Aromatic chloride to nitrile transformation: medicinal and synthetic chemistry†
Lyn H.
Jones
*,
Nicholas W.
Summerhill
*,
Nigel A.
Swain
and
James E.
Mills
Sandwich Chemistry, World Wide Medicinal Chemistry, Pfizer Ltd., Ramsgate Road, Sandwich, United Kingdom. E-mail: lyn.jones@pfizer.com; Fax: +44-1304-651821; Tel: +44-1304-644256
First published on 11th October 2010
Abstract
This review highlights the medicinal and synthetic chemistry relevance of replacing an aromatic chloride motif with an aromatic nitrile. We explore the desirable features that this transformation can bring in a drug design sense and the recent synthetic chemistry advances that effect this replacement in a single step.
Introduction
It is rare to find examples of ‘design rules’ in medicinal chemistry. We are often faced with situations where the incorporation of thematic learnings from previous medicinal chemistry programmes can lead to successful outcomes, but where specific ‘medicinal chemistry transformations’ often do not translate. This ambiguity adds to the challenges of effective drug discovery and suggests that a formulation of successful transformations, if possible, would increase our chances of success. This philosophy has been applied to the area of ‘bioisosteric’ replacements, where the key features of molecular interaction are mimicked in alternative structural motifs that can modify the overall medicinal chemistry profile of the molecule.1Aromatic chlorides are ubiquitous amongst high affinity protein binders and are often hits from high throughput screening efforts. There are presumably various reasons for this, including a high proportion of chloroaromatics in screening sets in the first place, possibly due to the synthetic expediency of their creation, and a variety of available binding modes, including strong hydrophobic interactions with the target protein driven by the lipophilicity of the residue (see below). Within Pfizer we have discovered through experience that replacing this moiety with an aromatic nitrile can often result in a compound with similar affinity/potency, although this transformation does not seem to have achieved widespread use, presumably in part due to the lack of publications that signpost this useful substitution. This review will explore the fundamental medicinal chemistry aspects of this switch and describe the aromatic nitrile containing drugs on the market. We will augment the analysis with data that support the generality of the conversion and the improvements seen in drug-type properties driven by modifications to the physicochemical properties of the resulting molecules. Moreover, the synthetic chemistry to effect this transformation in a single step has made significant progress in the last decade, making this a particularly apt time for review. Aromatic chlorides are in general inexpensive and much more readily available than bromides and iodides. Therefore, they are ideal starting materials for cross coupling reactions. Unfortunately, chlorides also constitute the most challenging substrates for these reactions, due to the high bond dissociation energy of the C(sp2)–Cl bond. For this reason the transition metal-catalysed cyanation reaction of aryl chlorides is an emerging but valuable field when aligned with the medicinal chemistry relevance of the conversion, and this review is anticipated to be of interest to a wide community of synthetic and medicinal chemists from both academia and industry.
Medicinal chemistry relevance and physicochemical properties
Drugs containing an aromatic nitrile
A search of Prous Science Integrity® (Thomson Reuters) revealed 12 launched drugs (10 structurally distinct compounds) that contain the aromatic nitrile moiety (Fig. 1), compared with 224 aromatic chloride-containing drugs. All but one (lodoxamide) of the aromatic nitriledrugs was found to possess structure–activity relationships (SARs) supporting a potent chloride partner, although the exact matched pair is not always available for nitrile containing drugs.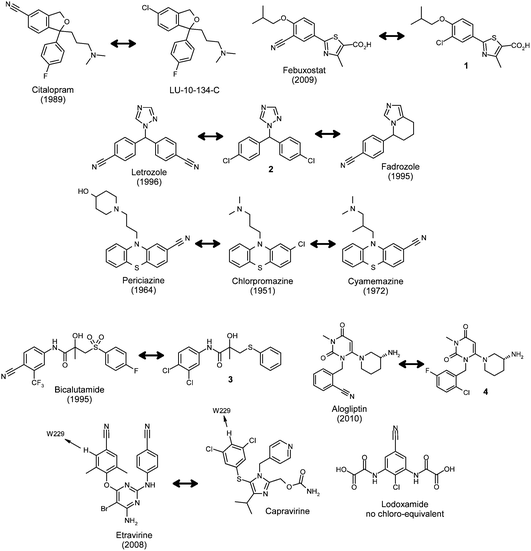 | ||
| Fig. 1 Aromatic nitrile-containing drugs and their corresponding chloride congeners (launch date in brackets). | ||
For the selective serotonin re-uptake inhibitor (SSRI) and antidepressantdrugcitalopram, there is a matching chloro-analogue with similar potency called LU-10-134-C (Fig. 1).2 Similarly, febuxostat, a non-purine selective xanthine oxidase inhibitor for the treatment of hyperuricemia, possesses a matching potent chlorinated derivative 1.3 Interestingly, a co-crystal structure of febuxostat with xanthine oxidase shows there to be a hydrogen bond between the nitrile moiety and the carboxamide residue of Asn768.4 In the case of the aromatase inhibitor and anticancer agent letrozole, both nitriles can derive from chlorine substituents (molecule 2).5 Additionally, structure–activity relationships map fadrozole to the chloro-equivalent molecule.6
Periciazine and cyamemazine are antipsychotic drugs from the phenothiazine class and although the exact matching pair where the nitrile is replaced by chlorine, are not known, both drugs derive from the prototypical chlorpromazine.7 The SAR of bicalutamide, an oral antiandrogen for the treatment of prostate cancer, suggests that a 4-CNgroup is superior to a chloro-substituent (compound 3).8 The crystal structure of bicalutamide with the antiandrogenreceptor ligand binding domain shows there to be a hydrogen bond between the nitrile moiety and the Arg752 residue.9
Similarly, the cyano-group in the dipeptidylpeptidase IV inhibitoralogliptin (for the oral treatment of type 2 diabetes) is able to hydrogen bond to Arg125 in the enzyme binding site.10 A corresponding chloro-derivative has also been described.11
The non-nucleosidereverse transcriptase inhibitor (NNRTI) and anti-HIV drugetravirine contains two aromatic nitrile moieties and structure–activity relationships suggest that at least one can be effectively replaced by a chlorine atom.12 A key structural component for resilience to drug resistant mutations in this class is an effective edge-to-face π-interaction with an immutable tryptophan residue in the allosteric reverse transcriptase (RT) binding pocket.13 Crystal structures of etravirine14 and capavirine15 with RT show that the hydrogen atom ortho- to the chlorine atoms in capravirine and nitrile moiety in etravirine is indeed interacting with the immutable Trp229 and both molecules possess excellent mutant viral profiles. We also have a matching pair of imidazole NNRTIs with excellent mutant profiles from our own work16 that supports these observations (compounds 5 and 6, Fig. 2). Additionally, the nitrile moiety in these ligands points through a hole in the enzyme towards solvent, an arrangement that is ideally suited to the shape and electrostatics of this residue (see below).17
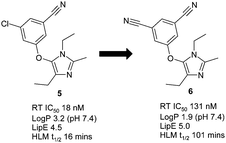 | ||
| Fig. 2 Potency, lipophilic efficiency (LipE) and metabolic stability of NNRTIs. | ||
High throughput screening hits
For the drugs highlighted above, it is likely that the preparation of the aromatic chlorides preceded that of the nitrile-containing drug in those corresponding chemotypes, judging from the relevant publications. This may result from the much higher likelihood of discovering an aryl chloride ‘hit’ or lead that could then persist through a drug discovery programme. For example, an analysis of 56 recent, diverse high throughput screens (HTS) at Pfizer (across enzyme, GPCR and ion channel classes) revealed that the proportion of actives (>40% activity at 10 μM) that contain an aromatic chloride is significantly higher than that for an aromatic nitrile (Fig. 3).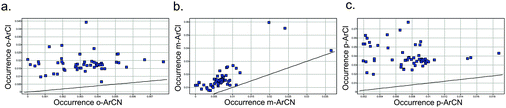 | ||
| Fig. 3 Proportion of HTS actives containing an active ArCl or ArCN, for a) ortho- b) meta- and c) para-derivatives (unity line shown). Each point represents an HTS. | ||
Additionally, the plots of the propensity of an aromatic chloride to yield an active versus an aromatic nitrile show that the chloride consistently outperforms the nitrile across the three substitutions (Fig. 4, also see Supporting Information for logarithmic plots).
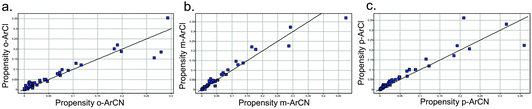 | ||
| Fig. 4 Propensity for an ArCl or ArCN to yield an active hit in an HTS for a) ortho- b) meta- and c) para-derivatives (unity line shown). Each point represents an HTS. | ||
Additionally, there is a significant difference in the number of commercially available functional building blocks for synthesis, that we refer to as monomers, containing aromatic chlorides versus nitriles (Table 1).18 It follows that an aromatic chloride hit or lead may later be converted in a medicinal chemistry sense to a nitrile residue through bespoke synthesis, but the lack of functional monomers, and large number of chloroaromatic monomers, may also help explain the greater number of drugs containing an aromatic chloride residue.
| Monomer | Chloride | Nitrile |
|---|---|---|
| RCO2H | 65705 | 3690 |
| RNH2 | 134063 | 7805 |
| ArOH | 36577 | 1538 |
| ArB(OR)2 | 52 | 7 |
| RCO2Me | 46506 | 1948 |
Electrostatic potential and dipole moment
Fig. 5 shows the electrostatic potential maps for chlorobenzene and benzonitrile which illustrate the similarity in the electron density of the benzene rings.19 The key differences reside as expected in the region of the substituent, with an overall negative potential at the –CN terminus, whilst an electropositive σ ‘hole’ is present on the outermost portion of the chlorine atom surface. This positive potential (present on chlorine, bromine and iodine) can interact non-covalently with the negative potential present on other atoms – this is referred to as ‘halogen bonding’. It may therefore be expected that if binding energies for a particular template are dominated through interactions with the benzene ring directly (e.g. π-stacking arrangements) then there may be little to choose between these groups regarding their affinity or potency, but that significant changes may result from interactions with the chlorine or nitrile substituents directly (see below).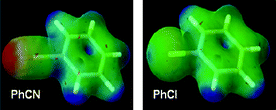 | ||
| Fig. 5 Electrostatic potential maps for chlorobenzene and benzonitrile (red = lowest, blue = highest electrostatic potential energy)19 | ||
The dipole moments of these groups are also substantially different: PhCl = 1.67 debye, PhCN = 4.44 debye and reflect the higher propensity for a dipole interaction with the aryl nitrile.20 Indeed, the high dipole moment and electronic polarisability of benzonitrile21 imparts interesting applications for this motif in linear and non-linear optical materials.22 These values would suggest that the preferred binding environments for the aromatic nitrile residue are likely to have a complementary polar nature, compared to that of the aryl chloride (see below). The hydrogen bonding to the aromatic nitriledrugsalogliptin, bicalutamide and febuxostat described above would appear to support this.
Binding site similarity and differences
Analysis of the polarity of the protein binding sub-pockets of 134 aromatic chloride and nitrile-containing paired ligands in the Protein Data Bank (PDB)23 and Pfizer internal co-crystal structures, and the degree to which these residues are buried in the protein, demonstrates some fundamental differences between these moieties (Fig. 6). Although this data set is relatively small, making firm conclusions difficult, it would appear that the preferred binding site of an aromatic chloride is more hydrophobic (highlighted by Fig. 6a) and buried (Fig. 6b) relative to that of an aromatic nitrile. This is in line with the propensity for the more lipophilic aromatic chloride motif to prefer hydrophobic regions of the binding protein (see discussion of lipophilicity below), which are likely to be more ‘buried’ due to the hydrophobic effect.24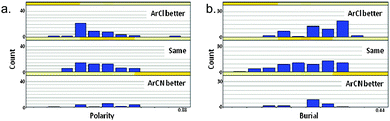 | ||
| Fig. 6 Sub-pockets predicted to contain ArCl and ArCN moieties (on the grounds that a co-crystallised ligand in the same pocket is at least 75% similar) were analysed using an in-house method to assess burial and polarity. For each such group, a regular array of 642 vectors is sprayed out from the centroid,25 each vector terminating at the closest point at which it meets the protein surface. The proportion of such vectors with a length < 5 Å gives a measure of sub-pocket burial and the proportion of vectors terminating at a polar protein atom gives a measure of the sub-pocket polarity. Each sub-pocket was defined as preferring ArCl, ArCN or neither using a 3-fold potency window. | ||
The nature of the sub-pocket in the immediate vicinity of the expected area of difference in electrostatic potential between the chlorine atom (positive electrostatic potential in this region) and nitrile residue (negative electrostatic potential in this region) was also explored. This was assessed by outputting the nature of the atom contributing the closest surface point to the vector passing through the C–Cl or C–CN bond) for para-substituted phenyl rings (for ortho- and meta-substituted groups, the precise location of the substituent in the pocket is ambiguous). Although the small numbers of observations makes it difficult to generate statistically significant results, it can be seen that sub-pockets containing H-bond acceptor, acid or π-system atoms in this position show a preference for Cl moieties and those sites that contain a donor or amphiprotic atom show more of a preference for CN moieties, again in line with the electrostatic potentials of these groups (Fig. 7).
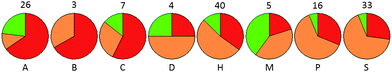 | ||
| Fig. 7 Relationship between preference and nature of binding pocket at the point (X) where the electrostatic potentials of ArCl and ArCN differ. Pockets were defined as either CN-preferring (green), Cl-preferring (red) or preferring neither (amber) using a 3-fold potency cutoff window. Pocket properties are described by the nature of the atom (A = acceptor, B=basic, C=acidic, D = donor, H=hydrophobic, M = amphiprotic, P = π-system atom, S=solvent-exposed i.e. no atom) closest to X. Number of observations giving rise to each pie chart is shown. | ||
Specifically relevant for the ArCl systems is halogen bonding (the non-covalent interaction between the electropositive σ-crown on the halogen and a Lewis base) and this has been reviewed regarding its importance in biologically-relevant interactions.26 Of particular interest in halogen bonding is the affinity of aromatic chlorides for π-systems, as illustrated by the serine protease factor Xainhibitors that interact in an edge-to-face manner through the chlorine atom of the inhibitor and Tyr228.27 Similarly, an analysis of the Cambridge Structural Database28 revealed 400 possible interactions between the chlorine atom on an aromatic ring and the π-system of a nearby aromatic residue (4.5 Å cut off).29
Since halogen bonding requires specific interactions with the electropositive crown on the chlorine atom, it is possible that a nitrile switch would result in an affinity reduction, particularly if a large component of the binding energy is derived from the halogen bond itself.27
Lipophilic efficiency
As we previously mentioned, the conversion of an aromatic chloride to a nitrile is a desirable modification of the drug molecule particularly when the potency is similar, or even improved (see analysis below). The main reason for this is that the lipophilicity of the molecule is significantly reduced, as reflected in the measured LogP (logarithm of 1-octanol/water partition coefficient) for chlorobenzene of 2.84,30 but only 1.5631 for benzonitrile (Rekker fragment32 for arylCN = −0.2; arylCl = 0.9; ΔLogP = 1.1). A reduction in lipophilicity can improve drug-type properties, such as improved metabolic stability,33 higher aqueous solubility34 and reduced toxicological outcomes35 (by reducing target promiscuity, amphiphilicity and detergent effects). For example, the NNRTI compounds shown in Fig. 2 illustrate the significant improvement in metabolic stability that can be achieved, as measured by half-life in human liver microsomes, on making the nitrile switch. Since lipophilicity can drive binding through non-specific hydrophobic collapse (a ΔLogP of 1 could translate to a ΔG difference of 1.4 kcal mol−1) we can normalise potency for lipophilicity by using the lipophilic efficiency parameter, LipE = −Log(IC50) – LogP.36 Calculating LipE for the NNRTIs in Fig. 2 shows the di-cyanobenzene derivative 6 outperforms the chloro-derivative 5, thus imparting a ‘true’ benefit to the molecule.37To further explore the LipE relevance of the aromatic chloride to nitrile switch, an analysis of the Targetbook database38 comparing the potency of 1074 distinctly matched ArCl-ArCN pairs across a multitude of target classes was undertaken. As expected, a direct comparison of the potency for matched pairs showed a spread of data from across enzyme, GPCR and ion channel targets (Fig. 8a). However upon normalising for lipophilicity, using calculated LogP, the plot showed a clear trend towards improved LipE for aromatic nitriles versus their corresponding aromatic chloride congener (Fig. 8b).39 Very similar results are obtained when the Pfizer data set is interrogated in this way (see ESI†).
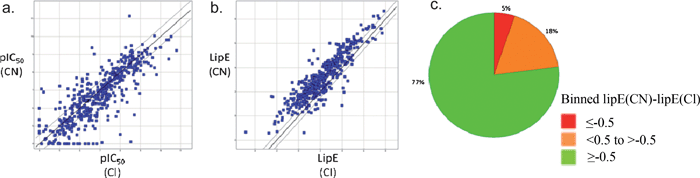 | ||
| Fig. 8 a) Potency of ArCNversusArCl; b) LipE of ArCNversusArCl (line of unity in bold, ± 3-fold ‘error’ IC50 lines either side, translating to a ± 0.5 shift in LipE); c) Pie chart plot of data in b) (green/amber/red = ArCN better/same/worse potency than ArCl). | ||
A pie chart analysis of these data (Fig. 8c) clearly shows an exceptionally high probabability of aryl nitriles displaying better lipophilic efficiency relative to the corresponding aryl chloride. These statistics make a compelling case for replacing an aromatic chloride with the nitrile derivative to improve the lipophilic efficiency, and by inference, the medicinal chemistry properties of the molecule.
An analysis of the Pfizer data set confirms the translation of improved LipE for the aromatic nitrile providing improved metabolic stability relative to the aromatic chloride. Fig. 9 shows that the aromatic nitrile out-performs the chloride congener as assessed by an improvement in metabolic stability in human liver microsomes.
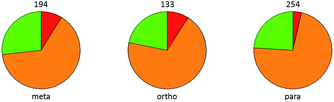 | ||
| Fig. 9 Assessment of metabolic stability in human liver microsomes for matched aromatic chloride-nitrile pairs. ArCN 2-fold more stable (green), ArCl 2-fold more stable (red) or the same stability (within 2-fold, amber). Number of observations giving rise to each pie chart is shown. | ||
Advances in the ArCl to ArCN synthetic transformation
Since the replacement of an aromatic chloride with an aromatic nitrile can impart desirable drug-type properties to the resulting molecule, synthetic methods that effect aromatic nitrile synthesis would facilitate their incorporation into drug designs. Several methods are available for the preparation of aromatic nitriles, including the Rosemund-von Braun reaction40 and diazotisation of anilines followed by the Sandmeyer reaction.41 On tonne scale the route of choice is often ammoxidation42 where very simple aryl nitriles are synthesised from the toluene derivative by reaction with oxygen and ammonia at 300–550 °C in the presence of heterogeneous fixed-bed catalysts. A more synthetically attractive method is the transition metal-catalysed cyanation of aryl halides, which has been reviewed in the literature.43 However, to date these reviews have focused almost exclusively on aryl bromide or iodide starting points.Aryl chlorides are in general much more readily available and inexpensive than bromides and iodides, and are ideal starting materials for cross-coupling reactions.44 Unfortunately, chlorides also constitute the most challenging substrates for these reactions, due to the high bond dissociation energy of the C(sp2)–Cl bond (C–Cl = 402, C–Br = 339, C–I = 272 kJ mol−1).45 For this reason the transition-metal catalysed cyanation reaction of aryl chlorides is an emerging field, and significant advances have been made in the last decade. The purpose of this synthetic review is to bring together the known methods of this transformation catalysed by palladium specifically (see below). The attractiveness of this synthetic transformation is highlighted by that of the medicinal chemistry described above, thus providing opportunities to improve drug-type properties in a single-step conversion.
Zinc metal-mediated cyanations
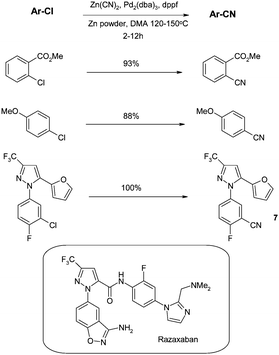
Prior to 2000, reports of transition-metal catalysed cyanations of aryl chlorides were infrequent. A few reports46 involved the use of Nicatalysts which have the disadvantage of being rather intolerant of air, moisture and a range of functionalities. The reports involving palladiumcatalysts were initially limited to those with strongly activated (heteroaryl) chlorides such as pyrazines and purines.47Therefore, there clearly remained a need for a more general procedure suitable for all chloroarenes. This need was first addressed by Jin and Confalone from the process group in DuPont, who published the first general set of conditions for the cyanation of aryl chlorides in 2000.48 4 mol% of the Pd complex was generated from tris(dibenzylideneacetone)dipalladium(0) (Pd2(dba)3) and 1,1′-bis(diphenyl-phosphino)ferrocine (dppf), zinc cyanide as the CN source, and catalyticZn powder at high temperature in dimethyl acetamide (DMA). The scope of the reaction was impressive as it was not limited to electron poor “activated” aromatic chlorides (see Scheme 1) and could be applied to the preparation of 7, an intermediate en route to the factor Xainhibitor razaxaban.49
The role of the zinc powder is intriguing and worthy of further comment. Early mechanistic studies by Takagi et al.50 showed that an excess of cyanide ions inhibits the catalytic cycle due to formation of inactive Pd(II) cyano compounds, that cannot be reduced to the catalytically active Pd(0) species; the Zn is key in reducing these compounds, thus allowing the cycle to continue. Cyanide ions can also react with Pd(0) giving species that cannot undergo oxidative addition to Ar–Cl as illustrated in red in Fig. 10. An analysis of the mechanisms by which palladium poisoning can occur in aromatic cyanations has been reported.51
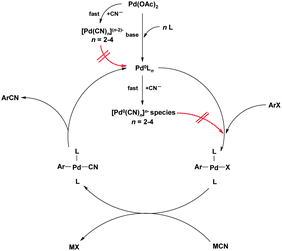
It is clear that the CN− anion concentration in the reaction mixture is crucial; too much and the catalyst is deactivated as above, too little and the transmetallation (and hence reductive elimination step) is retarded. These effects also lead to solvents playing a key role in the reaction due to variation in cyanide solubility and show the importance of having a relatively insoluble cyanide source. In the above case, zinc cyanide (originally introduced for cyanations by the Merck process group in 1994) is used, as its solubility in DMF (at 80 °C) is only 1.8 × 10−4 g mL−1.52 Using elegant NMR studies, Beller was able to isolate the oxidative addition complex and proved that this step is rate determining, the transmetallation and reductive elimination steps being much quicker.53
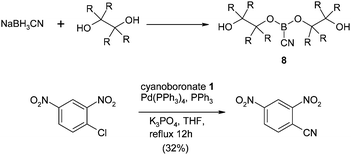
In the same year Jiang et al. described dialkylcyanoboronates 8 as a novel source of cyanide for the transformation.54 However, these reagents required pre-synthesis from a diol precursor and only reacted in moderate yield with activated chlorides as shown in Scheme 2.
Crown ether and amine additives
The main issue related to the procedure described above is the generation of zinc chloride waste, the disposal of which is problematic and expensive. To avoid this, Beller and co-workers turned to KCN as the nitrile source, using toluene as a sparingly soluble solvent.55 Using the knowledge that crown ethers have a positive effect on the reaction with other aryl halides,56 Beller experimented using 18-crown-6 with various bases and Pd ligands. This dramatically affected the yield on the model cyanation of 4-chlorobenzotrifluoride. Combination of these properties (chelation and base) into a single additive gave tetramethylethylenediamine (TMEDA) as the co-catalyst of choice, providing 91% yield for the model reaction compared with only 13% when this was absent (Scheme 3).55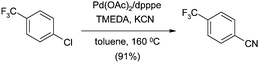
From a mechanistic perspective, the benefits of the crown ether or amine additive are two-fold: the concentration of CN− anions is controlled and the exchange of cyanide with other ligands is facilitated. Also, crucial to the success of the reaction is the use of chelating phosphine ligands, with 1,5-bis(diphenylphosphino)-pentane (dpppe) being optimal. However, under these conditions, non-activated chlorides such as simple chlorobenzene give poor yields. Therefore, Beller investigated alternative amine co-catalysts such as those shown in Fig. 3.531-1′-methylenediperidine (MDP) as an additive provided the best yields under these conditions (Fig. 11, Scheme 4).
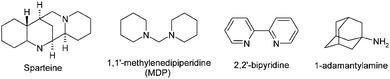 | ||
| Fig. 11 Amine additives used by Beller et al.53 | ||
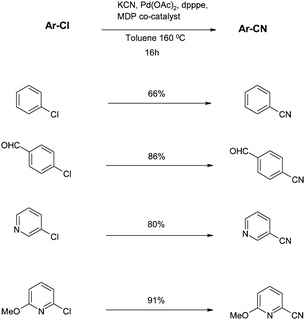
Noteworthy here is the smooth reaction of 4-chlorobenzaldehyde that may have been expected to undergo cyanide-catalysed benzoin condensation, reflecting the low cyanide concentration in the reaction. Also of interest are the cyanations of heteroaryl substrates, thus broadening the scope and relevance of this transformation.
Cyanohydrin as the nitrile source
Beller also investigated an alternative approach to controlling the concentration of CN− anions in the cyanation reaction.57 As previously mentioned, the main approach to this had been through judicious choice of solvent and/or additives such as crown ethers. This new approach instead used a liquid source of cyanide, acetone cyanohydrin which could be added in a controlled manner via a syringe pump. Although this method worked well for aryl bromides, allowing the reaction temperature to be dropped to 80 °C, the yields and temperatures with chlorides (shown in Scheme 5) are quite similar to those achieved with Beller's previous conditions.55 The main advantage of this protocol is the reduction of catalyst loading (down to 0.5–1%) but this has to be weighed against the operationally more complex use of a syringe pump combined with the toxicity of a liquid cyanide source. In a further publication Beller also used TMS cyanide as a liquid CN− source but the yields of the aromatic nitriles were inferior to those obtained using acetone cyanohydrin.58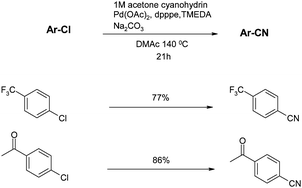 | ||
| Scheme 5 TMSCN cyanations (Beller et al.)57 | ||
Microwave cyanations
In two papers published in 2006, microwave technology had its first impact on the aromatic chloride nitrile transformation. In the first, Pitts and co-workers built on earlier work by Hallberg (showing that microwave irradiation increased the rate of cyanation of various bromides)59 and applied it to the last step of the synthesis of citalopram from the bromo precursor 9.60 Several parameters were varied, including ligand, additives, temperature and time, to provide Pd2(dba)3/Xantphos/TMEDA as the optimal conditions. These conditions also gave excellent yields for a narrow range of aryl chlorides (Scheme 6).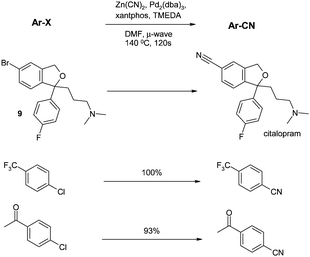 | ||
| Scheme 6 Microwave-assisted cyanations (Pitts et al.)60 | ||
In the second microwave paper, Chobanian and co-workers61 found that the key to a successful reaction in their case was the use of the Buchwald S-Phos ligand62 which was previously noted to possess “unprecedented activity” in the coupling of aryl chlorides with boronic acids and esters.63 Application of this ligand to cyanations under microwave conditions gave good results. For the first reaction shown below, use of dppf or dpppe gave no reaction at all, whereas S-Phos gave a 97% yield (Scheme 7).
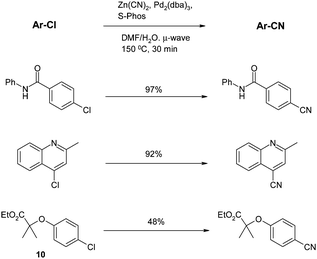 | ||
| Scheme 7 Microwave-assisted cyanations (Chobanion et al.)61 | ||
An important feature of this work is that the thermal conditions do not appear to give significantly different yields to the microwave under these conditions; even the length of reaction is comparable (30 min for MW, 1 h for thermal reaction). Also noteworthy is the poor yield in the reaction of the relatively electron rich chloride10, which, apart from an isolated example in the original Confalone work,48 is a common theme in the papers described thus far.
These conditions were recently used to convert the antitumour smoothened antagonists11 as a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of regioisomers into the nitrile containing derivatives 12 (Scheme 8),64 although X-Phos was the chosen ligand.65 Interestingly, the nitrile containing compounds appeared to have improved LipE, although it should be noted the biological activity was measured on the regioisomeric mixture.
:1 mixture of regioisomers into the nitrile containing derivatives 12 (Scheme 8),64 although X-Phos was the chosen ligand.65 Interestingly, the nitrile containing compounds appeared to have improved LipE, although it should be noted the biological activity was measured on the regioisomeric mixture.
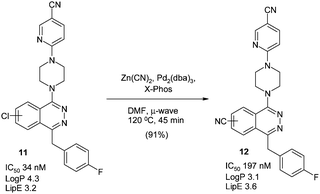 | ||
| Scheme 8 Smoothened antagonists and microwave-assisted cyanation64 | ||
Reducing reaction temperature
Another issue with the conditions described above is the high reaction temperature required for the transformation (120–160 °C), requiring the need for sealed tubes as solvents are used at temperatures exceeding their boiling points. The issues of high temperature and the lack of scope for electron rich chlorides were addressed by Littke and co-workers.66 All possible variables with the reaction were optimised and a new catalyst system of Pd(TFA)2 together with another Buchwald ligand (racemic 2-di-tert-butylphosphino-1,1′-binaphthyl)67 was proposed (catalyst system A). CatalyticZn was used as the additive and it was noted that on large scale it was essential to use fine zinc particles to ensure homogeneity. In a few cases it was found that Pd[P(t-Bu)3]2 was more effective (catalyst system B). Crucially these reactions proceed at a much more user-friendly temperature of 80–95 °C and work for a host of electron-rich substrates including the very challenging 2-chlorophenol and 4-chloroaniline. These conditions have by far the widest substrate scope seen, as illustrated in the number of examples shown in Scheme 9. Unprotected anilines, phenols, carboxylic acids and boronic acids are all tolerated. More impressively, a range of heterocycles can be cyanated, even those containing an unprotected NH.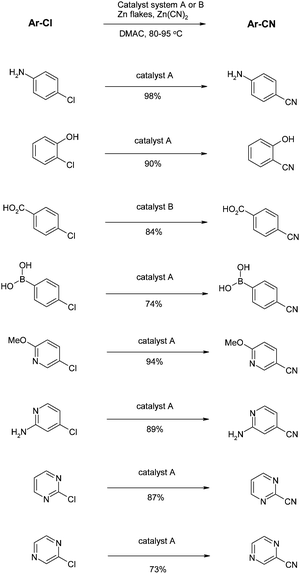 | ||
| Scheme 9 Reduced temperature cyanations (Littke et al.)66 | ||
Potassium ferrocyanide as the nitrile source
In the same year Beller addressed yet another issue with this reaction, namely the toxicity of the cyanide source. All cyanide reagents up to this point had been highly toxic, with LD50s (oral, human) all comparable with that of KCN (2.86 mg kg−1). Beller had previously introduced potassium ferrocyanide, K4[Fe(CN)6] as the most environmentally benign cyanation source known to date.68Potassium ferrocyanide is completely non-toxic and is even used in the food industry as an anti-agglutinating auxiliary for table salt - in fact, the LD50 is lower than that of NaCl. The reason for this is that it is an incredibly stable molecule, and will not liberate HCN, even when dissolved in dilute HCl and boiled. However, under the basic conditions typically used in cyanation reactions it will liberate CN− very slowly and is therefore an effective cyanating reagent, controlling the potentially detrimental high levels of the anion. Beller applied this reagent to the cyanation of activated and non activated chlorides.69 Using the cyanation of 2-chlorotoluene as a model reaction, the conditions were optimised and sterically hindered adamantyl phosphine ligands (such as the commercially available cataCXium®) gave the best results, and Pd(0) and Pd(II)catalysts were equally effective (Scheme 10).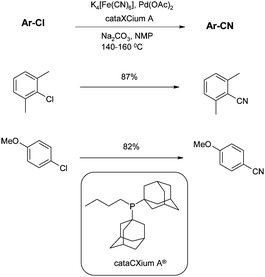 | ||
| Scheme 10 Potassium ferrocyanide and cataCXium-mediated cyanations (Beller et al.)69 | ||
The sterically hindered 2-chloro-1,3-dimethylbenzene and electronically deactivated 4-chloroanisole, both give good yields. Of particular note is the small amount of catalyst required, since reactions require only 0.2–0.5mol%. In fact, lower yields are obtained by raising the amount of catalyst; this is attributed to agglomeration of molecular palladium complexes to palladium black. A similar effect has been shown by de Vries et al. for Heck olefinations of aryl iodides.70 The low catalyst loading combined with the green credentials of the ferrocyanide suggest these conditions are ideal for larger, process scale reactions.
Finally, recent work by Beller and co-workers addresses some very demanding substrates that further expand the scope of the palladium-mediated cyanation reaction.71Learning from Littke's use of Pd(TFA)2, unprotected chloroanilines and chlorophenols can be cyanated with potassium ferrocyanide, provided the optimal ligand is chosen. However no single ligand satisfies all substrate examples, and reactions were optimised on a case-by-case basis. Additionally, the yields with these demanding substrates are only moderate (Scheme 11).
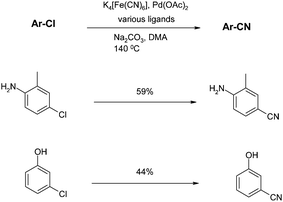 | ||
| Scheme 11 Potassium ferrocyanide-mediated cyanations (Beller et al.)71 | ||
Conclusions
The presence of an aromatic chloride in an HTS hit, or a lead molecule for further refinement into a clinical candidate, is a common occurrence. Indeed, it is likely that a significant share of the binding energy is due to the aryl chloride motif itself. It is therefore tempting to retain this function through a drug discovery programme to retain the desired affinity for the target protein. However, this review highlights the advantages that can be achieved through replacement of an aromatic chloride with an aromatic nitrile moiety. The statistics support the likelihood that there will be a resulting improvement in lipophilic efficiency and therefore switching an aromatic chloride for the nitrile congener is advisable at an early stage to assess the possibility of improved drug-type properties.Significant recent progress in the synthetic conversion of aromatic chlorides to nitriles has turned a difficult transformation into one that can be effected smoothly in one step and in high yield across a variety of substrates, that include sterically hindered and electronically deactivated aryl chlorides. These advances will provide the medicinal chemist with a wider synthetic repertoire of available chemistries to facilitate this desirable transformation and the opportunity to transform an aryl chloride drug-type molecule into the corresponding nitrile in a single step.
The ability to interconvert medicinal chemistry fragments using simple and mild synthetic techniques is a powerful and efficient strategy to provide structure–activity relationships. The available toolbox contains some very useful transformations (for example, phenol to trifluoromethoxybenzene,72 chloroaromatic to trifluoromethylbenzene,73benzene to fluorobenzene and various C–H activation chemistries74) that is augmented by the aryl chloride to nitrile conversion.
References
- C. A. Lipinski, Annu. Rep. Med. Chem., 1986, 21, 283 Search PubMed.
- A. J. Bigler, K. B. Bøgesø and A. Toft, Eur. J. Med. Chem, 1977, 12, 289 CAS.
- S. Kondo, H. Fukushima, M. Hasegawa, M. Tsuchimoto, I. Nagata, Y. Osada, K. Komoriya and H. Yamaguchi, WO9209279.
- K. Okamoto, B. Eger, T. Nishino, S. Kondo, E. F. Pai and T. Nishino, J. Biol. Chem., 2003, 278, 1848 CrossRef CAS (PDB code 1N5X).
- C. D. Jones, M. A. Winter, K. S. Hirsch, N. Stamm, H. M. Taylor, H. E. Holden, J. D. Davenport, E. V. Krumkalns and R. G. Suhr, J. Med. Chem., 1990, 33, 416 CrossRef CAS.
- M.-P. Lézé, M. Le Borgne, P. Pinson, A. Palusczak, M. Duflos, G. Le Baut and R. W. Hartmann, Bioorg. Med. Chem. Lett., 2006, 16, 1134 CrossRef CAS.
- T. A. Ban, Neuropsych. Disease Treat, 2007, 3, 495 Search PubMed.
- X. Zhang, G. Allan, T. Sbriscia, O. Linton, S. Lundeen and Z. Sui, Bioorg. Med. Chem. Lett., 2006, 16, 5763 CrossRef CAS.
- C. E. Bohl, W. Gao, D. D. Miller, C. E. Bell and J. T. Dalton, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 6201 CrossRef CAS (PDB code 1Z95).
- J. Feng, Z. Zhang, M. B. Wallace, J. A. Stafford, S. W. Kaldor, D. B. Kassel, M. Navre, L. Shi, R. J. Skene, T. Asakawa, K. Takeuchi, R. Xu, D. R. Webb and S. L. Gwaltney II, J. Med. Chem., 2007, 50, 2297 CrossRef CAS (PDB code 2ONC).
- J. Feng, S. L. Gwaltney, J. A. Stafford and Z. Zang, JP2005263780.
- J. Van Roey, M.-P. De Bethune and P. Stoffels, WO2003094920.
- (a) H. Pelemans, R. Esnouf, E. De Clercq and J. Balzarini, Mol. Pharm, 2000, 57, 954 CAS; (b) C. Fattorusso, S. Gemma, S. Butini, P. Huleatt, B. Catalanotti, M. Persico, M. De Angelis, I. Fiorini, V. Nacci, A. Ramunno, M. Rodriquez, G. Greco, E. Novellino, A. Bergamini, S. Marini, M. Coletta, G. Maga, S. Spadari and G. Campiani, J. Med. Chem., 2005, 48, 7153 CrossRef CAS.
- E. B. Lansdon, K. M. Brendza, M. Hung, R. Wang, S. Mukund, D. Jin, G. Birkus, N. Kutty and X. Liu, J. Med. Chem., 2010, 53, 4295 CrossRef CAS (PDB code 3MEC).
- J. Ren, C. Nichols, L. E. Bird, T. Fujiwara, H. Sugimoto, D. I. Stuart and D. K. Stammers, J. Biol. Chem., 2000, 275, 14316 CrossRef CAS (PDB code 1EP4).
- L. H. Jones, G. Allan, R. Corbau, D. Hay, D. S. Middleton, C. E. Mowbray, S. D. Newman, M. Perros, A. Randall, H. Vuong, R. Webster, M. Westby and D. Williams, ChemMedChem, 2008, 3, 1756 CrossRef CAS.
- L. H. Jones, G. Allan, O. Barba, C. Burt, R. Corbau, T. Dupont, T. Knöchel, S. Irving, D. S. Middleton, C. E. Mowbray, M. Perros, H. Ringrose, N. A. Swain, R. Webster, M. Westby and C. Phillips, J. Med. Chem., 2009, 52, 1219 CrossRef CAS.
- Scifinder® Version 2007.1, Copyright© American Chemical Society 2006 Search PubMed.
- Calculated in Linux environment using ab initio code Jaguar (Schrödinger), using Density Functional Theory (DFT) and the hybrid functional B3LYP (3–21G**).
- N. Pilpel, J. Am. Chem. Soc., 1955, 77, 2949 CrossRef CAS.
- Y. J. Alvarado, P. H. Labarca, N. Cubillan and A. Karam, ZNaturforsch, 2003, 58a, 68 Search PubMed.
- S. Howard, I. Fallis and D. Willock, Mol. Phys., 1999, 97, 913 CrossRef CAS.
- http://www.pdb.org/pdb/home/home.do .
- B. Matthews, Handbook of Proteins, 2007, 1, 45 Search PubMed.
- P. L. Chau and P. M. Dean, J. Mol. Graphics, 1987, 5, 97 CrossRef CAS.
- (a) Y. Lu, Y. Wang and W. Zhu, Phys. Chem. Chem. Phys., 2010, 12, 4543 RSC; (b) P. Auffinger, F. A. Hays, E. Westhof and P. S. Ho, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 16789 CrossRef CAS.
- H. Matter, M. Nazaré, S. Güssregen, D. W. Will, H. Schreuder, A. Bauer, M. Urmann, K. Ritter, M. Wagner and V. Wehner, Angew. Chem., Int. Ed., 2009, 48, 2911 CrossRef CAS.
- http://www.ccdc.cam.ac.uk/products/csd/ .
- S. Maignan, J. P. Guilloteau, Y. M. Choi-Sledeski, M. R. Becker, W. R. Ewing, H. W. Pauls, A. P. Spada and V. Mikol, J. Med. Chem., 2003, 46, 685 CrossRef CAS.
- J. Sangster Octanol-Water Partition Coefficients: Fundamentals and Physical Chemistry. New York: WIley; 1997 Search PubMed.
- A. Toulmin, J. M. Wood and P. W. Kenny, J. Med. Chem., 2008, 51, 3720 CrossRef CAS.
- R. Rekker, The Hydrophobic Fragmental Constant, Elsevier, Amsterdam, 1977 Search PubMed.
- H. Van Der Waterbreemd, D. A. Smith, K. Beaumont and D. K. Walker, J. Med. Chem., 2001, 44, 1313 CrossRef CAS.
- C. A. Lipinski, J. Pharmacol. Toxicol. Methods, 2000, 44, 235 CrossRef CAS.
- D. A. Price, J. Blagg, L. H. Jones, N. Greene and T. Wager, Expert Opin. Drug Metab. Toxicol., 2009, 5, 921 Search PubMed.
- (a) T. Ryckmans, M. P. Edwards, V. A. Horne, A. Correia, D. R. Owen, L. R. Thompson, I. Tran, M. F. Tutt and T. Young, Bioorg. Med. Chem. Lett., 2009, 19, 4406 CrossRef CAS; (b) P. D. Leeson and B. Springthorpe, Nat. Rev. Drug Discovery, 2007, 6, 881 CrossRef CAS.
- K. D. Freeman-Cook, C. Autry, G. Borzillo, D. Gordon, E. Barbacci-Tobin, V. Bernardo, D. Briere, T. Clark, M. Corbett, J. Jakubczak, S. Kakar, E. Knauth, B. Lippa, M. J. Luzzio, M. Mansour, G. Martinelli, M. Marx, K. Nelson, J. Pandit, F. Rajamohan, S. Robinson, C. Subramanyam, L. Wei, M. Wythes and J. Morris, J. Med. Chem., 2010, 53, 4615 CrossRef CAS.
- L. Harland and A. Gaulton, Expert Opin. Drug Discovery, 2009, 4, 857 Search PubMed.
- When ortho-, meta- and para-substituted aromatic substitutions are viewed separately, practically identical plots are obtained – see Supplementary Information.
- (a) K. W. Rosenmund and E. Struck, Chem. Ber., 1919, 52, 1749 CrossRef; (b) J. von Braun and E. Anton, Chem. Ber., 1934, 67, 1051.
- T. Sandmeyer, Ber. Dtsch. Chem. Ges., 1884, 17, 2650 CrossRef.
- A. Martin, N. V. Kalevaru, B. Lucke and J. Sans, Green Chem., 2002, 4, 481 RSC.
- (a) G. P. Ellis and T. M. Romney-Alexander, Chem. Rev., 1987, 87, 779 CrossRef CAS; (b) M. Sundermeier, A. Zapf and M. Beller, Eur. J. Inorg. Chem., 2003, 3513 CrossRef CAS.
- A. F. Littke and G. C. Fu, Angew. Chem., Int. Ed., 2002, 41, 4176 CrossRef CAS.
- V. V. Grushin and H. Alper, Chem. Rev., 1994, 94, 1047 CrossRef CAS.
- (a) L. Cassar, M. Foa, F. Montanari and G. P. Marinelli, J. Organomet. Chem., 1979, 173, 335 CrossRef CAS; (b) L. Cassar, J. Organomet. Chem., 1973, 54, C57–C58 CrossRef CAS.
- (a) Y. Akita, M. Shimazaki and A. Ohta, Synthesis, 1981, 974 CrossRef CAS; (b) L. L. Gundersen, Acta Chem. Scand., 1996, 50, 58 CrossRef CAS.
- F. Jin and P. N. Confalone, Tetrahedron Lett., 2000, 41, 3271 CrossRef CAS.
- M. L. Quan, P. Y. S. Lam, Q. Han, D. J. P. Pinto, M. Y. He, R. Li, C. D. Ellis, C. G. Clark, C. A. Teleha, J.-H. Sun, R. S. Alexander, S. Bai, J. M. Luettgen, R. M. Knabb, P. C. Wong and R. R. Wexler, J. Med. Chem., 2005, 48, 1729 CrossRef CAS.
- K. Takagi, T. Okamoto, Y. Sakakibara, A. Ohno, S. Oka and N. Hayama, Bull. Chem. Soc. Jpn., 1976, 49, 3177 CAS.
- S. Erhardt, V. V. Grushin, A. H. Kilpatrick, S. A. Macgregor, W. J. Marshall and D. C. Roe, J. Am. Chem. Soc., 2008, 130, 4828 CrossRef CAS.
- D. M. Tschaen, R. Desmond, A. O. King, M. C. Fortin, B. Pipik, S. King and T. R. Verhoeven, Synth. Commun., 1994, 24, 887 CAS.
- M. Sundermeier, A. Zapf, S. Mutyala, W. Baumann, J. Sans, S. Weiss and M. Beller, Chem.–Eur. J., 2003, 9, 1828 CrossRef CAS.
- B. Jiang, Y. Kan and A. Zhang, Tetrahedron, 2001, 57, 1581 CrossRef CAS.
- M. Sundermeier, A. Zapf, J. Sans and M. Beller, Tetrahedron Lett., 2001, 42, 6707 CrossRef CAS.
- T. Okano, M. Iwahara and J. Kiji, Synlett, 1998, 243 CAS.
- M. Sundermeier, A. Zapf and M. Beller, Angew. Chem., Int. Ed., 2003, 42, 1661 CrossRef CAS.
- M. Sundermeier, A. Zapf, S. Mutyala, A. Spannenberg and M. Beller, J. Organomet. Chem., 2003, 684, 50 CrossRef CAS.
- M. Alterman and A. J. Hallberg, J. Org. Chem., 2000, 65, 7984 CrossRef CAS.
- M. R. Pitts, P. McCormack and J. Whittall, Tetrahedron, 2006, 62, 4705 CrossRef CAS.
- H. R. Chobanian, B. P. Fors and L. S. Lin, Tetrahedron Lett., 2006, 47, 3303 CrossRef CAS.
- T. E. Barder, S. D. Walker, J. R. Martinelli and S. L. Buchwald, J. Am. Chem. Soc., 2005, 127, 4685 CrossRef CAS.
- R. A. Altman and S. L. Buchwald, Nat. Protoc., 2007, 2, 3115 Search PubMed.
- K. Miller-Moslin, S. Peukert, R. K. Jain, M. A. McEwan, R. Karki, L. Llamas, N. Yusuff, F. He, Y. Li, Y. Sun, M. Dai, L. Perez, W. Michael, T. Sheng, H. Lei, R. Zhang, J. Williams, A. Bourret, A. Ramamurthy, J. Yuan, R. Guo, M. Matsumoto, A. Vattay, W. Maniara, A. Amaral, M. Dorsch and J. F. Kelleher III, J. Med. Chem., 2009, 52, 3954 CrossRef CAS.
- X. Huang, K. W. Anderson, D. Zim, L. Jiang, A. Klapars and S. L. Buchwald, J. Am. Chem. Soc., 2003, 125, 6653 CrossRef CAS.
- A. Littke, M. Soumeillant, R. F. Kaltenbach, R. J. Cherney, C. M. Tarby and S. Kiau, Org. Lett., 2007, 9, 1711 CrossRef CAS.
- K. E. Torraca, S.-I. Kuwabe and S. L. Buchwald, J. Am. Chem. Soc., 2000, 122, 12907 CrossRef CAS.
- T. Schareina, A. Zapf and M. Beller, Chem. Commun., 2004, 1388 RSC.
- T. Schareina, A. Zapf, W. Magerlein, N. Muller and M. Beller, Tetrahedron Lett., 2007, 48, 1087 CrossRef CAS.
- A. H. M. De Vries, J. M. C. A. Mulders, J. H. M. Mommers, H. J. W. Henderickx and J. G. De Vries, Org. Lett., 2003, 5, 3285 CrossRef CAS.
- T. Schareina, R. Jackstell, T. Schulz, A. Zapf, A. Cotte, M. Gotta and M. Beller, Adv. Synth. Catal., 2009, 351, 643 CrossRef CAS.
- Y. Matsuya, K. Sasaki, H. Ochiai and H. Nemoto, Bioorg. Med. Chem., 2007, 15, 424 CrossRef CAS.
- E. J. Cho, T. D. Senecal, T. Kinzel, Y. Zhang, D. A. Watson and S. L. Buchwald, Science, 2010, 328, 1679 CrossRef CAS.
- T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary figures. See DOI: 10.1039/c0md00135j |
| This journal is © The Royal Society of Chemistry 2010 |