α-synuclein are not directly involved in the coordination of Zn2+. For example, a mutant with triple Met → Leu substitutions fibrillates faster than WT protein but would be expected to fibrillate slower or not at all if the methionine sulfoxides were involved in strong intermolecular metal interactions. Interaction with Zn2+ decreased the propensity of the Leu-substituted α-synucleins to oligomerize.115 It was also concluded that the effectiveness of Zn2+ in accelerating fibrillation of both oxidized wild type and mutated α-synucleins indicated that the presence of Zn2+ caused partitioning in favor of the fibrillation pathway. Therefore, Zn2+ overcame the factors that lead to population and stabilization of the soluble oligomers.115 Zinc was also one of the several metals shown to effectively promote interaction of α-synuclein with herbicides and to dramatically accelerate the herbicide-induced fibrillation of this protein.44
Concluding remarks: Multiple metal binding modes of α-synuclein
Significant knowledge is already accumulated in the field of α-synuclein metalloproteomics and metal toxicology. It is recognized now that metals play a crucial role in α-synuclein homeostasis, modulating its structural properties and aggregation propensity. In general, there are three major types of interactions of the non-oxidized α-synuclein with metal ions, non-specific, low-affinity and high-affinity binding. The non-specific binding is driven by the electrostatic interactions of the metal ions with the negatively charged side chains of α-synuclein. Although monovalent cations mostly provide the non-specific screening, polyvalent cations can additionally bridge two or more carboxylates, playing the role of structural cross-linkers.33 Many metals can interact with α-synuclein non-specifically, resulting in the effective neutralization of the Coulombic charge–charge repulsion and stimulates partial folding of α-synuclein, therefore favoring the formation of the aggregation-prone or even amyloidogenic species.33Low affinity (in the millimolar range) and high affinity (in the micromolar range) metal binding sites are located in the C-terminal and N-terminal domains of α-synuclein, respectively.116 In fact, besides purely non-specific sites of electrostatic interactions, the C-terminus of α-synuclein contains the primary binding site, 119DPDNEA124 motif, suggesting that metal binding is not only driven by electrostatic interactions but is also determined by some residual structure of the α-synuclein C-terminus.40 The C-terminal low affinity sites can interact with many different metal ions, including copper and iron. Furthermore, the low affinity metal ion binding to the C-terminal domain can be modulated by phosphorylation.42 In fact, α-synuclein phosphoryled at Tyr125 possesses remarkable selectivity for trivalent metal ions Tb3+, Fe3+, and Al3+. Here, the phosphoester group on tyrosine acts as a metal-binding anchor that is supplemented by the carboxylates at positions 119, 121, and 126 to establish a multidentate ligand, whereas two glutamic acid residues at positions 130 and 131 contribute to binding additional trivalent ions.42
Although the majority of metal ions interact with α-synuclein mostly non-specifically or bind with low affinity, this protein possesses relatively high affinity to Cu2+ and Fe3+. These metal ions interact with the C-terminal low affinity sites and are also able to coordinate with high affinity to the N-terminal region of α-synuclein. The higher-affinity site is assigned to the N-terminal amino group of Met1 and slightly lower-affinity site is ascribed to the imidazol ring of the sole histidine residue.73 This second Cu2+ binding site involves His50 as the anchoring residue and other nitrogen/oxygen donor atoms in a square planar or distorted tetragonal geometry.70 Being redox-active metal ions, Cu2+ and Fe3+ may also induce chemical modifications of the protein in vitro and in the reducing intracellular environment.116
There is a noticeable correlation between the propensity of a metal ion to induce partial folding of the non-oxidized α-synuclein and its ability to promote fibrillation of this protein.33 Intriguingly, among the most effective stimulators of protein partial folding and subsequent fibrillation were Cu2+ and Fe2+, which were shown to interact with α-synuclein specifically, together with non-specifically interacting Al3+, Co3+, and Mn2+.33 An important consequence of Mn3+ interaction with α-synuclein is the immediate formation of di-tyrosines.33 This suggests that in addition to Cu2+ and Fe2+ this cation is also responsible for the metal-induced oxidation of the protein. Since di-tyrosines can be form both intra- and intermolecularly, this modification represents an additional structure- and aggregation-promoting factor.
Methionine-oxidized α-synuclein possesses a different mechanism of interaction with metal ions. This mechanism involves metal coordination by methionine sulfoxides, which are the very abundant products of the α-synuclein oxidation at mild oxidative conditions. In fact, certain metal ions have a high propensity to strongly coordinate with sulfoxides, being potentially bridged between two (or more) sulfoxides. Such intermolecular or intramolecular coordination of two (or more) methionine sulfoxides could significantly affect both the structure and fibrillation of α-synuclein. Different metals posed different potency to induce fibrillation of the methionine-oxidized α-synuclein. In fact, the fibrillation rates were very close for oxidized and non-oxidized α-synuclein in the presence of Zn2+ and Pb2+. However, fibrillation was still inhibited in the presence of Hg2+, Cu2+, and Ca2+.46 It has been proposed that intermolecular coordination of two (or more) methionine sulfoxides by certain metal ions (such as Al3+, Zn2+ or Pb2+) could significantly promote fibrillation. On the other hand, some other metals (e.g. Hg2+, Ca2+, and Cu2+), being able to form sulfoxide bridges, may be limited to intramolecular coordination due to different ligand bonding.46
Since there are several different modes of metal ion interactions with α-synuclein it is not surprising that metal binding induces a wide range of conformational changes in this natively unfolded protein. Metal-induced structural changes vary from relatively minor gain of residual ordered structure in the presence of some monovalent metals to relatively more pronounced transformation into the pre-molten globule-like conformation promoted by interaction with some polyvalent ions.33 The partially folded conformation can also be cross-linked by some polyvalent metal ions. Different partially folded monomeric species can assemble into different oligomers some of which can finally form morphologically distinct fibrils. Illustrative examples of this variability of the metal-stabilized oligomeric forms include 0.8–4 nm spherical particles induced by Cu2+, Fe3+, and Ni2+; larger 5–8 nm spherical oligomers induced by Mg2+, Cd2+ and Zn2+; 22–30 nm in diameter Co2+; and 70–90 nm annular oligomers stabilized by Ca2+.58 The morphology of the amyloid fibrils and the propensity of amorphous aggregate formation were shown to be strongly dependent on the nature of the metal ion too. For example, interaction of the wild-type of α-synuclein with Cu2+ promoted formation of thin long network-like fibrils, whereas the mutant forms of the protein assembled into the amorphous aggregates with no fibrillar species in the presence of this metal ion. On the other hand, Fe3+ induced short and thick fibrils with both wild type and mutant forms of α-synuclein.89 Such metal-specific structural and morphological variability of monomeric and oligomeric species together with the metal-specific fibril morphology should be taken into account in the analysis of the molecular mechanisms of neurodegeneration and the role of metals in this process.
Future perspectives
The field of α-synucelin metalloproteomics and metal toxicology attracted significant attention primarily because of the link between the α-synuclein misfolding and aggregation and a number of neurodegenerative diseases and because of the potential link between the exposure to heavy metals and the pathogenesis of some of the synucleinopathies. Much remains to be learned in order to better understand this intriguing interconnection between metals, α-synuclein and pathogenesis of neurodegeneration. One of the perspective directions in the field is elucidation of the effect of protein posttranslational modifications (e.g., phosphorylation) and oxidative modifications on peculiarities of α-synuclein interaction with various metals. Synergistic effects of metals and other environmental toxins (such as pesticides and herbicides) on α-synuclein structure and aggregation propensity should be analyzed in more detail. The effect of macromolecular crowding on the peculiarities of α-synuclein interaction with various metals should be investigated. Detailed structural characterization of α-synuclein complexes with various metals and other environmental agents by high-resolution techniques (e.g., NMR) will shed more light on the molecular mechanisms of synucleinopathies. Better connection between the extensive in vitro data on the effect of metal ions on α-synuclein structure and aggregation and the cellular processes triggered by the exposure to heavy atoms should be established. Other interesting developments are expected in nanotechnology, where α-synuclein fibrils can be used as templates for the bottom-up device fabrication. In fact, α-synuclein fibrils have already been used as a scaffold for palladium, gold and copper nanoparticle chains synthesis, generating metal-coated fibers with reproducible average diameters between 50 and 200 nm.117Acknowledgements
This work was supported in part by the grants R01 LM007688-01A1 and GM071714-01A2 from the National Institute of Health, the grant EF 0849803 from the National Science Foundation, and the Programs of the Russian Academy of Sciences for the “Molecular and Cellular Biology”. We also gratefully acknowledge the support of the IUPUI Signature Centers Initiative.References
- X. Chen, H. A. de Silva, M. J. Pettenati, P. N. Rao, P. St George-Hyslop, A. D. Roses, Y. Xia, K. Horsburgh, K. Ueda and T. Saitoh, Genomics, 1995, 26, 425–427 CrossRef CAS.
- L. Maroteaux, J. T. Campanelli and R. H. Scheller, J. Neurosci., 1988, 8, 2804–2815 CAS.
- R. Jakes, M. G. Spillantini and M. Goedert, FEBS Lett., 1994, 345, 27–32 CrossRef CAS.
- A. Iwai, E. Masliah, M. Yoshimoto, N. Ge, L. Flanagan, H. A. de Silva, A. Kittel and T. Saitoh, Neuron, 1995, 14, 467–475 CrossRef.
- J. E. Galvin, V. M. Lee and J. Q. Trojanowski, Arch. Neurol., 2001, 58, 186–190 CrossRef CAS.
- V. N. Uversky, J. Neurochem., 2007, 103, 17–37 CAS.
- V. N. Uversky, Curr. Protein Pept. Sci., 2008, 9, 507–540 CrossRef CAS.
- J. Q. Trojanowski and V. M. Lee, NeuroToxicology, 2002, 23, 457–460 CrossRef CAS.
- P. H. Weinreb, W. Zhen, A. W. Poon, K. A. Conway and P. T. Lansbury Jr., Biochemistry, 1996, 35, 13709–13715 CrossRef CAS.
- V. N. Uversky, J. Li and A. L. Fink, J. Biol. Chem., 2001, 276, 10737–10744 CrossRef CAS.
- R. Bussell Jr. and D. Eliezer, J. Biol. Chem., 2001, 276, 45996–46003 CrossRef CAS.
- D. Eliezer, E. Kutluay, R. Bussell Jr. and G. Browne, J. Mol. Biol., 2001, 307, 1061–1073 CrossRef CAS.
- V. N. Uversky, J. Biomol. Struct. Dyn., 2003, 21, 211–234 CAS.
- C. M. Tanner, R. Ottman, S. M. Goldman, J. Ellenberg, P. Chan, R. Mayeux and J. W. Langston, JAMA, J. Am. Med. Assoc., 1999, 281, 341–346 Search PubMed.
- C. M. Tanner, Trends Neurosci., 1989, 12, 49–54 CrossRef CAS.
- J. M. Gorell, C. C. Johnson, B. A. Rybicki, E. L. Peterson, G. X. Kortsha, G. G. Brown and R. J. Richardson, Neurotoxicology, 1999, 20, 239–247 CAS.
- E. Altschuler, Med. Hypotheses, 1999, 53, 22–23 CrossRef CAS.
- J. Zayed, G. Campanella, J. C. Panisset, S. Ducic, P. Andre, H. Masson and M. Roy, Rev. Epidemiol. Sante. Publique, 1990, 38, 159–160 Search PubMed.
- J. Zayed, S. Ducic, G. Campanella, J. C. Panisset, P. Andre, H. Masson and M. Roy, Can. J. Neurol. Sci., 1990, 17, 286–291 Search PubMed.
- B. A. Rybicki, C. C. Johnson, J. Uman and J. M. Gorell, Movement Disord., 1993, 8, 87–92 Search PubMed.
- J. M. Gorell, C. C. Johnson, B. A. Rybicki, E. L. Peterson, G. X. Kortsha, G. G. Brown and R. J. Richardson, Neurology, 1997, 48, 650–658 CAS.
- E. C. Hirsch, J. P. Brandel, P. Galle, F. Javoy-Agid and Y. Agid, J. Neurochem., 1991, 56, 446–451 CrossRef CAS.
- P. Riederer, E. Sofic, W. D. Rausch, B. Schmidt, G. P. Reynolds, K. Jellinger and M. B. Youdim, J. Neurochem., 1989, 52, 515–520 CrossRef CAS.
- D. T. Dexter, A. Carayon, F. Javoy-Agid, Y. Agid, F. R. Wells, S. E. Daniel, A. J. Lees, P. Jenner and C. D. Marsden, Brain, 1991, 114(4), 1953–1975 CrossRef.
- D. T. Dexter, F. R. Wells, A. J. Lees, F. Agid, Y. Agid, P. Jenner and C. D. Marsden, J. Neurochem., 1989, 52, 1830–1836 CrossRef CAS.
- H. S. Boudreau, K. M. Krol, J. K. Eibl, L. D. Williams, J. P. Rossiter, V. P. Palace and G. M. Ross, Aquat. Toxicol., 2009, 92, 258–263 CrossRef CAS.
- E. Oestreicher, G. J. Sengstock, P. Riederer, C. W. Olanow, A. J. Dunn and G. W. Arendash, Brain Res., 1994, 660, 8–18 CrossRef CAS.
- E. Kienzl, L. Puchinger, K. Jellinger, W. Linert, H. Stachelberger and R. F. Jameson, J. Neurol. Sci., 1995, 134(Suppl), 69–78 CrossRef.
- E. B. Montgomery Jr., Toxicology, 1995, 97, 3–9 CrossRef CAS.
- A. I. Bush, Curr. Opin. Chem. Biol., 2000, 4, 184–191 CrossRef CAS.
- S. R. Paik, H. J. Shin, J. H. Lee, C. S. Chang and J. Kim, Biochem. J., 1999, 340(3), 821–828 CrossRef CAS.
- S. R. Paik, J. H. Lee, D. H. Kim, C. S. Chang and J. Kim, Arch. Biochem. Biophys., 1997, 344, 325–334 CrossRef CAS.
- V. N. Uversky, J. Li and A. L. Fink, J. Biol. Chem., 2001, 276, 44284–44296 CrossRef CAS.
- Y. Goto and A. L. Fink, Biochemistry, 1989, 28, 945–952 CrossRef CAS.
- Y. Goto, L. J. Calciano and A. L. Fink, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 573–577 CAS.
- Y. Goto, N. Takahashi and A. L. Fink, Biochemistry, 1990, 29, 3480–3488 CrossRef CAS.
- A. L. Fink, L. J. Calciano, Y. Goto, T. Kurotsu and D. R. Palleros, Biochemistry, 1994, 33, 12504–12511 CrossRef CAS.
- V. N. Uversky, A. S. Karnoup, D. J. Segel, S. Seshadri, S. Doniach and A. L. Fink, J. Mol. Biol., 1998, 278, 879–894 CrossRef CAS.
- A. Khan, A. E. Ashcroft, V. Higenell, O. V. Korchazhkina and C. Exley, J. Inorg. Biochem., 2005, 99, 1920–1927 CrossRef CAS.
- A. Binolfi, R. M. Rasia, C. W. Bertoncini, M. Ceolin, M. Zweckstetter, C. Griesinger, T. M. Jovin and C. O. Fernandez, J. Am. Chem. Soc., 2006, 128, 9893–9901 CrossRef CAS.
- Bharathi and K. S. Rao, Biochem. Biophys. Res. Commun., 2007, 359, 115–120 CrossRef.
- L. L. Liu and K. J. Franz, JBIC, J. Biol. Inorg. Chem., 2007, 12, 234–247 CrossRef CAS.
- V. N. Uversky, J. Li, K. Bower and A. L. Fink, NeuroToxicology, 2002, 23, 527–536 CrossRef CAS.
- C. Andre, T. T. Truong, J. F. Robert and Y. C. Guillaume, Electrophoresis, 2005, 26, 3256–3264 CrossRef CAS.
- L. A. Munishkina, A. L. Fink and V. N. Uversky, Protein Pept. Lett., 2008, 15, 1079–1085 CrossRef CAS.
- G. Yamin, C. B. Glaser, V. N. Uversky and A. L. Fink, J. Biol. Chem., 2003, 278, 27630–27635 CrossRef CAS.
- M. H. Polymeropoulos, C. Lavedan, E. Leroy, S. E. Ide, A. Dehejia, A. Dutra, B. Pike, H. Root, J. Rubenstein, R. Boyer, E. S. Stenroos, S. Chandrasekharappa, A. Athanassiadou, T. Papapetropoulos, W. G. Johnson, A. M. Lazzarini, R. C. Duvoisin, G. Di Iorio, L. I. Golbe and R. L. Nussbaum, Science, 1997, 276, 2045–2047 CrossRef CAS.
- M. G. Spillantini, M. L. Schmidt, V. M. Lee, J. Q. Trojanowski, R. Jakes and M. Goedert, Nature, 1997, 388, 839–840 CrossRef CAS.
- T. Ly and R. R. Julian, J. Am. Soc. Mass Spectrom., 2008, 19, 1663–1672 CrossRef CAS.
- M. P. Mattson, Aging Cell, 2007, 6, 337–350 CrossRef CAS.
- U. Wojda, E. Salinska and J. Kuznicki, IUBMB Life, 2008, 60, 575–590 Search PubMed.
- J. B. Schulz, Parkinsonism Relat. Disord., 2007, 13(Suppl 3), S306–308 CrossRef.
- A. Adamczyk and J. B. Strosznajder, NeuroReport, 2006, 17, 1883–1886 CrossRef CAS.
- I. Bezprozvanny and M. P. Mattson, Trends Neurosci., 2008, 31, 454–463 CrossRef CAS.
- M. S. Nielsen, H. Vorum, E. Lindersson and P. H. Jensen, J. Biol. Chem., 2001, 276, 22680–22684 CrossRef CAS.
- N. T. Hettiarachchi, A. Parker, M. L. Dallas, K. Pennington, C. C. Hung, H. A. Pearson, J. P. Boyle, P. Robinson and C. Peers, J. Neurochem., 2009, 111, 1192–1201 CrossRef CAS.
- E. V. Mosharov, K. E. Larsen, E. Kanter, K. A. Phillips, K. Wilson, Y. Schmitz, D. E. Krantz, K. Kobayashi, R. H. Edwards and D. Sulzer, Neuron, 2009, 62, 218–229 CrossRef CAS.
- R. Lowe, D. L. Pountney, P. H. Jensen, W. P. Gai and N. H. Voelcker, Protein Sci., 2004, 13, 3245–3252 CrossRef CAS.
- D. L. Pountney, R. Lowe, M. Quilty, J. C. Vickers, N. H. Voelcker and W. P. Gai, J. Neurochem., 2004, 90, 502–512 CrossRef CAS.
- D. L. Pountney, N. H. Voelcker and W. P. Gai, Neurotoxic. Res., 2005, 7, 59–67 Search PubMed.
- S. Tamamizu-Kato, M. G. Kosaraju, H. Kato, V. Raussens, J. M. Ruysschaert and V. Narayanaswami, Biochemistry, 2006, 45, 10947–10956 CrossRef CAS.
- E. Gaggelli, H. Kozlowski, D. Valensin and G. Valensin, Chem. Rev., 2006, 106, 1995–2044 CrossRef CAS.
- M. A. Lovell, J. D. Robertson, W. J. Teesdale, J. L. Campbell and W. R. Markesbery, J. Neurol. Sci., 1998, 158, 47–52 CrossRef CAS.
- E. Tiffany-Castiglion and Y. Qian, NeuroToxicology, 2001, 22, 577–592 CrossRef CAS.
- T. Grune, T. Reinheckel and K. J. Davies, J. Biol. Chem., 1996, 271, 15504–15509 CrossRef CAS.
- T. Kowalik-Jankowska, A. Rajewska, E. Jankowska, K. Wisniewska and Z. Grzonka, J. Inorg. Biochem., 2006, 100, 1623–1631 CrossRef CAS.
- T. Kowalik-Jankowska, A. Rajewska, E. Jankowska and Z. Grzonka, Dalton Trans., 2008, 832–838 RSC.
- S. R. Paik, H. J. Shin and J. H. Lee, Arch. Biochem. Biophys., 2000, 378, 269–277 CrossRef CAS.
- M. Sandal, F. Valle, I. Tessari, S. Mammi, E. Bergantino, F. Musiani, M. Brucale, L. Bubacco and B. Samori, PLoS Biol., 2008, 6, e6 CrossRef.
- R. M. Rasia, C. W. Bertoncini, D. Marsh, W. Hoyer, D. Cherny, M. Zweckstetter, C. Griesinger, T. M. Jovin and C. O. Fernandez, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 4294–4299 CrossRef CAS.
- J. C. Lee, H. B. Gray and J. R. Winkler, J. Am. Chem. Soc., 2008, 130, 6898–6899 CrossRef.
- S. C. Drew, S. L. Leong, C. L. Pham, D. J. Tew, C. L. Masters, L. A. Miles, R. Cappai and K. J. Barnham, J. Am. Chem. Soc., 2008, 130, 7766–7773 CrossRef.
- A. Binolfi, G. R. Lamberto, R. Duran, L. Quintanar, C. W. Bertoncini, J. M. Souza, C. Cervenansky, M. Zweckstetter, C. Griesinger and C. O. Fernandez, J. Am. Chem. Soc., 2008, 130, 11801–11812 CrossRef CAS.
- M. S. Jackson and J. C. Lee, Inorg. Chem., 2009, 48, 9303–9307 CrossRef CAS.
- Y. H. Sung, C. Rospigliosi and D. Eliezer, Biochim. Biophys. Acta, Proteins Proteomics, 2006, 1764, 5–12 CrossRef CAS.
- D. R. Brown, Biochem. Biophys. Res. Commun., 2009, 380, 377–381 CrossRef CAS.
- J. A. Wright, X. Wang and D. R. Brown, FASEB J., 2009, 23, 2384–2393 CrossRef CAS.
- M. B. Youdim, D. Ben-Shachar and P. Riederer, Eur. Neurol., 1991, 31(Suppl 1), 34–40 CrossRef.
- D. Berg, M. Gerlach, M. B. Youdim, K. L. Double, L. Zecca, P. Riederer and G. Becker, J. Neurochem., 2001, 79, 225–236 CrossRef CAS.
- M. E. Gotz, K. Double, M. Gerlach, M. B. Youdim and P. Riederer, Ann. N. Y. Acad. Sci., 2004, 1012, 193–208 CrossRef.
- S. V. Kalivendi, S. Cunningham, S. Kotamraju, J. Joseph, C. J. Hillard and B. Kalyanaraman, J. Biol. Chem., 2004, 279, 15240–15247 CrossRef CAS.
- S. Arawaka, Y. Saito, S. Murayama and H. Mori, Neurology, 1998, 51, 887–889 CAS.
- J. E. Galvin, B. Giasson, H. I. Hurtig, V. M. Lee and J. Q. Trojanowski, Am. J. Pathol., 2000, 157, 361–368 Search PubMed.
- M. Neumann, S. Adler, O. Schluter, E. Kremmer, R. Benecke and H. A. Kretzschmar, Acta Neuropathol., 2000, 100, 568–574 CrossRef CAS.
- J. E. Duda, V. M. Lee and J. Q. Trojanowski, J. Neurosci. Res., 2000, 61, 121–127 CrossRef CAS.
- B. Wolozin and N. Golts, Neuroscientist, 2002, 8, 22–32 CrossRef CAS.
- N. Ostrerova-Golts, L. Petrucelli, J. Hardy, J. M. Lee, M. Farer and B. Wolozin, J. Neurosci., 2000, 20, 6048–6054 CAS.
- B. J. Tabner, S. Turnbull, O. M. El-Agnaf and D. Allsop, Free Radical Biol. Med., 2002, 32, 1076–1083 CrossRef CAS.
- Bharathi, S. S. Indi and K. S. Rao, Neurosci. Lett., 2007, 424, 78–82 CrossRef.
- K. M. Danzer, D. Haasen, A. R. Karow, S. Moussaud, M. Habeck, A. Giese, H. Kretzschmar, B. Hengerer and M. Kostka, J. Neurosci., 2007, 27, 9220–9232 CrossRef CAS.
- M. Kostka, T. Hogen, K. M. Danzer, J. Levin, M. Habeck, A. Wirth, R. Wagner, C. G. Glabe, S. Finger, U. Heinzelmann, P. Garidel, W. Duan, C. A. Ross, H. Kretzschmar and A. Giese, J. Biol. Chem., 2008, 283, 10992–11003 CrossRef CAS.
- A. S. Hillmer, P. Putcha, J. Levin, T. Hogen, B. T. Hyman, H. Kretzschmar, P. J. McLean and A. Giese, Biochem. Biophys. Res. Commun., 2009 Search PubMed.
- A. Gaeta and R. C. Hider, Br. J. Pharmacol., 2005, 146, 1041–1059 CrossRef CAS.
- F. Molina-Holgado, R. C. Hider, A. Gaeta, R. Williams and P. Francis, BioMetals, 2007, 20, 639–654 Search PubMed.
- R. C. Hider, Y. Ma, F. Molina-Holgado, A. Gaeta and S. Roy, Biochem. Soc. Trans., 2008, 36, 1304–1308 CrossRef CAS.
- A. Alimonti, B. Bocca, A. Pino, F. Ruggieri, G. Forte and G. Sancesario, J. Trace Elem. Med. Biol., 2007, 21, 234–241 CrossRef CAS.
- P. Zuo, W. Qu, R. N. Cooper, R. A. Goyer, B. A. Diwan and M. P. Waalkes, Toxicol. Sci., 2009, 111, 100–108 CrossRef CAS.
- R. J. Uitti, A. H. Rajput, B. Rozdilsky, M. Bickis, T. Wollin and W. K. Yuen, Can. J. Neurol. Sci., 1989, 16, 310–314 Search PubMed.
- M. Yasui, T. Kihira and K. Ota, Neurotoxicology, 1992, 13, 593–600 CAS.
- B. Bocca, A. Alimonti, O. Senofonte, A. Pino, N. Violante, F. Petrucci, G. Sancesario and G. Forte, J. Neurol. Sci., 2006, 248, 23–30 CrossRef CAS.
- K. Oyanagi, E. Kawakami, K. Kikuchi-Horie, K. Ohara, K. Ogata, S. Takahama, M. Wada, T. Kihira and M. Yasui, Neuropathology, 2006, 26, 115–128 CrossRef.
- W. Hoyer, T. Antony, D. Cherny, G. Heim, T. M. Jovin and V. Subramaniam, J. Mol. Biol., 2002, 322, 383–393 CrossRef CAS.
- N. Golts, H. Snyder, M. Frasier, C. Theisler, P. Choi and B. Wolozin, J. Biol. Chem., 2002, 277, 16116–16123 CrossRef CAS.
- T. Hashimoto, K. Nishi, J. Nagasao, S. Tsuji and K. Oyanagi, Brain Res., 2008, 1197, 143–151 CrossRef CAS.
- J. Couper, Br. Ann. Med. Pharmacol., 1837, 1, 41–42 Search PubMed.
- G. C. Cotzias, Physiol. Rev., 1958, 38, 503–532 CAS.
- A. Barbeau, Neurotoxicology, 1984, 5, 13–35 CAS.
- D. B. Calne, N. S. Chu, C. C. Huang, C. S. Lu and W. Olanow, Neurology, 1994, 44, 1583–1586 CAS.
- M. L. Bleecker, Neurotoxicol. Teratol., 1988, 10, 475–478 CrossRef CAS.
- Y. Matsuoka, M. Vila, S. Lincoln, A. McCormack, M. Picciano, J. LaFrancois, X. Yu, D. Dickson, W. J. Langston, E. McGowan, M. Farrer, J. Hardy, K. Duff, S. Przedborski and D. A. Di Monte, Neurobiol. Dis., 2001, 8, 535–539 CrossRef CAS.
- C. Pifl, M. Khorchide, A. Kattinger, H. Reither, J. Hardy and O. Hornykiewicz, Neurosci. Lett., 2004, 354, 34–37 CrossRef CAS.
- H. Fujiwara, M. Hasegawa, N. Dohmae, A. Kawashima, E. Masliah, M. S. Goldberg, J. Shen, K. Takio and T. Iwatsubo, Nat. Cell Biol., 2002, 4, 160–164 CAS.
- L. L. Liu and K. J. Franz, J. Am. Chem. Soc., 2005, 127, 9662–9663 CrossRef CAS.
- T. D. Kim, S. R. Paik, C. H. Yang and J. Kim, Protein Sci., 2000, 9, 2489–2496 CrossRef CAS.
- M. J. Hokenson, V. N. Uversky, J. Goers, G. Yamin, L. A. Munishkina and A. L. Fink, Biochemistry, 2004, 43, 4621–4633 CrossRef CAS.
- M. Bisaglia, I. Tessari, S. Mammi and L. Bubacco, Neuromolecular Med., 2009 Search PubMed.
- R. Colby, J. Hulleman, S. Padalkar, J. C. Rochet and L. A. Stanciu, J. Nanosci. Nanotechnol., 2008, 8, 973–978 CAS.
| This journal is © The Royal Society of Chemistry 2010 |