Strategies to prolong the plasma residence time of peptidedrugs
Lisa
Pollaro
and
Christian
Heinis
*
Institute of Chemical Sciences and Engineering, Ecole Polytechnique Fédérale de Lausanne, CH-1015 Lausanne, Switzerland. E-mail: christian.heinis@epfl.ch
First published on 21st October 2010
Abstract
Peptides are an attractive class of molecules for the development of therapeutics because they combine unique properties such as high binding affinity, excellent target specificity, low toxicity and a relatively small mass. However, the short in vivo half-life of peptides which is typically only a few minutes had hampered the development of a larger number of peptide leads into drugs. The main reasons for the fast elimination of peptides from the circulation are enzymatic degradation and/or fast renal clearance. To prolong the half-life of peptides, their proteolytic stability can be improved by chemical modification strategies and the rate of clearance can be reduced by conjugating the peptides to molecules that prevent their elimination through the kidney. In this article we review the latter class of strategies that aims at prolonging the in vivo plasma residence time of peptides. Techniques including peptidedrug linkage to large polymers, fusion to long-lived proteins such as albumin or the Fc fragment of immunoglobulin and conjugation to small molecule albumin-binding tags are discussed and the peptide-conjugate half-lives achieved are compared.
1. Introduction
Peptides are an attractive class of molecules that combine favourable properties of small molecule drugs and protein therapeutics.1 As small molecule drugs, peptides are accessible to chemical synthesis which allows the facile modulation of properties such as stability, solubility and others. Also as small molecules, peptides have shown to penetrate tissue, although generally to a lesser extent due to the slightly larger mass. As protein therapeutics, peptides typically bind to their targets with high affinity and target specificity and they are capable of disrupting protein–protein interactions. And although peptides are generally considered to be cell-impermeable, strategies are now available that facilitate their cellular uptake.2,3 However, despite their favourable properties, still relatively few peptides are approved as new drugs every year.1,2,4One of the main limitation of peptides as drug candidates has been the short half-life in the circulation which is caused mainly by proteolytic degradation and/or fast renal clearance.5 To prevent enzymatic peptide degradation, a whole range of strategies based on the chemical modification of the peptides have proved to be effective and generally applicable. These strategies include backbone modification, side chain substitution, use of D-amino acids, cyclisation, termini modification and others.5,6 To prevent the fast clearance of peptides by the renal route, strategies were developed that hinder the peptides from being filtered through the glomeruli of the kidney and they are subject of this review article. In a first section, the clearance mechanisms of peptides are briefly reviewed and in the further sections half-life extension strategies based on polymer fusion, linkage to long-lived proteins and albumin-binding tags are discussed. While many of the reviewed strategies were applied to both, peptide and proteindrugs,5,7,8 this article focuses on examples of peptidedrugs but exceptionally also refers to examples of proteins with extended half-lives.
2. Clearance mechanisms of peptidedrugs
Typically peptides are cleared from the bloodstream within minutes after intravenous administration.9 The major site of peptide elimination is the kidney, where glomerular ultrafiltrate is pressed out of the plasma. The glomeruli have a pore size of around 8 nm and molecules with a mass below 5 kDa such as peptides are filtered out completely. Larger polypeptides are less rapidly eliminated and proteins with a mass above a threshold which is between 50 and 70 kDa (corresponding to a diameter of approximately 90 Å in the case of globular proteins) are efficiently retained in the circulation. The renal clearance of some peptides and peptidedrugs is reduced due to binding to accessible membraneproteins or serum proteins. For example the cyclic peptidedrug ocreotide (brand name: Sandostatin) has a particularly long half-life of more than an hour due to binding to lipoproteins.10 Other, generally less relevant routes of peptide clearance, are endocytosis and degradation by the proteasome as well as clearance by the liver.9The lifetime of molecules in the circulation is expressed mostly with the plasma half-life, the time taken to half the concentration of molecules through elimination. When a molecule is administered by a rapid intravenous injection into the vascular system, a disappearance from blood almost always occurs in a biphasic fashion. In a first phase, the drug concentration rapidly declines because of distribution to peripheral tissue. In a second phase, the drug concentration declines less rapidly through elimination. The term ‘plasma half-life’ generally refers to the second phase and it is therefore also termed ‘elimination half-life’ or ‘terminal half-life’.11 In the following sections we compare mostly the ‘terminal half-life’ of peptides and peptide-conjugates. Since these values vary significantly in different species, the terminal half-lives determined in different animals should not be directly compared. Pharmacokinetics in humans may be predicted based on data from animal experiments by allometric scaling.12
3. Linking peptides to large polymers to prevent renal filtration
Based on the observation that proteins larger than 50 to 70 kDa are not rapidly filtered out by the kidney, peptides and small proteins have been conjugated to long hydrophilic synthetic or natural polymers, recombinant polymer mimetics or carbohydrates that increase their hydrodynamic volume.7,8 The polymer that had most widely been employed to increase the size of proteins is polyethyleneglycol (PEG), a molecule built of repeating ethylene oxide (CH2–CH2–O) units (Fig. 1; 1).13,14 One or multiple PEG chains ranging from approximately 5 to 40 kDa are linked to random or specific amino acid positions of peptides or proteins.15 Since one PEG monomer unit binds multiple water molecules, the hydrodynamic radius of a PEG chain is much larger than the one calculated from its molecular weight. The half-life extension of proteins and peptides through PEGylation varies but is typically between 10 and 100-fold. In addition to reducing the renal clearance, PEG was also found to stabilize peptides from proteolytic degradation, to increase their solubility and to decrease the immunogenicity in some cases. The first PEGylated protein, adenosine deaminase, entered the market in 199016 and the efficacy and safety of several other PEGylated proteins have since then been confirmed.13,17,18 Some of the best selling examples of PEGylated proteindrugs are pegfilgrastim (trade name: Neulasta), a human granulocyte colony-stimulating factor (G-CSF) linked via its N-terminus to a 20 kDa PEG unit with a 12-fold extended terminal half-life (42 h)19 or peginterferon α-2a (trade name: Pegasys), an interferon linked to a 40kDa branched PEG unit with a 20-fold prolonged half-life (60 h).20 Potential problems that were reported to occur with PEGylation strategies are the inhibition of protein function and the renal toxicity if applied over longer time periods.21 In contrast to proteins, none of the PEGylated peptides had yet reached the market but several are currently in development. For example peginesatide (brand name: Hematide), a conjugate of two erythropoietin mimetic peptides that are coupled to a PEG chain. The peptide portion of peginesatide is a completely novel sequence of 20 amino acids isolated by phage display from a random peptide library.22 The peginesatide has a half-life between 21.5 and 59.7 h in rat and monkey when applied intravenously23 and the drug is currently tested in a phase 3 trial. Another example of a PEGylated peptide is HM-3, a synthetic 17 amino acid integrin-binding peptide that showed anti-angiogenesis activity in rats. Attachment of a 10 kDa PEG unit to the N-terminus extended its terminal half-life in rats by a factor 6 (from 28 to 162 min).24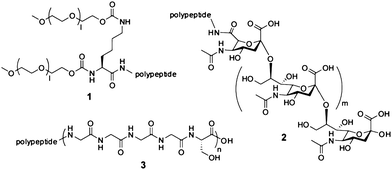 | ||
| Fig. 1 Natural and synthetic polymers. (1) Structure of a dimeric PEG unit. A 40 kDa polymer of this format with l = 420–510 was linked to the N-terminus of interferon α-2a.20 (2) Structure of polysialic acid. N-acetylneuraminic acid units are linked via α-(2 → 8) glycosidic linkages. (3) Structure of a homo-amino-acidpolymer with repeating Gly4Ser units. Polypeptides of this type with n = 40 and 80 were linked to an antibody Fab fragment.28 | ||
Alternatively to the conjugation with synthetic PEG polymers, proteins and peptides were linked to natural polymers such as polysialic acid (PSA) or hydroxyethylstarch (HES). Polysialic acidpolymers are negatively charged and highly hydrated carbohydrate chains (Fig. 1; 2) that have been linked to various proteins and peptides.25Conjugation of 22 or 39 kDa PSA units to insulin extended its glucose-lowering activity in mice suggesting a longer drug half-life (the half-lives of the conjugates were not determined).26Hydroxyethylstarch (HES) is a branched polymer composed of hydroxyethylated D-glucose units that has extensively been used as plasma volume expander. Conjugation of the safe polymer to proteins has shown to prolong their half-lives and suggests that the same effect should be found for peptides.27
Recently, recombinant PEG mimetics based on long unstructured peptides that are expressed as a genetic fusion with the protein or peptidedrug were developed.28–30 Skerra, A. and co-workers had developed a glycine-rich homo-aminoacidpolymer (HAP) composed of multiple pentapeptide (Gly4Ser) repeats (Fig. 1; 3). Linkage of such a 200 amino acid sequence to an antibody Fab fragment prolonged its terminal half-life from around 2 h to 6 h.28 Stemmer, P. C. and co-workers developed a longer unstructured polypeptide comprising 864 amino acids, called XTEN. A fusion polypeptide of XTEN and exendin-4, a 39-amino-acidpeptidehomologue of the glucagon-like peptide-1 (GLP-1) found in saliva of the Gila monster, had a terminal half-life of 12, 32 and 60 h in mouse, rat and monkey, respectively.29 This is 71, 65 and 125-fold longer than the half-life of extendin-4 alone (0.17, 0.49 and 0.48 h in mouse, rat and monkey, respectively) which is already used as a drug for the treatment of type 2 diabetes (exenatide, brand name: Byetta). Fusion of a shorter, 288 amino acid XTEN polymer to the peptide glucagon increased its half-life in monkey more than 53-fold to 9 h. The recombinant PEG mimetics have the advantage of not requiring a chemical conjugation step and of yielding a homogenous product. Also, as PSA and HES polymers, the polypeptides are biodegradable and no toxicity was so far been reported. Yet another unstructured polypeptide developed by Skerra, A. and co-workers is based on peptide sequences rich in proline, alanine and serine (PAS) and has shown to extend the half-life of fused polypeptides.31 None of the recombinant PEG mimetics had yet been tested in clinic and information about immunogenicity in humans is hence not available.
4. Covalent linkage to plasma proteins with long serum half-lives
The plasma proteins albumin and immunoglobulin display inherently long circulation half-lives of 19 (albumin) and up to 21 days (immunoglobulin; depending on the isotype) in humans. The large size of albumin (67 kDa) and immunoglobulin (e.g. 150 kDa for IgG) prevents fast renal clearance. Additionally, the elimination through pinocytosis by the vascular epithelium is reduced because the plasma proteins bind to neonatal Fcreceptor (FcRn), a MHC class I-related receptor, which recycles them back into circulation where they dissociate at the higher pH.32,33 Towards the extension of their half-life, peptides have been coupled to albumin and immunoglobulin fragments.Peptide drugs were linked to human serum albumin either through chemical conjugation or the recombinant expression of peptide-albumin fusion proteins (Table 1). When chemically linked, the peptides are preferentially tethered to the free thiol group of cysteine-34 on the surface of albumin. For example, exendin-4 was conjugated with a chemical linker via its carboxyterminal end to cysteine-34 of albumin. The exendin-4-albumin conjugate (CJC-1134-PC) has a half-life of around 8 days in humans and is currently tested in a phase 1/2 clinical study.34 Other peptidedrugs that have also been chemically linked to albumin with the same strategy are an analogue of GLP-1, an HIV entry inhibitor (C34 peptide) and insulin.35 Examples for a peptide-albumin conjugate that were generated by genetic fusion are albiglutide (formerly known as albugon) and albuBNP. Albiglutide is a 29 amino acid dipeptidyl-protease-IV-resistant GLP-1 analogue that is fused as a dimer to human albumin.36,37 The fusion peptide has a half-life of 6–8 days in humans when applied subcutaneously and is currently tested in a phase 3 clinical trial.38 AlbuBNP is a 32 amino acidB-type natriuretic peptide (BNP) fused to albumin. Linkage to albumin extended the half-life of the peptide from 3 min to 12–19 h (around 300-fold) in mice.39
| Drug/molecule | Peptide | Peptide conjugate | MW | Stage, company | t1/2 conjugate | Ref. |
|---|---|---|---|---|---|---|
| CJC-1134-PC | Exendin-4 (peptide hormone from a lizard), 39 aa | Albumin (conjugation to Cys34) | 71 kDa | Phase 1/2, Conjuchem | 8 daysa | 34 |
| Albiglutide (albugon) | Glucagon-like peptide 1 (GLP-1 analogue, 30 aa | Albumin (genetic fusion) | 70 kDa | Phase 3, GSK | 6–8 daysa,* | 36,38 |
| AlbuBNP | B-type natriuretic peptide, 32 aa | Albumin (genetic fusion) | 70 kDa | Preclinical, Human Genome Sciences | 12–19 hb | 39 |
| Romiplostim (AMG 531, brand name: Nplate) | Thrombopoietin analogue, 41 aa | Fc (genetic fusion) | 59 kDa | Launched 2008, Amgen | 3.5 daysa, * | 41–43 |
| AMG 386 (2xCon4[C]) | Angiopoietin-neutralizing peptide, 20 aa | Fc (genetic fusion) | 63 kDa | Phase 2, Amgen | 3.4–4.1 daysa | 44,45 |
| CNTO 528 | Erythropoietin mimetic, 20 aa | Fc (genetic fusion) | 59 kDa | Phase 1, Centocor | 5.9 daysa | 46 |
Immunoglobulins of the subclasses IgG1, IgG2 and IgG4 have the longest half-lives of 2–3 weeks and are therefore most suited as carriers of peptides to extend their circulation time. Most peptides were linked to only a portion of IgGs, the 50 kDa Fc fragment which is a homodimer of the IgG heavy chain domains CH2 and CH3.40 A first peptide drug-Fc conjugate, romiplostim (earlier known as AMG 531) has recently been approved for the treatment of chronic immune thrombocytopenia purpura (brand name: NPlate).41,42 In romiplostim two consecutive copies of a peptide mimetic of thrombopoietin are fused via a glycine linker N-terminally to each monomer of the Fc fragment. The average half-life of romiplostim is 3.5 days when applied subcutaneously.43 A range of other peptide-Fc domain fusions have been developed40 and are currently tested in clinical trials including Fc fusion with an angiopoietin-mimicking peptide (AMG 386),44,45 a B-lymphocyteinhibitor (AMG 623),40 an erythropoietin mimetic peptide of 20 amino acids that was isolated from phagepeptide libraries (CNTO 528, CNTO 530)46 and a GLP-1-analogue (CNTO 3649)40 (Table 1).
5. Conjugation to albumin-binding molecules
As an alternative to the covalent linkage with albumin, peptides have been conjugated to albumin-binding molecules to indirectly tether them to the long-lived serum protein (Table 2). Upon intravenous injection, the peptide conjugates bind to the highly abundant plasma albumin (45 mg mL−1, 600 μM) and are less rapidly filtered out of the blood stream. As albumin-binding molecules, small organic compounds, peptides and whole proteins have been used wherein the molecules with smaller molecular masses have several advantages. Firstly, the site-specific chemical linkage of tags to peptides and the subsequent purification of the conjugates are much facilitated with small molecules compared to large polymers or proteins. Secondly, the conjugation of small tags to peptides does not significantly increase their molecular size which can be advantageous to achieve good tissue penetration (e.g. diffusion into a tumour). If tissue diffusion should be achieved, a fine balance between the free and albumin-bound peptide conjugate must be maintained. Thirdly, the conjugation of peptides to small molecules yields a better activity to mass ratio and generally a more stable drug which together with the small size enables a wider choice of application routes.| Peptide | Tag | Affinity of tag for albumin (Kd) | t1/2peptide | t1/2 conjugate | Factor | Reference |
|---|---|---|---|---|---|---|
| Insulin analogue, 50 aa (conjugate: insulin detemir, NN304, brand name: Levemir) | Myristoyl (C14 fatty acid) | 4 μMa (myristoyl linked to insulin) | 1.5 ha,* | 5–7 ha,* | 4a,* | 47,58 |
| Glucagon-like peptide 1 (GLP-1) analogue, 31 aa (conjugate: liraglutide, NN2211, brand name: Victoza) | Palmitoyl (C16 fatty acid) | No data available | 1 ha,* | 11–15 ha,* | 15a,* | 48 |
| E76 (fVIIa-inhibiting peptide), 18 aa | Naphthalene acylsulfonamide | No data available | 7.6 minb | 24 minb | 3b | 49 |
| Fab D3H44 (TF-inhibiting antibody, 50 kDa) | Peptide SA21, 18 aa, cyclic | 467 nMa | 0.88 hb | 32.4 hb | 37b | 53 |
| 320 nMb | 0.4 hd | 10.4 hd | 26d | |||
| 266 nMc | ||||||
| E76 (fVIIa-inhibiting peptide), 18 aa | Diphenylcyclo-hexanol phosphate ester | No data available | 4.2 minb | 3.7 hb | 50b | 50 |
| scFv F8 (anti-EDA domain of fibronectin antibody fragment, homodimer, 51 kDa) | 6-(4-(4-iodophenyl)butanamido)hexanoate, ‘Albu’-tag | 3.2 μMa | 20–30 mind | 16.7 hd | 50e | 51,52 |
| 3.6 μMd |
Early trials to extend the half-life of peptides with small albumin-binding tags were performed with insulin. Saturated fatty acids that bind with weak affinities (10−4-10−5 M) to serum albumin were linked to the C-terminal lysine side chain (Lys B29) of an insulin analogue47 (Fig. 2; 4). A first molecule of this type, the insulin detemir (NN304) is a conjugate of an insulin analogue and a myristic acid (a fatty acid with 14 carbon atoms) and has been approved for clinical use in 2005 (brand name: Levemir). In patients, insulin detemir is usually applied subcutaneously as an oligomer (insulin forms a hexamer upon addition of zinc) to be slowly released and dissociated into biologically active monomers. The albumin-binding fatty acid tail further extends the half-life of the drug to 5–7 h. A second peptide that was acylated with a fatty acid is a GLP-1 analogue. The 31 amino acidpeptide was linked to palmitic acid (16 carbon atoms) and the resulting conjugate, liraglutide (NN2211), has recently (2009) reached the market (brand name: Victoza). The terminal half-life of subcutaneously applied liraglutide is 11–15 h which is around 15 times longer than the half-life of the peptide alone.48 Due to the poor solubility of the resulting conjugates, the strategy of peptideacylation with fatty acids is not generally applicable to all peptidedrugs.49
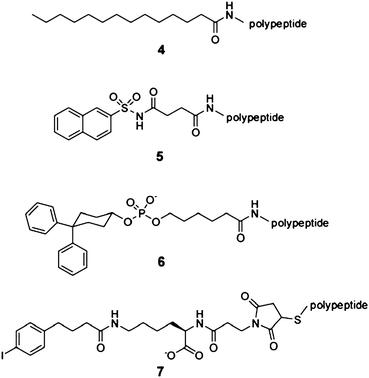 | ||
| Fig. 2 Chemical structures of albumin-binding small molecules. (4) Myristic acid,47 (5) naphthalene acylsulfonamide,49 (6) diphenylcyclohexanol phosphate ester,50 and (7) 6-(4-(4-iodophenyl) butanamido) hexanoate (‘Albu’-tag).51,52 | ||
Searching for small organic molecules that could extend the half-life of peptides without impairing their solubility, researchers at Genentech linked a small set of compounds with structural similarities to known albumin-binding drugs to a cyclic coagulation factor VIIa-inhibiting peptide. A naphthalene sulfoneacylamide moiety (Fig. 2; 5) extended the terminal half-life of the peptide from 7.6 to 30 min in rabbits.49 Along this line, a small albumin-binding moiety (a diphenylcyclohexanol phosphate ester; Fig. 2; 6) of a contrast agent used in magnetic resonance imaging, was linked to the same peptide and increased its half-life from 4.2 to 222 min in rabbits.50 Screening of larger libraries comprising more than 600 small organic molecules by Neri, D. and co-workers yielded several albumin-binding compounds including the 6-(4-(4-iodophenyl)butanamido hexanoate (‘Albu’-tag; Fig. 2; 7) with a Kd for albumin in the low micromolar range.51Conjugation of the tag to fluorescein and a scFvantibody fragment extended their half-lives in mice from 4.6 and 20–30 min to 495 and 1000 min, respectively.51,52 Although not tested yet with peptides, the ‘Albu’-tag is likely to extend equally well the plasma half-life of peptides. No information about the toxicity of the newer small molecule albumin-binding tags is yet known.
A number of albumin-binding molecules based on peptides and proteins have also been developed. For example the cyclic 18 amino acidpeptide SA21 isolated by phage selection53 binds to rat, rabbit and human albumin with dissociation constants of 266, 320 and 467 nM, respectively. Conjugation of SA21 to a Fab antibody fragment extended its terminal half-life from 53 min to 32.4 h in rabbits and from 24 min to 10.4 h in mice.53 Fab fragments fused to truncated peptide variants of SA21 had lower affinities for albumin and the weaker binding correlated with shorter half-lives in mouse, rat and rabbit.54 Examples for larger albumin-binding tags are domain antibodies (e.g. dAbh8: Kd mouse, rat and human albumin = 34–600 nM),55 Fab antibody fragments (e.g. bispecific F(ab′)2: Kd rat albumin = 26 nM)56 and albumin binding domains (e.g. ABD035: Kd = 50–500 fM).57 Due to their high albumin-binding affinities, all of these albumin binding proteins showed half-lives longer than 24 h in the various animals.
6. Conclusion
Several strategies are now available to extend the elimination half-life of peptides from minutes to several hours or even days. While some of these strategies have shown to be effective in preclinical tests, others have already been applied for the development of clinically approved therapeutics. Despite these successes, intensive research activities are ongoing to develop new or improved half-life prolongation strategies. For example, in the last few years multiple approaches based on biodegradable recombinant polymer mimetics were developed28–30 and new companies such as Amunix and XL-protein that exploit these new technologies were founded. Or much progress has recently been made towards the development of small-molecular-weight albumin-binding molecules. As discussed above, due to their small size, these tags offer several advantages over the large polymers and proteins. Different laboratories had developed ligands to albumin that have increasing affinities for the serum protein and show longer retention in the circulation.49–52 It can be expected that the engineering of half-life prolongation strategies will advance in the future with a similar pace and that new strategies and molecule tags will be developed to improve the therapeutic efficiency of existing and new peptidedrugs.Acknowledgements
The financial contribution from the Swiss National Science Foundation (SNSF Professorship PP00P3_123524/1 to C.H.) is gratefully acknowledged.References
- C. L. Stevenson, Curr. Pharm. Biotechnol., 2009, 10, 122–137 CAS.
- A. K. Sato, M. Viswanathan, R. B. Kent and C. R. Wood, Curr. Opin. Biotechnol., 2006, 17, 638–642 CrossRef CAS.
- L. D. Walensky, A. L. Kung, I. Escher, T. J. Malia, S. Barbuto, R. D. Wright, G. Wagner, G. L. Verdine and S. J. Korsmeyer, Science, 2004, 305, 1466–1470 CrossRef CAS.
- D. P. McGregor, Curr. Opin. Pharmacol., 2008, 8, 616–619 CrossRef CAS.
- M. Werle and A. Bernkop-Schnurch, Amino Acids, 2006, 30, 351–367 CrossRef CAS.
- C. Adessi and C. Soto, Curr. Med. Chem., 2002, 9, 963–978 CrossRef CAS.
- A. Constantinou, C. Chen and M. P. Deonarain, Biotechnol Lett, 32, pp. 609–622 Search PubMed.
- R. E. Kontermann, BioDrugs, 2009, 23, 93–109 Search PubMed.
- A. J. Banga, Therapeutic Peptides and Proteins, Taylor & Francis, New York, 2006 Search PubMed.
- P. Chanson, J. Timsit and A. G. Harris, Clin. Pharmacokinet., 1993, 25, 375–391 CrossRef CAS.
- P. L. Toutain and A. Bousquet-Melou, J. Vet. Pharmacol. Ther., 2004, 27, 427–439 CrossRef CAS.
- I. Mahmood, Adv. Drug Delivery Rev., 2007, 59, 1177–1192 CrossRef CAS.
- S. Jevsevar, M. Kunstelj and V. G. Porekar, Biotechnol J, 5, pp. 113–128 Search PubMed.
- F. M. Veronese and G. Pasut, Drug Discovery Today, 2005, 10, 1451–1458 CrossRef CAS.
- M. J. Roberts, M. D. Bentley and J. M. Harris, Adv. Drug Delivery Rev., 2002, 54, 459–476 CrossRef CAS.
- Y. Levy, M. S. Hershfield, C. Fernandez-Mejia, S. H. Polmar, D. Scudiery, M. Berger and R. U. Sorensen, J. Pediatr., 1988, 113, 312–317 CrossRef CAS.
- J. M. Harris and R. B. Chess, Nat. Rev. Drug Discovery, 2003, 2, 214–221 CrossRef CAS.
- G. Pasut and F. M. Veronese, Drugs Today, 2009, 45, 687–695 Search PubMed.
- G. Molineux, Curr. Pharm. Des., 2004, 10, 1235–1244 CrossRef CAS.
- S. J. Matthews and C. McCoy, Clin. Ther., 2004, 26, 991–1025 CrossRef CAS.
- A. Bendele, J. Seely, C. Richey, G. Sennello and G. Shopp, Toxicol. Sci., 1998, 42, 152–157 CrossRef CAS.
- N. C. Wrighton, F. X. Farrell, R. Chang, A. K. Kashyap, F. P. Barbone, L. S. Mulcahy, D. L. Johnson, R. W. Barrett, L. K. Jolliffe and W. J. Dower, Science, 1996, 273, 458–464 CrossRef CAS.
- Q. Fan, K. K. Leuther, C. P. Holmes, K. L. Fong, J. Zhang, S. Velkovska, M. J. Chen, R. B. Mortensen, K. Leu, J. M. Green, P. J. Schatz and K. W. Woodburn, Exp. Hematol., 2006, 34, 1303–1311 CrossRef CAS.
- K. Zhou, X. Zheng, H. M. Xu, J. Zhang, Y. Chen, T. Xi and T. Feng, Bioconjugate Chem., 2009, 20, 932–936 CrossRef CAS.
- G. Gregoriadis, S. Jain, I. Papaioannou and P. Laing, Int. J. Pharm., 2005, 300, 125–130 CrossRef CAS.
- S. Jain, D. H. Hreczuk-Hirst, B. McCormack, M. Mital, A. Epenetos, P. Laing and G. Gregoriadis, Biochim. Biophys. Acta, Gen. Subj., 2003, 1622, 42–49 Search PubMed.
- Fresenius Kabi, Hesylation, http://www.fresenius-kabi.com.
- M. Schlapschy, I. Theobald, H. Mack, M. Schottelius, H. J. Wester and A. Skerra, Protein Eng., Des. Sel., 2007, 20, 273–284 CrossRef CAS.
- V. Schellenberger, C. W. Wang, N. C. Geething, B. J. Spink, A. Campbell, W. To, M. D. Scholle, Y. Yin, Y. Yao, O. Bogin, J. L. Cleland, J. Silverman and W. P. Stemmer, Nat. Biotechnol., 2009, 27, 1186–1190 CrossRef CAS.
- P. Alvarez, C. A. Buscaglia and O. Campetella, J. Biol. Chem., 2003, 279, 3375–3381 CrossRef.
- XL-protein: PASylation, http://www.xl-protein.com.
- C. L. Anderson, C. Chaudhury, J. Kim, C. L. Bronson, M. A. Wani and S. Mohanty, Trends Immunol., 2006, 27, 343–348 CrossRef CAS.
- D. C. Roopenian and S. Akilesh, Nat. Rev. Immunol., 2007, 7, 715–725 CrossRef CAS.
- L. L. Baggio, Q. Huang, X. Cao and D. J. Drucker, Gastroenterology, 2008, 134, 1137–1147 CrossRef CAS.
- F. Kratz, J. Controlled Release, 2008, 132, 171–183 CrossRef CAS.
- L. L. Baggio, Q. Huang, T. J. Brown and D. J. Drucker, Diabetes, 2004, 53, 2492–2500 CrossRef CAS.
- M. A. Bush, J. E. Matthews, E. H. De Boever, R. L. Dobbins, R. J. Hodge, S. E. Walker, M. C. Holland, M. Gutierrez and M. W. Stewart, Diabetes, Obes. Metab., 2009, 11, 498–505 CrossRef CAS.
- J. E. Matthews, M. W. Stewart, E. H. De Boever, R. L. Dobbins, R. J. Hodge, S. E. Walker, M. C. Holland and M. A. Bush, J. Clin. Endocrinol. Metab., 2008, 93, 4810–4817 CrossRef CAS.
- W. Wang, Y. Ou and Y. Shi, Pharm. Res., 2004, 21, 2105–2111 CrossRef CAS.
- C. Huang, Curr. Opin. Biotechnol., 2009, 20, 692–699 CrossRef CAS.
- J. E. Frampton and K. A. Lyseng-Williamson, Drugs, 2009, 69, 307–317 CrossRef CAS.
- B. Wang, J. L. Nichol and J. T. Sullivan, Clin. Pharmacol. Ther., 2004, 76, 628–638 CrossRef CAS.
- D. Hubulashvili and N. Marzella, Pharmacy and Therapeutics, 2009, 34, 482–485 Search PubMed.
- A. C. Mita, C. H. Takimoto, M. Mita, A. Tolcher, K. Sankhala, J. Sarantopoulos, M. Valdivieso, L. Wood, E. Rasmussen, Y. N. Sun, Z. D. Zhong, M. B. Bass, N. Le and P. LoRusso, Clin Cancer Res, 16, pp. 3044–3056 Search PubMed.
- J. Oliner, H. Min, J. Leal, D. Yu, S. Rao, E. You, X. Tang, H. Kim, S. Meyer, S. J. Han, N. Hawkins, R. Rosenfeld, E. Davy, K. Graham, F. Jacobsen, S. Stevenson, J. Ho, Q. Chen, T. Hartmann, M. Michaels, M. Kelley, L. Li, K. Sitney, F. Martin, J. R. Sun, N. Zhang, J. Lu, J. Estrada, R. Kumar, A. Coxon, S. Kaufman, J. Pretorius, S. Scully, R. Cattley, M. Payton, S. Coats, L. Nguyen, B. Desilva, A. Ndifor, I. Hayward, R. Radinsky, T. Boone and R. Kendall, Cancer Cell, 2004, 6, 507–516 CrossRef CAS.
- J. J. Perez-Ruixo, W. Krzyzanski, E. Bouman-Thio, B. Miller, H. Jang, S. A. Bai, H. Zhou, J. Yohrling, A. Cohen, J. Burggraaf, K. Franson and H. M. Davis, Clin. Pharmacokinet., 2009, 48, 601–613 CrossRef CAS.
- P. Kurtzhals, S. Havelund, I. Jonassen, B. Kiehr, U. D. Larsen, U. Ribel and J. Markussen, Biochem J, 1995, 312(Pt 3), 725–731 CAS.
- B. Elbrond, G. Jakobsen, S. Larsen, H. Agerso, L. B. Jensen, P. Rolan, J. Sturis, V. Hatorp and M. Zdravkovic, Diabetes Care, 2002, 25, 1398–1404 CrossRef CAS.
- M. F. Koehler, K. Zobel, M. H. Beresini, L. D. Caris, D. Combs, B. D. Paasch and R. A. Lazarus, Bioorg. Med. Chem. Lett., 2002, 12, 2883–2886 CrossRef CAS.
- K. Zobel, M. F. Koehler, M. H. Beresini, L. D. Caris and D. Combs, Bioorg. Med. Chem. Lett., 2003, 13, 1513–1515 CrossRef CAS.
- C. E. Dumelin, S. Trussel, F. Buller, E. Trachsel, F. Bootz, Y. Zhang, L. Mannocci, S. C. Beck, M. Drumea-Mirancea, M. W. Seeliger, C. Baltes, T. Muggler, F. Kranz, M. Rudin, S. Melkko, J. Scheuermann and D. Neri, Angew. Chem., Int. Ed., 2008, 47, 3196–3201 CrossRef CAS.
- S. Trussel, C. Dumelin, K. Frey, A. Villa, F. Buller and D. Neri, Bioconjugate Chem., 2009, 20, 2286–2292 CrossRef CAS.
- M. S. Dennis, M. Zhang, Y. G. Meng, M. Kadkhodayan, D. Kirchhofer, D. Combs and L. A. Damico, J. Biol. Chem., 2002, 277, 35035–35043 CrossRef CAS.
- A. Nguyen, A. E. Reyes, 2nd, M. Zhang, P. McDonald, W. L. Wong, L. A. Damico and M. S. Dennis, Protein Eng., Des. Sel., 2006, 19, 291–297 CrossRef CAS.
- L. J. Holt, A. Basran, K. Jones, J. Chorlton, L. S. Jespers, N. D. Brewis and I. M. Tomlinson, Protein Eng., Des. Sel., 2008, 21, 283–288 CrossRef CAS.
- B. J. Smith, A. Popplewell, D. Athwal, A. P. Chapman, S. Heywood, S. M. West, B. Carrington, A. Nesbitt, A. D. Lawson, P. Antoniw, A. Eddelston and A. Suitters, Bioconjugate Chem., 2001, 12, 750–756 CrossRef CAS.
- A. Jonsson, J. Dogan, N. Herne, L. Abrahmsen and P. A. Nygren, Protein Eng., Des. Sel., 2008, 21, 515–527 CrossRef CAS.
- S. Havelund, A. Plum, U. Ribel, I. Jonassen, A. Volund, J. Markussen and P. Kurtzhals, Pharm. Res., 2004, 21, 1498–1504 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |