Boronic acids in medicinal chemistry: anticancer, antibacterial and antiviral applications
Paul C.
Trippier
* and
Christopher
McGuigan
Welsh School of Pharmacy, Cardiff University, Redwood Building, King Edward VII Avenue, Cardiff, CF10 3NB. E-mail: trippierp@Cardiff.ac.uk
First published on 20th August 2010
Abstract
In 2003, bortezomib, a first-in-class therapeutic, gained approval from the US Federal Drug Administration for the treatment of relapsed multiple myeloma and mantle cell lymphoma. Approval in the UK, for multiple myeloma, followed in 2006. Bortezomib contains a boronic acid, a functional group that has become increasingly more commonplace within the medicinal chemistry literature. The introduction of this drug has sparked a renewed interest in the investigation of boronic acids as drugs for a wide range of diseases. This review will guide the reader through the most recent developments in this field, by considering in turn, the biological target's amenable to the action of boronic acids.
1. Introduction
Boronic acids have been known in the literature since 1860,1 developing over the years into a widely studied area of chemistry. The ease of synthesis and stability of the group has led to its use in a variety of important synthetic reactions, from hydroboration2 (C–H, C–OH bonds), Suzuki–Miyaura coupling3 (C–C bonds) and the Chan–Lam coupling4 (C–N and C–O bonds). Initially, the use of boronic acids in medicinal chemistry was limited to formation of intermediates for synthesis towards, or further elaboration of, a lead compound. For reviews on synthetic uses of boronic acids see ref. 5–7.Boron Neutron Capture Therapy (BNCT) for the treatment of cancers, represents an important façade of boron in medicinal chemistry. For reviews on BNCT see ref. 8.
Boron is contained in a number of natural products isolated from bacteria, such as the polyether-macrolide antibiotic boromycin,9 aplasmomycin, an antibiotic that possess activity versus Gram positive bacteria, plasmodia10 and tartrolon B.11 These natural products illustrate that boron is tolerated in biological systems. Efforts towards the total synthesis of these natural products have resulted in just one reported12 synthesis. In addition, boron has been shown to have an important role in plant cell wall synthesis along with further implications for plant health and cell signalling.13
Boronic acids act as strong Lewis acids. Boron can form reversible covalent complexes with, amongst others, sugars and amino acids exhibiting vicinal (1,2) or (1,3) di-Lewis base donors (alcohols or amines). The pKa range of boronic acids is approx. 8–10, thereby, allowing them to remain protonated under physiological conditions. In addition, boronic acids are remarkably stable despite their high reactivity and have long been established as presenting a very low toxicology profile.14 Boronic acids are considered a bioisotere of carboxcylic acids, occupying the same period as carbon.
The ability of boronic acids to act as pH dependant, competitive enzyme inhibitors has been known for some time.15 In initial studies, aryboronic acids were found to be strong, competitive inhibitors of subtilisin and chymotrypsin, both, serine proteases. The boronic acid motif acts as a transition state analogue, forming both, hydrogen- and covalent bonds in the enzyme active site. Inhibition constants of chymotrypsin at pH 7, for simple phenylboronic acid, where calculated at 1.96 × 10−4 M, whilst the comparable, benzyl alcohol, lacking the boronic acid motif, displayed Ki = 1.0 × 10−2 M. Evidence that the inhibition activity is due solely, to the boronic acid. Slight changes in pH can result in release of the inhibitor from the active site, allowing for the use of boronic acids as biological probes, as well as, potential therapeutics.
The primary mode of action, involves the formation of a tetravalent boronate complex, from the initial trigonal boronic acid. This results from co-ordination of an electron-pair donating moiety16 (hydroxy, imidazole or carboxyl) of an active site amino acid residue, to the boronic acid, providing a complex that resembles the intermediate for amidolysis. Additional modes of action include the formation of covalent adducts with the serine or histidine residues of the active site.16
Boronic acids have received much attention17 as saccharide binders, for both probing biological systems and as potential indicators for identifying metabolites in the disease pathology of such afflictions as diabetes.18 Several excellent reviews exist in this area, in particular a review by Wang et al. detailing design considerations for the construction of such binding agents.19 Recently, the use of carbohydrate binding agents has been proposed as a potential therapeutic route to the treatment of infections attributed to viruses possessing a highly glycosylated envelope such as HIV.20
In a related area, phenylboronic acid has seen attention in the development of drug delivery systems. Kitano et al. described21 a glucose responsive polymer complex containing a phenylboronic acid moiety that acted as a novel drug delivery polymer.
This review will focus on the recent developments of boronic acids applied to antiviral, antibacterial and anticancer therapy. Each sub-section will examine the latest developments categorised by biological target. Previous reviews and texts have detailed the chemistry of boron analogues of biomolecules22 and the developments in the medicinal chemistry of boronic acids pre-200323 and pre-2005.5 Therefore, this review will consider developments from 2005 to present.
2. Anticancer applications
2.1 Tubulin polymerisation inhibitors
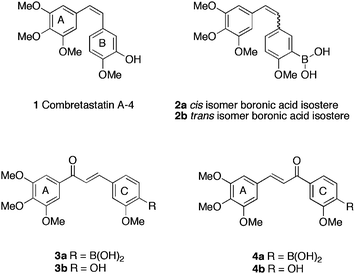
The targeting of the microtubule system of eukaryotic cells has been an anticancer strategy of great importance24 for many years. Tubulin plays a key role in cellular division, consisting of 50 kDa α- and β- subunits, previous studies have identified three unique binding sites for antimitotic action; the taxane, vinca alkaloid and cholchicine binding sites. Antimitotic compounds perturb the normal microtubule polymerisation/depolymerisation pathway by binding to one of the three possible binding sites.Kong et al.25 undertook studies on Combretastatin A-4261, a natural product exhibiting potent antimitotic activity when bound to the cholchicine binding site, however, high lipophillicity precluded clinical development. The team utilised boronic acid to mimic the C-ring hydroxy functionality, reasoning, through predicted interactions of the boronic acid with the tubulin heterodimer, that antimitotic activity would be retained or enhanced, whilst increasing the water solubility. As predicted by a docking model, 2a was identified as a potent inhibitor of the binding of cholchicine to β tubulin, but less so, than the parent natural product 1 (Table 1). The cis isomer 2a, exhibited greater inhibition ability (79% inhibition at 5 μM in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio with [3H] colchicine) of tubulin polymerisation, possessed higher cytotoxicity towards MCF-7 human breast cancer cells (IC50 17 ± 5 nM) and was active versus several human cancer cell lines in the National Cancer Institute (NCI) cancer cell panel27 (inhibiting growth in 43 of 53 cancer cell lines with GI50 <10 nM). Solubility measurements at physiological pH showed comparable data for 1 and 2a however, at pH 2, the cis boronic acid compound 2a was more soluble, by more than two-fold. The same group reported28 the synthesis and activity of two boronic acid containing chalcone analogues 3a and 4a of combretastatin A-4. Chalcone, flavokawain A, is a naturally occurring biphenyl, obtained from kava extracts, exhibiting anti-proliferative and anticancer activity.29 Literature precedent existed for the development of boronic acid based chalcone analogues as antitumour agents.30 Chalcone analogues 3a, b and 4a, b were designed as carbonyl-expanded analogues of chalcone. Evaluation (Table 1), identified 3a as a potent inhibitor of cancer in cell proliferation and angiogenesis, albeit, with modest improvements on the parent chalcone. Screening in the NCI human cancer cell screen highlighted activity below 10 nM in colon cancer HCT-15, CNS cancer SNB-19 and ovarian cancer SK-OV-3 cell lines. Boronic acid 4a displayed the same inactivity as its parent compound, suggestive, that the backbone structure, not the boronic acid, was the reason for this inactivity. The attenuated activity in inhibition of tubulin, yet potent cytotoxicity, suggests that 3a possess an alternate mechanism of action than its chalcone parent.
:1 ratio with [3H] colchicine) of tubulin polymerisation, possessed higher cytotoxicity towards MCF-7 human breast cancer cells (IC50 17 ± 5 nM) and was active versus several human cancer cell lines in the National Cancer Institute (NCI) cancer cell panel27 (inhibiting growth in 43 of 53 cancer cell lines with GI50 <10 nM). Solubility measurements at physiological pH showed comparable data for 1 and 2a however, at pH 2, the cis boronic acid compound 2a was more soluble, by more than two-fold. The same group reported28 the synthesis and activity of two boronic acid containing chalcone analogues 3a and 4a of combretastatin A-4. Chalcone, flavokawain A, is a naturally occurring biphenyl, obtained from kava extracts, exhibiting anti-proliferative and anticancer activity.29 Literature precedent existed for the development of boronic acid based chalcone analogues as antitumour agents.30 Chalcone analogues 3a, b and 4a, b were designed as carbonyl-expanded analogues of chalcone. Evaluation (Table 1), identified 3a as a potent inhibitor of cancer in cell proliferation and angiogenesis, albeit, with modest improvements on the parent chalcone. Screening in the NCI human cancer cell screen highlighted activity below 10 nM in colon cancer HCT-15, CNS cancer SNB-19 and ovarian cancer SK-OV-3 cell lines. Boronic acid 4a displayed the same inactivity as its parent compound, suggestive, that the backbone structure, not the boronic acid, was the reason for this inactivity. The attenuated activity in inhibition of tubulin, yet potent cytotoxicity, suggests that 3a possess an alternate mechanism of action than its chalcone parent.
| Compound | Tubulina IC50 ± SD (μM) | MCF-7b IC50 ± SD (μM) | A-10c EC50 (μM) |
|---|---|---|---|
| a Inhibition of tubulin polymerisation. Tubulin was at 10 μM. b Inhibition of growth of MCF-7 human breast carcinoma cells. c The percentage of cellular microtubule loss was estimated visually over a range of concentrations. Dose-response curves were generated and EC50 values calculated. The values represent the results of two independent experiments. | |||
| cis-2a | 1.5 ± 0.2 | 0.017 ± 0.005 | 0.058 |
| trans-2b | 7.8 ± 1.4 | 0.47 ± 0.14 | >15 |
| 3a | 31 ± 3.4 | 0.9 ± 0.18 | 16.5 |
| 3b | 2.6 ± 0.2 | 0.11 ± 0.024 | 0.98 |
| 4a | >40 | >2.5 | >15 |
| 4b | >40 | >2.5 | 18 |
| 1 | 2.0 ± 0.1 | 0.032 ± 0.021 | <0.025 |
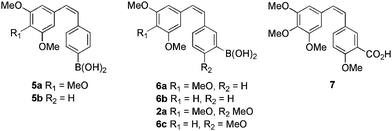
Nakamura et al.31 investigated a number of boronic acid containing cis-stilbenes (Table 2) for their apoptotic activity. Structures 5a, b containing a para- boronic acid moiety displayed some activity versus B-16 and 1–87 cell lines yet, were surpassed in activity when the boronic acid was moved to the meta- ring position. Notably, 2a was identified as the most potent structure, with IC50 values of 6.3 nM, 13 nM and 22 μM versus B-16, 1–87 cell lines and tubulin polymerisation inhibition respectively. Carboxylic acid derivative 7 (classically considered an isotere of boronic acid) displayed 10,000-fold less cell growth inhibition than its boronic acid analogue. The reason for this inactivity could lie with the Lewis acid character of the boronic acid. Compound 2a also displayed enhanced inhibitory activity in HT-29, AS49, OVCAR-4, OVCAR-5 and PC-3 cell lines. Apoptotic activity of 2a was 10-fold less than that of combrestatin A-4, inducing apoptosis in 24% of cells at >10−7 M. The lack of tubulin polymerisation inhibition, yet, retention of cytotoxicity led to the thesis that 2a, like 3a, acts on cellular processes other than inhibition of tubulin polymerisation.
| Compound | B-16 cellsa IC50 ± SD (μM)b | 1–87 cellsc IC50 ± SD (μM) | Tubulind IC50 ± SD (μM) |
|---|---|---|---|
| a Mouse B-16 melanoma cell line. b Values represent mean ± SD of an average of three experiments. c Human lung carcinoma 1–87 cell line. d IC50 is the drug concentration needed for 50% inhibition of tubulin polymerisation following incubation for 30 min at 37 °C. Values represent mean ± SD of an average of three experiments. | |||
| 5a | 2.6 ± 0.14 | 3.2 ± 0.12 | >100 |
| 5b | 12 ± 0.72 | 9.1 ± 0.3 | 79 ± 7.0 |
| 6a | 0.49 ± 0.019 | 2.1 ± 0.13 | >100 |
| 6b | 1.8 ± 0.13 | 2.0 ± 0.12 | >100 |
| 2a | 0.0063 ± 0.0015 | 0.013 ± 0.0044 | 22 ± 2.1 |
| 6d | 0.019 ± 0.0032 | 0.028 ± 0.0052 | 21 ± 2.6 |
| 7 | 160 ± 15 | 110 ± 13 | >100 |
| 1 | 0.0046 ± 0.0005 | 0.0085 ± 0.0011 | 1.8 ± 1.0 |
In summary, whilst the boronic acids discussed above were initially designed as inhibitors of tubulin polymerisation it is acknowledged that the high cytotoxic profiles do not correspond with the relatively weak tubulin polymerisation inhibition of the compounds. Thus a distinctly different method of action requires consideration.
2.2 Proteosome inhibitors
Bortezomib 832 (Fig. 1) was the first boronic acid containing compound and the first proteasome inhibitor33 to gain approval by the US FDA and European authorities. The compound is a first-in-class proteasome inhibitor possessing an α-amino boronic acid34 moiety linked to a dipeptide backbone. The α-amino boronic acid is very similar in structure and properties to the dipeptide chain upon which it is based, with, the replacement of the carboxyl group with the bioisoteric boronic acid the only difference. For a comprehensive review of α-amino boronic acids see ref. 34. Bortezomib was targeted for use in antineoplastic therapy versus multiple myeloma and possess a unique mode of action; inhibition of the 26S proteasome, (the key regulator of intracellular protein degradation, found in the nucleus and cytosol of all eukaryotic cells, forming part of the critical ubiquitin-proteasome system). The 26S proteasome is comprised of the barrel-like 20S proteasome capped at both ends by the regulatory 19S proteasome. Inhibition, results in disruption of homeostatic mechanisms within the cell that can lead to cell death.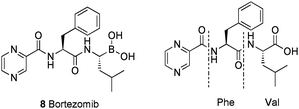 | ||
| Fig. 1 Structure of bortezomib in comparison with the corresponding α-amino acid dipeptide (for illustrative purposes only). | ||
The specific mode of action, is attributed to the boronic acid moiety complexing the threonine hydroxy group in the chymotrypsin-like active site of the 26S proteasome. Thereby, inhibiting the degradation of proteins essential for regulation of cell proliferation. This disruption also deregulates signalling molecules critical to binding between the myeloma cell and the bone marrow stromal cells, leading to growth arrest and apoptosis.35 Bortezomib has been reported36 as an effective and well tolerated treatment combined with other chemotherapeutics prior to bone marrow transplant. Development of proteasome inhibitors has received a great deal of attention in the literature, with many academic and industrial groups producing work in this area.
Kumar et al.37 synthesised a series of boronic-chalcone derivatives to investigate their activity against human breast cancer. Murine double minute 2 (MDM2) oncogene is amplified or overexpressed, in human breast cancer cells. MDM2 inhibits the tumor suppressor protein p53, via binding to its transactivation domain. Disruption of the p53/MDM2 complex may be effective in anticancer therapy.38 Chalcones have been shown to disrupt this complex, however, toxicity to both cancer and normal breast cells was observed. The group theorised that, at neutral pH, replacement of the carboxylic acid moiety with a boronic acid may result in a stronger salt bridge formation (due to the electron deficient nature of the boronic acid functionality and the electron donating nature of the amino group) with lysine51(K51) and/or lysine94(K94) of MDM2. Thereby, simultaneously breaking the salt bridge with glutamic acid25 (E25) of the MDM2 oncogene, which would introduce selectivity to cancerous cells expressing MDM2 over normal cells with no, or reduced, MDM2 expression. Several compounds synthesised (Fig. 2), inhibited the growth of human breast cancer cell lines, whilst possessing significantly less activity versus normal breast epithelial cells.
 | ||
| Fig. 2 Boronic-chalcone analogues designed to target breast cancer. | ||
The compounds inhibited37 cell growth in human breast cancer cell lines MDA-MB-435, MDA-MB-231 and wt-MCF7, whilst growth in normal breast epithelial cell lines MCF-10A and MCF-12A was less inhibited. Methylene boronic acid 9d, exhibited greatest potency with IC50 (μM) values of 18, 11, 9.5, 38, 100 respectively for the cell lines described above.
A series of bis-boronic acid analogues39 were synthesised (Table 3) to investigate the correlation of additional boronic acid functionalities with activity. The bis-boronic acids displayed a higher in vitro efficacy than the corresponding mono-boronic acids. Bis-chalcones 10a and 10c inhibited all cell lines at a low micromolar to nanomolar range, albeit, showing little selectivity over cancerous- and normal epithelial cell lines. Reduction of the ketone group to provide compounds 11a–b, resulted in entirely selective inhibition of cancer cell line growth. Bis-boronic acid 11a displayed complete selectivity for wt-MCF7 and MDA-MB-231 cell lines at 16.5 and 15 μM respectively. Greater efficacy for the bis-boronic acid than the dichloro- analogue 11b, highlighted the importance of the boronic acid moiety in the mode of action of these therapeutic candidates. In support of the anticipated mechanism of action, it was found that compound 10c showed marginally greater activity in a pair of isogenic HCT116 colon cancer cell lines that differed only in p53 expression. Cells with wild type p53 were more sensitive (IC50 = 2.8 μM) to the cytotoxic effects of 10c than p53 deficient cells (IC50 = 5.5 μM).
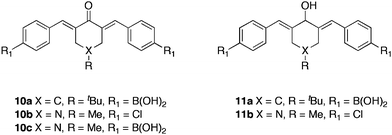
| Compound | wt-MCF7a (μM) | MDA-MB-231a (μM) | MCF-10Ab (μM) | MCF12Ab (μM) |
|---|---|---|---|---|
| a Human breast cancer cell lines. b Normal human breast epithelial cells. | ||||
| 10a | 1.3 ± 0.94 | 0.7 ± 0.25 | 7.0 ± 2.75 | 8.0 ± 1.77 |
| 10b | 2.0 ± 2.12 | 0.4 ± 0.14 | 2.5 ± 1.87 | 4.8 ± 2.40 |
| 10c | 0.35 ± 3.33 | 0.35 ± 1.05 | 1.5 ± 0.58 | 4.0 ± 2.42 |
| 11a | 16.5 ± 1.06 | 15 ± 1.06 | >25 | >25 |
| 11b | 20 ± 3.89 | 20.3 ± 2.12 | 22.3 ± 0.35 | >25 |
Further studies on boronic acid 10c corroborated the finding that greater cytotoxicity was observed in p53 enriched cells at relatively low concentrations (0.3–10 μM), compared to p53 deficient cells, which required a higher concentration (30 μM). This is consistent with the behavior of known40 proteasome inhibitors such as bortezomib. Biochemical analysis highlighted that 10c significantly inhibited the activity of the 20S proteasome in vitro (IC50 = approx. 1 μM). Compound 9e, also exhibited preferential activity towards wt-p53 cells and was observed to cause accumulation of p53 and p21 proteins, without disruption of the MDM2-p53 interaction.
It was found that 10c exhibited a greater than additive effect with ionizing radiation in p53 deficient cells (approx. 4-fold growth inhibition versus10c alone). The group postulated that in p53 deficient cells, accumulation of the ubiquitinated proteins, due to inhibition of the proteasome by a low concentration of 10c, is insufficient to trigger cell death in the absence of p53. However, such inhibition may have the effect of rendering the cells particularly vulnerable to ionizing radiation.
Boronic acid analogues of peptides have garnered much attention, building upon the success of bortezomib. Zhu et al.41 had previously developed a series of tripeptide aldehydes that possessed potent activity versus the 20S proteasome. In fact, the aldehyde moiety proved too active, with far too short a half life to convey therapeutic benefit. Searching for a possible isotere, the group turned to boronic acids. The boronic acid and aldehyde moieties were expected to possess the same mode of action, in reacting with the hydroxy group of the N-terminal threonine, of the β5 subunit to form a tetrahedral structure, a mechanism supported by X-ray diffraction.42
A wide range of tripeptides were synthesised with varying amino acids (phenylalanine, leucine, tyrosine, unnatural amino acids) and aryl constituents (Table 4). Building upon a prior report43 that the pinanediol ester of boronic acids exhibited almost identical activity and selectivity, these moieties were also included in the SAR library. Little significant difference between the activities of free boronic acids 12a–d and there corresponding esters 13a–d was found. Bulky and negative moieties at the R1 position, along with hydroxy substitution (leading to greater hydrogen bond interactions with the proteasome), increased the proteasome-inhibitory potency in line with theoretical models, whilst those at the R2 position deviated from these models. Most of the compounds screened41 showed activity at sub nanomolar concentration, with 13d identified as the most potent (IC50 = 0.079 nM), twice as active as bortezomib (IC50 = 0.161 nM). Furthermore, several of these boronic acids were evaluated in a series of human cancer cell lines, exhibiting particular activity in two haematological tumor cell lines, HL-60 (promyelocytic leukemia) and U266 (multiple myeloma). The most potent compound was identified as 13d, inhibiting both cell lines with IC50 values below 10 nM, outperforming bortezomib (IC50 = 6.9–12.2 nM).
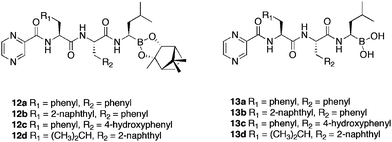
| Compound | CT-La | T-Lb | PGPHc | HL-60d | U266e |
|---|---|---|---|---|---|
| a Chymotrypsin-like proteasome subunit. b Trypsin-like proteasome subunit. c Post-glutamyl peptide hydrolysis proteasome subunit. d Promyelocytic leukemia cell line. e Multiple myeloma cell line. | |||||
| 12a | 0.40 ± 0.02 | >20 | 8.33 ± 2.49 | n.d. | n.d. |
| 13a | 0.55 ± 0.08 | >20 | 10.91 ± 3.21 | n.d. | n.d. |
| 12b | 0.22 ± 0.11 | >20 | >20 | n.d. | n.d. |
| 13b | 0.42 ± 0.07 | >20 | 4.43 ± 1.41 | 10 | 53.0 |
| 12c | 0.34 ± 0.03 | >20 | 5.39 ± 0.37 | n.d. | n.d. |
| 13c | 0.39 ± 0.04 | >20 | 4.01 ± 1.47 | 552.5 | 250.0 |
| 12d | 0.38 ± 0.14 | >20 | 3.91 ± 1.72 | n.d. | n.d. |
| 13d | 0.08 ± 0.01 | >20 | 3.63 ± 1.67 | 4.6 | 9.9 |
The peptide-like natural compound belactosin C4414, exhibits in vitro antiproliferative activity against HeLa S3 cells, with IC50 values of 200 μM and inhibitory activity towards the β5 chymotrypsin-like proteasome subunit at an IC50 of 0.21 μM. The mode of action is attributed to inhibition of the chymotrypsin-like activity of rabbit 20S proteasome, via nucleophilic ring opening of the β-lactam by the hydroxy group of the N-terminal threonine residue, forming a covalent bond, leading to irreversible binding. Nakamura et al.45 developed a series (Fig. 3) of belactosin C analogues containing a boronic acid moiety in place of the β-lactam, thus allowing reversible binding. Of the analogues synthesised, 16a displayed significant cell growth inhibition (IC50 = 0.35 μM) and showed the greatest inhibition of the chymotrypsin-like activity (IC50 = 0.28 μM), albeit, 10-fold lower than that reported for bortezomib (IC50 = 0.02 μM). The benzyl derivative 15a, displayed significant inhibition of chymotrypsin-like activity (IC50 = 0.54 μM), despite possessing relatively poor cell growth inhibition (IC50 = 5.11 μM). Both 16a and 15a were found to induce apoptosis of HeLa cells in over 10% of the population at 1–10 μM and 10 μM concentrations respectively, indicating, that inhibition of cell growth by boronate peptides is mediated by induction of G2/M cell cycle arrest and apoptosis.
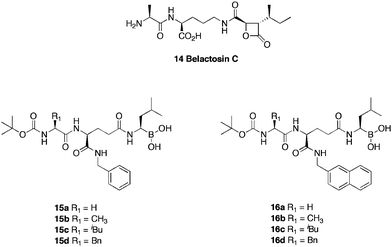 | ||
| Fig. 3 Structures of belactosin C and boronic analogues. | ||
Boronic acid derivatives of tyropeptin were synthesised by Watanabe et al.46 in response to a paper by Adams et al.47 where the enhancement in activity, 2,500-fold, of bortezomib, over the respective aldehyde was attributed to the slow dissociation of oxygen from boron, thereby, allowing a greater period of inhibition. Watanabe, targeted the boronic acid analogue of an aldehydic compound previously synthesised (17a), along with the corresponding leucine (17b) and phenylalanine (17c) amino acid analogues (Table 5), in an effort to clarify the mode of interaction between the S1 cavity of the proteasome and P1 side chains of the inhibitors. In addition, to evaluate the influence of the acyl group at the N-terminus of the proteasome on the inhibitory effect, the tripeptide HCl salts (18a–c) were also synthesised. Results highlighted compound 17c, with an aliphatic side chain, as a more potent inhibitor (IC50 = 0.0026 μM) of the chymotrypsin-like activity of the proteasome than bortezomib (IC50 = 0.024 μM). The lower activity of 17a–b suggests that the S1 pocket of the proteasome favours aliphatic side chains, over benzylic. Excision of the 1-naphthylacetyl group (18a–c), resulted in substantial loss of activity, whilst all of the tyropeptin analogue's analysed, displayed selectivity towards the chymotrypsin-like activity.
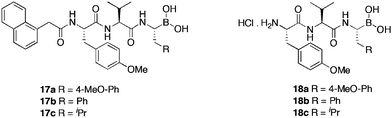
| Compound | CT-La IC50 μM | T-Lb IC50 μM | PGPHc IC50 μM |
|---|---|---|---|
| a Chymotrypsin-like proteasome subunit. b Trypsin-like proteasome subunit. c Post-glutamyl peptide hydrolysis proteasome subunit. | |||
| 17a | 0.0063 | 5.6 | >40 |
| 17b | 0.0044 | 4.6 | 7.4 |
| 17c | 0.0026 | 2.8 | 0.94 |
| 18a | 0.13 | 3.7 | 16 |
| 18b | 0.10 | 4.0 | 7.7 |
| 18c | 0.021 | 1.9 | 1.4 |
| bortezomib | 0.024 | >40 | 0.73 |
Zhu et al.48 set out to establish a comprehensive structure–activity relationship of dipeptidyl boronate proteasome inhibitors, based upon previous49 3D-QSAR studies of boronate dipeptides. Through the synthesis of >40 compounds of the type 19 (Fig. 4), varying P1−4 substituents, the group succeeded, not only, in establishing a detailed SAR for this kind of structure, but also identifying two highly potent proteasome inhibitor compounds 20a and 20b. The synthesised compounds were screened versus CT-L activity of human 20S proteasome. Several compounds were synthesised as the pinanediol protected boronic acid and evaluated without subsequent deprotection. This technique was based on the observation that both bortezomib and its pinanediol protected form, exhibit very similar data versus CT-L activity (IC50 = 1.92 nM and 1.90 nM respectively), suggestive of cleavage in vitro.
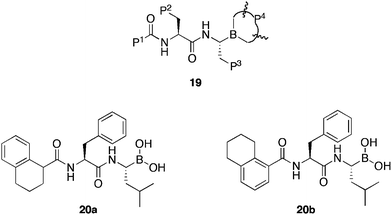 | ||
| Fig. 4 General structures of boronic acids synthesised for SAR studies 19 and active compounds 20a and 20b. | ||
Results of the SAR study provided data for substitutents at each of the four positions:
P1: Electronegative and aromatic substituents are required to improve inhibitory activity. S-isomers provide greater inhibitory activity than the corresponding R-isomers, whilst racemic mixtures provide a median value.
P2: Electronegative and aromatic groups, lacking 4-hydroxy functionality, with careful balancing of steric bulk, improved inhibitory activity.
P3: On one hand, bulky groups contribute more to the activity than smaller ones. Whilst, on the other, positive alkyl groups instead of negative aryl groups increase inhibitory activity. However, incorporation of an electron withdrawing group in the para position of a phenyl ring led to a significant increase in potency. In general, activities of the opposite isomer at P3 were reduced.
P4: Free boronic acid was very similar in activity to the corresponding pinanediol.
Free boronic acids 20a and 20b exhibited (IC50 = 1.90 nM), marginally better activity than bortezomib against CT-L proteasome inhibition, whilst, both displayed nearly the same potency as bortezomib for the two haematological tumor cell lines HL-60 (IC50 = 9.55 nM and 9.25 nM compared with 5.50 nM respectively) and U266 (IC50 = 2.28 nM and 2.95 nM compared with 2.45 nM respectively). In solid tumor cells, (IC50 < 100 nM) these compounds outperformed bortezomib. As a result of these studies, boronic acids 20a and 20b have been taken forward for further evaluation.
A further study,50 investigating the SAR of novel dipeptidyl boronic acids, composed of β-amino acids of the type 21 (Fig. 5) was conducted. Reasoning, that in several reported51 cases, the substitution of α-amino acids, with their β-amino acid counterparts in biologically active peptides, resulted in increased activity and enzymatic stability.
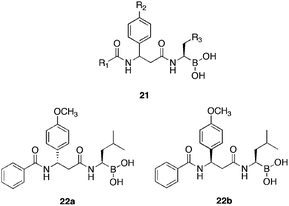 | ||
| Fig. 5 General 21 and active isomer 22a–b structures of dipeptidyl boronic acids comprised of β-amino acids. | ||
Detailed SAR studies were conducted to include;
R1: aromatic rings with or without N atoms.
R2: electron donating or electron withdrawing substituents.
R3: aliphatic or aromatic groups.
An order of activity for substituents at R1 was established, of the order; phenyl > nicotin-3-yl > pyrazin-2-yl > 5,6,7,8-tetrahydronaphalen-1-yl with substituents at R2 and R3 as the 4-methoxy and isopropyl groups respectively. Electron donating groups at R2, were found to exert greater activity than electron withdrawing groups. The R3 position showed a correlation with aliphatic moieties and increased potency. Overall, proving consistent with the previous discussion with regard to α-amino acids. Of the compounds surveyed, 22b (Fig. 5), was highlighted as the most active versus human 20S proteasome (IC50 = 9.6 nM), possessing 36-fold greater activity than its epimer 22a (IC50 = 350 nM), with the mixture displaying intermediate activity (IC50 = 12.5 nM). This result suggested a mode of inhibition requiring a stereoselective relationship between such compounds and the 20S proteasome, a theory further supported by docking studies. The three compounds were subjected to assay in 10 human cancer cells (2 haematologic and 8 solid tumour). As with the previous study, the two haematologic cells (HL-60 and U266) showed particular sensitivity, with 22b showing greatest activity (IC50 = 0.14 and 0.35 μM respectively). Furthermore, the two epimers were tested for toxicity in zebrafish embryos (a low cost, highly human similar tool for measuring drug response52) as a whole-organism screen, inducing no morphological changes at 1 and 10 μM concentrations over 24 h. A result, superior to that of bortezomib, indicting a reduced toxicity potential for 22b. Pharmacokinetic studies suggested a longer half-life and greater plasma exposure for boronic acid 22b, than for bortezomib.
The search for a second generation proteasome inhibitor is at an advanced stage with an orally bioavailable threonine-derived 20S human proteasome inhibitor CEP-18770 23 (Fig.. 6), identified as the most promising. This boronic acid was advanced from a study of dipeptides53 with varying aryl moieties. CEP-18770 exhibited excellent enzyme, cellular and antiprolifertion activities: IC50 = 3.8 nM versus 20S human erythrocyte chymotrypsin-like proteasome (comparable with bortezomib) and EC50 = 13.5 nM against the human leukemia cell line Molt-4 (bortezomib EC50 = 21 nM). Overall CEP-1877054 displayed a 2- to 11-fold lower cytotoxic potency compared to bortezomib, in solid tumor cell lines.
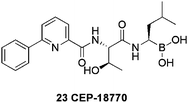 | ||
| Fig. 6 Structure of the orally active ‘second generation’ proteasome inhibitor CEP-18770. | ||
Thus, boronic acids, have established a niche in this crucial new target for cancer therapy, proteasome inhibition. Whilst further second generation proteasome inhibitors55 based on structural motifs other than boronic acids are under development, the field was developed through boronic acids and the functionality remains a mainstay in many compounds.
2.3 Enzyme and protein inhibitors
The ability of boronic acids to act as transition state analogues and to form both hydrogen- and covalent bonds in an enzyme's active site engenders this structural motif to inclusion in enzyme inhibitors.16,56 As previously discussed, the primary mode of action of these inhibitors centres around formation of a tetravalent boronate complex, via co-ordination of an electron-pair donating group, of a suitably disposed active site amino acid. Further modes of action have been posited in the literature17 and will be presented in the discussion of each individual inhibitor.Prostate specific antigen (PSA) is a serine protease produced in high quantities by prostate cancer cells. PSA is enzymatically active in the extracellular fluid surrounding prostate cancers and is not produced in any significant concentrations by any other tissue in the human male. This selectivity makes PSA a valid target for antineoplastic agents. Denmeade et al.57 exploited this target in the design of boronic acid inhibitors. Based on a peptide aldehyde substrate previously developed by the group,58 Denmeade designed a boronic acid isotere prodrug inhibitor of PSA. The most potent inhibitor identified, comprised the sequence Cbz-Ser-Ser-Lys-Leu-(boro)Leu, possessing a Ki for PSA of 65 nM, with a 60-fold higher Ki for chymotrypsin than for PSA (despite the ability of chymotrypsin to cleave peptide sequences after leucine residues). This peptide inhibitor was studied via GOLD docking calculations, illustrating the possibility of the deprotonated oxygen of the boronic acid, forming a hydrogen bond with the protonated nitrogen of the His41 residue, whilst, the remaining boronic hydroxy group is orientated towards Gly187 in the oxyanion hole. This adopted orientation is in close agreement to the high-resolution crystal structure of serine proteases crystallised with a boronic acid containing, transition state analogue.59 The inhibitor was assessed for cell growth inhibition in a panel of six PSA-positive and negative prostate cancer cell lines. At a concentration of 1 μM, all cell lines, irrespective of PSA production, had growth inhibited between 10–25%, suggestive that the mode of action may not be PSA dependant. The modeling studies provided insight into additional possible modifications that could optimize the inhibitor for in vivo applications, with studies currently underway.
Histone deacetylases (HDACs) control the acetylation of lysine residues in nucleosomal histones. Inhibition of HDAC causes histone hyperacetylation and leads to transcriptional activation of genes such as Gadd45, FAS and caspase 3, which are associated with growth arrest and apoptosis in tumor cells. Using the known inhibitor SAHA 25 as a scaffold and the proposed mechanism of action, Miyata et al.60 synthesised a family (Table 6) of α-boronic acid containing HDAC inhibitors, in which the acetamide of the acetylated lysine substrate is replaced with boronic acid. As the boron atom has a vacant p-orbital, it could be attacked by a water molecule in the active site. This boronic acid-water ‘ate’ complex could bind the zinc ion, forming two hydrogen bonds with Tyr303 and His140 which would lead to inhibition. Of the compounds assayed in 5 HDAC isoforms, (S)-24a–c proved the most potent (HDAC (nuclear extract) IC50 = 1.6, 1.4 and 0.92 respectively). The corresponding (R)-isomer being at least 10-fold less active. As the stereochemistry of the (S)-compounds is the same as natural lysine, it would suggest that these boronic acids act in the active site of HDAC. A molecular modeling study of the HDAC8/(S)-24c complex, suggested that the hydrated boronic acid interacts with the zinc ion, Tyr and His residue at the enzyme active site. Boronic acids 24a–c also displayed cancer cell growth arrest (MKN45 stomach cancer cells: GI50 = 9.3, 5.5, 3.5 μM respectively), equipotent with SAHA 25 (GI50 = 7.3 μM) (Table 6).
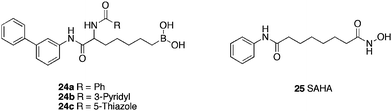
| Compound | HDAC IC50 (μM) | HDAC1 IC50 (μM) | HDAC2 IC50 (μM) | HDAC8 IC50 (μM) | HDAC6 IC50 (μM) |
|---|---|---|---|---|---|
| a Values are the mean of at least three experiments. b 38% inhibition at 10 μM. | |||||
| (S)-24a | 1.6 | >10b | 3.6 | >100 | 1.1 |
| (R)-24a | 17 | >100 | >100 | >100 | 24 |
| (S)-24b | 1.4 | 2.8 | 0.83 | 23 | 0.32 |
| (R)-24b | 15 | >100 | 99 | >100 | 14 |
| (S)-24c | 0.92 | 2.2 | 0.53 | 6.6 | 0.11 |
| (R)-24c | 24 | >100 | >100 | >100 | 5.6 |
| 25 | 0.28 | 0.35 | 0.25 | 1.7 | 0.028 |
Carbonic anhydrase (CA) exists as various isoforms, each with involvement in various critical physiological processes in humans. Their inhibition, largely with sulfonamides, provides many therapeutic effects such as diuretics, antiepileptics and antitumor drugs. The lack of selectivity for the inhibition of therapeutically significant isoforms in the many CAs present in organs, provides a challenge to design more potent and selective inhibitors. The boronic acid group was theorised to act as a zinc binding group, binding of the boronic acid group to the Zn(II) ion within the active site of CA, leading to a tetrahedral geometry of the metal ion and of the B(III) derivative, effectively inhibiting the enzyme. Based on a previous61 study, Winum et al.62 expanded the search for CA inhibitors to commercially available, substituted aryl, arylalkenyl and arylalkyl boronic acids. The synthesised compounds were assayed for activity versus four isoforms of human CA (hCA); hCA I and hCA II, targets for anti-glaucoma drugs and hCA IX and hCA XII (transmembrane, tumor-associated enzymes), targets for antineoplastic drugs. hCA IX and hCA XII were effectively inhibited by most of the aromatic boronic acids (Ki = 7.6–12.3 μM), whilst the arylalkenyl and arylalkyl boronic acids displayed less potent activity (Ki's of 34–531 μM). The study provided proof-of-concept that the boronic acid group represents a novel zinc-binding group, effective in the inhibition of therapeutically relevant CA inhibitors, thereby, lending the motif to further investigation in the design of CA isoform selective inhibitors.
Steroid sulfatase (STS) catalyses the hydrolytic desulfation of steroidal sulfates, such as oestrone sulfate to the corresponding steroids and inorganic sulfate. This sulfated species is considered to be the storage form of oestrone in breast tumor and other cells. Approximately 33% of breast tumors in post-menopausal women require stimulation by oestrogens for growth. Considerable interest in the inhibition of STS as a potential treatment for oestrogen-dependant breast cancer has arisen. Taylor et al.,63 synthesised several steroidal and non-steroidal boronic acids as potential STS inhibitors. The study found that the boronic acid moiety must be attached to a skeleton, closely resembling, the natural substrate. Oestrone boronic acid 26, possessing the boronic acid in the 3-position (Fig. 7), demonstrated competitive STS inhibition (Ki = 2.8 μM at pH 7.0 and 6.8 μM at pH 8.8). The significance of this activity at basic pH, provides, for the possibility of obtaining a crystal structure of the inhibitor in the active site of STS, since the enzyme is known to crystallize at pH 8.5. Boronic acid 27a shows potent, reversible, non-competitive STS inhibition activity (Ki = 250 nM), however, this activity is identical to the 3-hydroxy analogue 27b, suggestive that both compounds prefer to bind in a hydrophobic tunnel close to the entrance of the active site.
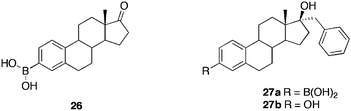 | ||
| Fig. 7 Structures of boronic acid and hydroxy STS inhibitors. | ||
Epidermal growth factor receptor (EGFR) tyrosine kinase plays an important role in cell proliferation. Deregulation of signaling pathway has been detected in many forms of tumors, possible due to uncontrolled activation of EGFR-mediated signaling from over expression of the receptor. Work on the inhibition of EGFR tyrosine kinase has been underway for some time, with 4-anilinoquinazolin based drugs64 in the clinic, as reversibly binding inhibitors, displaying therapeutic effects primarily against non-small-cell lung cancer (NSCLC). Whilst beneficial, these drugs eventually suffer from a build up of acquired resistance in the cancerous cell. Nakamura et al.65 sought to introduce a boronic acid moiety to the 6-position of the 4-anilinoquinazolin, reasoning, that such a motif would provide a covalent bond interaction between the boron atom and active site Cys797 or Asp800 residues, if positioned in the correct orientation, through the judicious design of a suitable linker. Of the compounds synthesised compounds 28–30 (Table 7) proved the most potent (IC50's = 0.20, 0.27 and 0.85 μM respectively) at reducing the EGF-mediated phosphorylation of EGFR tyrosine kinase and its downstream kinases. Cell growth being inhibited via arrest of G1 cell cycle, which, in turn, induced apoptosis. Of note is the prolonged activity of compound 30, active after 5 h post cell wash. These compounds also arrested growth in A431 cells (GI50 = 0.54, 0.68, 1.82 μM respectively). Boronic acids, due to their favorable steric and hydrophilic properties, displayed more potent inhibition, compared to the corresponding ester, as the boron moiety may be located in the hydrophilic region surrounded by Cys797 and Asp800 in the binding site. Docking studies suggested the formation of a covalent B–O bond with Asp800 and hydrogen bonds to Cys797 and Asp800 with the tetrahedral boron anion, which may be a cause of the prolonged inhibition by compound 30. These studies suggest that boron has potential in medicinal chemistry for the design of drugs with prolonged inhibition times through covalent bond formation.
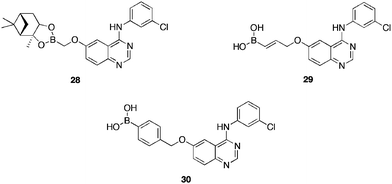
| Compound | EGFR inhibition at 1 μM/IC50 (μM)a |
|---|---|
| a The drug concentrations are those required to inhibit the phosphorylation of the poly(Glu:Tyr) substrate by 50% (IC50). They are determined from semilogarithmic dose-response plots, and represent the mean ± s.d. of triplicate samples. | |
| 28 | 65%/0.20 ± 002 |
| 29 | 63%/0.27 ± 0.02 |
| 30 | 55%/0.85 ± 0.03 |
| Tarceva63 | 82%/0.06 ± 0.07 |
The same group66 conducted studies on the inhibition of Hypoxia-inducible factors (HIF). These heterodimeric (α/β) transcriptional factors are major physiological stimuli for expression of angiogenesis factors (the formation of new blood vessels from existing host capillaries in tumor cells). The levels of HIF-1α are low under normal oxygen levels but increase in response to hypoxia, such as in the environment of a tumor. Thus, HIF-1α presents a valid antineoplastic target.
A series of boronic acid containing phenoxyacetanilide derivatives were synthesised as possible inhibitors. Carboranylphenoxyacetanilide 31 (Fig. 8) was identified as the most potent member of the series, inhibiting the hypoxia-induced HIF-1 transcriptional activity in HeLa cells with a IC50 = 0.74 μM. The compound suppressed hypoxia-induced HIF-1α accumulation and vascular endothelial growth factor mRNA, in a concentration-dependant manner, without affecting HIF-1α mRNA.
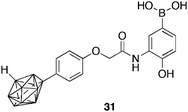 | ||
| Fig. 8 Structure of carboranylphenoxyacetanilide HIF-1α inhibitor 31. | ||
The novel compound contains a carboranyl (C2B10H14) moiety. Boron clusters, in particular carboranes, adopt an icosahedral geometry and are exceptionally hydrophobic, which allows for their potential67 use as hydrophobic pharmacophores that interact with target proteins. Boronic acid 31 represents a new class of HIF-1α inhibitors worthy of further exploration.
3 Antibacterial activity
3.1 β-lactamase inhibitors
Bacteria developing resistance to β-lactam antibiotics has been established as a major threat to human health in the twenty-first century, with the rise of the media dubbed ‘superbug’ infections (of which methicillin-resistant Staphylococcus aureus (MRSA) has achieved notoriety) responsible for approx. 18,000 deaths a year in the US alone.68The principle mechanism of resistance to penicillins and cephalosporins is the production of β-lactamase enzymes by bacteria. β-lactamases,69 are part of the serine-amidase class of enzymes, that have been extensively mapped and studied, with the primary mode of action being hydrolysis of the β-lactam ring of the antibiotic, leading to ring opening and inactivation of the drug.
Two strategies have evolved to counter this resistance; the synthesis of new β-lactam antibiotics that can resist enzymatic hydrolysis and subsequent deactivation; combined with the development of β-lactamase inhibitors, to be delivered in concert with the current arsenal of antibiotics.
In the latter strategy, boronic acids acting as transition state inhibitors, have proven the most effective reversible inhibitors,70 exhibiting a greater intracellular efficacy than phosphonate inhibitors. The boron atom is known to act as an electrophile that mimics the carbonyl functionality of the β-lactam, the boron forms a tetrahedral geometry with the catalytic serine, mimicking the transition state of the enzyme-adduct complex, and thereby, blocking access to the active site of the β-lactam ring71 of a drug molecule.
The group of Shoichet has published extensively72 in the field of boronic acid based β-lactamase inhibitors and based upon this previous experience, set out to identify a new, more potent inhibitor. Cephalothin (Table 8), a widely used Cephalosporin antibiotic, possess a Ki of 320 nM versus AmpC type β-lactamases. Shoichet et al. illustrated that the closer the boronic acid resembles the natural substrate, the better the potency. The more the structure mimics the β-lactam of cephalothin, the greater the inhibition, with boronic acid 32 (Ki = 0.32 μM)71a the weakest. Addition of a stereogenic phenyl group 33, mimicking the dihydrothiazine of cephalothin, provides a 10-fold increase in potency (Ki = 0.035 μM). The insertion of a meta-carboxyphenyl moiety further improved the affinity for AmpC type β-lactamase to provide the most potent inhibitor yet described, boronic acid 34 (Ki = 0.001 μM).72b
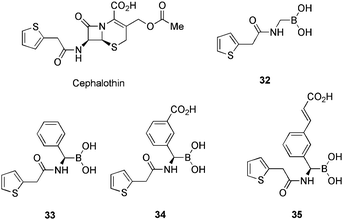
Analysis of the crystal structure of 34, in complex with AmpC β-lactamase provided several important insights into the binding of the inhibitor, including a hydrogen bond between the carboxylate group and the amide of Asn289. This was not anticipated and as the Asn289 is not required for enzyme activity, could easily provide a route for mutation and resistance. The targeted site for the carboxylate binding was designed to be the Asn343, Asn346 and Thr316 carboxylates.73 It was with this in mind, that the group synthesised a family of boronic acids74 with lengthened carboxylic acid side chains to exploit the hydrogen bonding of these residues and acquire a greater affinity inhibitor.
Thus, boronic acid 35 (Table 8) was obtained as the most potent inhibitor (Ki = 0.001 μM) of the generated family, albeit, equipotent with compound 34. The X-ray crystal structure of 34/Amp C β-lactamase confirmed the presence of a hydrogen bond, formed with Asn343 and an absence of the Asn289 hydrogen bond from the N289A mutant enzyme structure. The interaction energy of the two boronic acids 34 and 35 were essentially identical, despite the better fit of 35. Two possibilities exist to explain these data; the structure-based design may not have succeeded in every detail to generate a ‘perfect’ fit, with even small discrepancies taking a large toll on energetic values; the theory may have been incorrect, with the Asn343 interaction offering no advantage over the Asn289 residue.
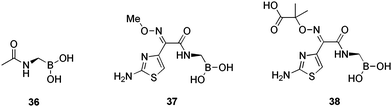
Thomson et al.75 utilised previously synthesised,71 boronic acid transition state inhibitors (BASTIs), as chemical probes (Fig. 9) to determine the changes in extended-spectrum β-lactamase (ESBLs) phenotypes responsible for antibiotic resistance. The reference compound 36, exhibited poor activity for all enzymes analysed, confirming the importance of the side chains for activity. Generally, the SHV ESBLs exhibited greater affinity for the cefotaxime BATSI 37 than did wild-type SHV-1, lowering Ki values from 8.9 ± 0.9 μM for SHV-1 to 1.1 ± 0.2 μM for D104K G238S variant. This observation provided evidence for the suggestion that the G238S substitution is a ‘cefotaximase’ specific mutation in SHV, leading to resistance. The ceftazidime BATSI 38 possessed a similar range of binding activities (Ki = 6.8 ± 0.9 μM for SHV-2 (G238S) and 0.7 ± 0.1 μM for the D104K variant). These data supported the group's previous study76 on the importance of the D104K mutation for substrate hydrolysis by SHV, creating a widening of the active site cavity. It is evident, that the Lys104 residue plays an important role in oxyimino-cephalosporin binding by SHV. As shown in this study, the β-lactamase containing the 104 K substitution, shows greater affinity for the BATSIs. Thus, boronic acids are providing insights into the mechanism of resistance development in β-lactamases.
3.2 Other antibacterial targets
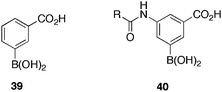
Penicillin binding proteins77 (PBPs) catalyse steps in the biosynthesis of bacterial cell walls. β-lactam antibiotics react irreversibly with the nucleophilic serinyl residue of the PBP, resulting in irreversible inhibition. Inglis et al.78 initiated a programme to identify boronic acid containing PBP inhibitors. The binding kinetics of peptidyl boronic acid inhibitors of PBP3, 4 and 5 have been reported,79 and a crystal structure of a PBP5/boronic acid inhibitor complex isolated.80 Preliminary studies,81 have revealed that commercially available phenylboronic acid possess modest inhibitor activity versus PBP3. D,D-Carboxypeptidase R39, a low molecular weight PBP that is related to PBP4 from Escherichia coli. and a number of other PBP subtypes were incorporated into the assay.To identify an initial lead, the group tested more than 20 commercially available phenylboronic acids and pinacol esters. Phenyl boronic acid 39 (Table 9) was identified as the most active (IC50 = 400 ± 19 μM), with the corresponding pinacol ester displaying similar activity, suggestive that the pinocol ester is hydrolysed82 under the assay conditions. These initial studies suggested that phenylboronic acids with a carboxylic acid at the meta- position can inhibit R39. With the carboxylate group in the para- position, or excised, activity was absent. Docking studies were performed, assuming the boronic acid reacted with the nucleophillic serine (ser49) in the R39 active site, forming a covalent tetrahedral complex. These results suggested the carboxylate group binds in a similar manner as the known inhibitor nitrocefin.
| Compound | R | IC50 (μM)a |
|---|---|---|
| a Values calculated based on three replicate experiments and adjusted to account for standard error of regression of about 50% inhibition. b Not determined due to low activity. | ||
| 39 | — | 400 ± 19 |
| 40a | PhCH2 | 88 ± 2.6 |
| 40b | Ph | 34 ± 0.3 |
| 40c | o-OMeC6H12 | 23 ± 1.0 |
| 40d | o,o-(OMe)2C6H12 | n.d.b |
| 40e | Thiophene | 32 ± 2.2 |
Progressing with these data and lead, compounds of general structure 40 (Table 9) where synthesised and assayed. Meta- substituted compounds showed improved activity against R39, over their ortho- substituted counterparts. The most active compounds displayed activities in the range 20–90 μM, with the most potent 2-methoxybenzoyl derivative 40c, exhibiting IC50 = 23 μM, a significant improvement over the lead compound 39. Important additional SAR data was generated for further compound design. The number of atoms between the aromatic side chain and carbonyl group is important, with a methylene spacer decreasing activity by 50% or more (40a IC50 = 88 μM), compared to no spacer (40b IC50 = 34 μM). Interestingly, given the potent activity of the monomethoxy derivate, the 2,6-dimethoxybenzoyl derivative 40d exhibited poor activity, indicating that the side chain is too bulky to fit into the adjacent pocket in the active site. This study generated a 16-fold more potent inhibitor of R39, than the lead compound, which also displayed some activity against higher weight PBPs. This initial study will provide a basis for the development of even more potent boronic acid based PBP inhibitors, exhibiting a broad spectrum of antibacterial activity.
Inhibitors of bacterial quorum sensing represent a class of important chemical probes and potential therapeutic agents. Bacterial quorum sensing can be defined as the regulation of gene expression in response to changes in the number and/or species present in a community and in the environment. Bacteria regulate quorum sensing through the secretion and detection of molecules called autoinducers (AIs), as a form of communication. Autoinducer-2 (AI2) mediates quorum sensing in both Gram positive and Gram negative bacteria. Quorum sensing plays an important role in the virulence of bacterial infections. Ni et al.83 identified several potent boronic acid based AI2 antagonists using V. harveyi as the model organism, which, upon autoinducer stimulation, produces bioluminescence. AI-2 41 (Fig. 10) can exist in different forms in equilibrium and can complex with boric acid, with complex 42 existing as the biologically active form.84 Stabilisation of the negative charge is thought to be through interaction with two arginine residues within the receptor binding site.
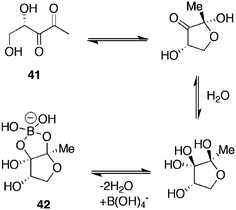 | ||
| Fig. 10 AI-2 equilibrium forms and complexation with boric acid. | ||
The group tested 50 boronic acids for activity versus AI-2; 17 displayed no activity; five were identified as exhibiting significant inhibitory activity (Fig. 11); of these, 4-(benzyloxy)phenylboronic acid 45 (IC50 = 4 ± 1 μM) and 2-fluoro-4-methylphenylboronic acid 46 (IC50 = 4 ± 1 μM) were the most potent. SAR data generated from this study highlighted that phenylboronic acids with minimal substituents, except at the para-position, proved the most active. Compounds displaying additional ionisable functions, tended to be less active. All inactive compounds possessed a boronic acid directly bound to a sp3 carbon. Thus, the relatively high pKa of such boronic acids could attribute for the exhibited inactivity. As the AI-2-boric acid complex exists in the anionic form, boronic acids with low pKa have the best chance of achieving molecular mimicry, due to their high tendency to exist as the anionic tetrahedral form.
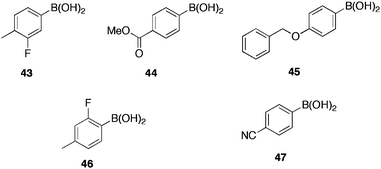 | ||
| Fig. 11 Boronic acid inhibitors of bacterial quorum sensing. | ||
In addition to this significant activity, no general cytotoxicity was detected at the concentrations tested in this study. The study drew initial structure–activity relationships for the generation of single digit micromolar range inhibitors of AI-2, the first boronic acid containing molecules with quorum sensing antagonistic ability.
The group of Benkovic et al.85 reported a novel class of borinic quinoline esters, targeted against bacterial methyltransferases. Using a mechanism-based approach, the group developed inhibitors against the Caulobacter crescentus cell cycle regulated methytransferase, CcrM, an essential DNA methyltransferase enzyme, found in most α-proteobacteria, exerting a key role in cell progression and virulence. A series of borinic esters of types 48 and 50 were designed as inhibitors of CcrM. These inhibitors showed activity against both Gram positive and Gram negative bacteria with minimum inhibitory concentrations (MIC) in the low mg/mL range (Table 10). As CcrM is not present in Gram positive bacteria the group identified the potential target as MenH via cloning, overexpressing and developing an assay for B. subtilis MenH.
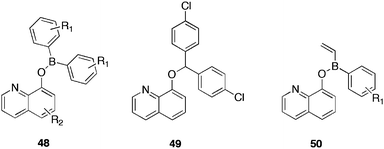
| Compound | R1 | R2 | S.aureus ATCC 12228 Gram +ve | S.epidermidis ATCC 12228 Gram +ve | B.subtilis ATCC 23857 Gram +ve | M.catarrhalis Gram -ve |
|---|---|---|---|---|---|---|
| 48a | 3-Cl | H | 1 | 2 | 2 | 2 |
| 48b | 4-Cl | 2-Me | 16 | 8 | — | — |
| 48c | 4-Cl | 5-Cl | 0.5 | 0.25 | 32 | ≤0.125 |
| 49 | — | — | >64 | >64 | >64 | — |
| 50a | 3-Cl | H | 2 | 1 | 4 | 4 |
| 50b | H | H | 2 | 1 | — | 2 |
Several borinic esters synthesised, displayed inhibitory activity against Gram negative and Gram positive bacteria, in particular 48c (Table 10), showed MIC vales of below 1 μg/mL against three out of seven Gram positive bacteria. The two compounds most selective for CcrM in vitro, 48a and 48b, showed different specificities; 48a exhibiting activity against both Gram positive and Gram negative bacteria, whilst 48b was active only against Gram negative bacteria. The carbon analogue 49, as expected, showed no activity, confirming the crucial role of the boron atom in inhibitory activity. The borinic esters reported, displayed comparable activity to clinically used antibiotics such as erythromycin (MIC90v. B. anthracis 1.5 μg/mL). Compounds of type 50 were tested to probe the SAR of this class of compound against the MenH strain. This strain proved 64-fold more sensitive to the 3-chlorophenyl vinyl borinic ester 50a. When the chlorine substituent was removed, to provide 50b, the activity was lost entirely. Additional analogues, with the chlorine substituent occupying differing positions on the ring, or with differing functionalities, all provided a reduced activity.
The study illustrated the broad spectrum antibiotic nature of these boron containing compounds, towards several pathogenic Gram positive and Gram negative bacteria and highlighted the intrinsic role of boron in providing activity. These borinic esters could be useful starting points for the development of inhibitors of bacterial growth and methyltransferase enzymes.
The group detailed86 a related class of boronic acid picolinate esters, targeted for dual antibacterial and anti-inflammatory activity, as a potential therapeutic against atopic dermatitis, or eczema. Both are inflammatory diseases, presenting as an itchy flaky rash which, when scratched, often provides opportunity for bacterial infection. In 90% of cases, the Gram positive bacteria S. aureus is present. Boronic ester 51 (Fig. 12) was identified as the most potent of the series with a MIC versus S. aureus of 1 μg/mL, comparable with that for Erythromycin (MIC = 0.5 1 μg/mL). Removal of the boronic acid resulted in elimination of all activity, the 3-chloro functionality, was also key, with substitution by fluoro, removing all activity and repositioning about the ring leading to reduced activity. At time of writing 51 had been advanced to preclinical trials.
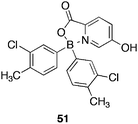 | ||
| Fig. 12 Structure of boronic acid picolinate ester 51 in preclinical development for dermatological conditions. | ||
The preceding section has highlighted the opportunity for boronic acids and borinic esters to contribute to the identification of new antibiotics to combat ever more resistant bacteria.
4 Antiviral activity
4.1 NS3 protease inhibitors
Hepatitis C virus (HCV), is the cause of chronic HCV infection, leading to liver cirrhosis and hepatocellular carcinoma. The HCV genome encodes a large polyprotein of around 3,000 amino acids. Proteolytic cleavage by NS3 protease, produces mature virons. Thus, inhibition of NS3 protease presents itself as a valid therapeutic target. HCV NS3 protease is a serine protease, which enables the lesion of a Cys-Ser bond with the assistance of the cofactor NS4A. Hydrolysis of the amide bond proceeds by attack of Ser-139, followed by the stabilisation of the resultant tetrahedral transition state, by His-57 and Asp-81 residues.Boronic acid moities have been applied87 to recently developed ketoamide containing inhibitors. The boronic acid analogue of the lead ketoamide was synthesised 52a (Table 11), the binding constant for this boronic acid was determined at ki = 380 nM, less potent than the ketoamide lead. The replacement of the R1 methyl group with a lipophilic cyclobutane enhanced the binding of both the free boronic acid 52b (ki = 10 nM) and the corresponding boronic ester 52c (ki = 10 nM). Further studies demonstrated efficacy properties of varying the R3 group and to this end, the tert-butyl was replaced with a sulfonamide derivative to generate the boronic ester 52d (ki = 0.5 nM) and the corresponding free acid 52e (ki = 0.2 nM), the first identified inhibitor with picomolar level binding. Replicon cellular assay (EC90 = >5.0 μM) suggested that cell permeability may be limiting and responsible for this demonstrated poor cellular activity, further studies to improve this situation continue. The X-ray structure of boronic acid 52e, bound to HCV NS3, revealed insights into the binding attributes and the confirmation of the tetrahedral boron environment through the oxygen of Ser-139.
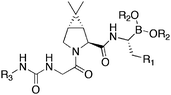
| Compound | R1 | R2 | R3 | Ki (nM) |
|---|---|---|---|---|
| 52a | CH3 | Pinene diol | tButyl | 380 |
| 52b | Cyclobutane | Pinene diol | tButyl | 10 |
| 52c | Cyclobutane | H | tButyl | 10 |
| 52d | Cyclobutane | Pinene diol | Mesyl | 0.5 |
| 52e | Cyclobutane | H | Mesyl | 0.2 |
A series of peptidomimetic boronic acid analogues where synthesised,88 those containing polar groups, exhibited good tolerance in the NS3/4A enzyme, however, considerable loss of cellular penetration was encountered. Boronic acid 53 (Fig. 13), containing the same N-methyl sulfonamide terminal group as compound 52e, displayed the best replicon assay activity (EC50 = 25 nM, EC90 = 117 nM), contrasting to the relatively poor demonstrated cellular activity of 52e. In addition, boronic acid 53, exhibited excellent genotype coverage of all clinically relevant HCV genotypes, demonstrating very low-fold IC50 increases across a range of mutant NS3/4A proteases, common in HCV patients, providing a resilient therapeutic candidate that is undergoing additional evaluation.
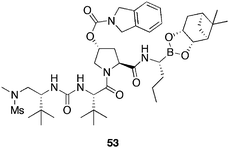 | ||
| Fig. 13 Structure of HCV-NS/4A boronic acid inhibitor 53. | ||
A protected tetrapeptidic boronic acid89 of the sequence Bz-Nle-Lys-Lys-Arg-B(OH)2 was found to be active (Ki = 0.05 μM), against Yellow Fever Virus NS3 protease, expressed in E. coli. The group conducted docking and SAR studies to elucidate the optimum structures for inhibition, concluding that the P1 and P2 positions were most important for activity. The study revealed the NS3 domain is highly conserved in Dengue, West Nile Virus and Yellow Fever Virus suggesting a pan-flavivirus NS3 protease drug may be a possibility in treatment of flaviviral diseases.
4.2 Viral entry inhibitors
Balzarini90 advanced a hypothesis that carbohydrate-binding agents (CBAs), capable of binding the glycans of HIV-1 gp120, represent a promising class of viral entry inhibitors. The CBAs are foreseen as possessing two modes of action; in not only, binding to the gp120, to prevent binding to CD4+ T-lymphocytes and proliferation of the virus; but also, in engendering mutation of the virus to escape the drug pressure, thereby, exposing the ‘deprotected’ gp120 to the action of the host immune system. Phenylboronic acid is known91 to form reversible covalent bonds with 1,2- and 1,3- diols expressed in saccharides, to form five or six membered cyclic esters, a key factor in the important role occupied by boronic acids in saccharide recognition. This condensation reaction is reversible and is highly dependant92 on the pH and chemical structure of the boronic acid and diol.93 Conversion of the boronic acid to the tetrahedral boronate anion forms a more stable complex, resistant to hydrolysis compared to the trigonal ester.Utilising this inherent ability of boronic acids Jay et al.94 sought to develop a novel microbicide, a woman-controlled prophylactic for sexually transmitted infections. Whilst protein lectins are known to act as entry inhibitors, their production and formulation present economical and resource problems for the worlds poorest nations, a polymer based prophylactic would be amenable for use in such resource-poor nations. Berube et al.95 synthesised a water soluble polymer CBA that contains multiple benzoboroxole moieties capable of binding gp120 at physiological pH. A polymer backbone of type 54 (Fig. 14) was developed, with benzoboroxole loadings of 25, 50 and 75 mol %. In vivo testing for ability to neutralise HIV-1 entry for two HIV-1 clades demonstrated activity against all viral strains tested, with EC50 values = 15000 nM (1500 μg mL−1) for the 25 mol % polymer, increasing to 11 nM (1 μg mL−1) for the 75 mol % polymer. These data are consistent with the observation that the probability of a binding event occurring between the benzoboroxole and the gp120 mannose residues increases in proportion to the number of sites available for binding in the polymer. A theory supported by work from within our own laboratory96 in which a range of monophenylboronic acids failed to exhibited any significant gp120 binding activity, despite their ability to undergo binding to mannose in the laboratory. In addition, the nature and length of the linker separating multiple boronic acids, has demonstrated97 an effect in the binding selectivity of the molecule. All polymers exhibited minimal cytotoxicity after 24 h exposure to human vaginal cell lines. Studies continue in the hope of developing new classes of economically and formulation simple, polymer-based entry inhibitors for use as vaginal microbicides that will display broad-spectrum activity against HIV-1.
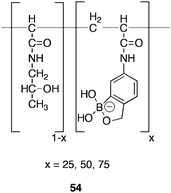 | ||
| Fig. 14 Structure of benzoboroxole functionalized polymer 54. | ||
The use of boronic acids in antiviral therapy is, in respect to the other areas covered in this review, still in the development stages. Investigations from numerous academic and industrial groups continue to focus on the development of boronic acid containing molecules for the treatment of a number of viruses, most notably HIV.
Conclusions
As can be seen from the work described herein, the boronic acid motif plays a recurring role in the design of novel therapeutics and the medicinal chemistry of this group continues to be developed and revised.The ability of boronic acid esters to undergo hydrolysis under assay conditions to liberate the free boronic acid has potential both in the design of synthetic routes to act as a protecting group and as the possibility of development as a prodrug delivery system, elevating cell wall transfer due to the low polarity of many boronic esters, especially pinacol esters. It is worthy of note, of the interest generated by boronic acids, that whilst this review pertains to the published journal literature a similar amount of effort, if not more, is described in the patent literature. Thus, establishing the presence of boronic acids in the oncology, antiviral, antibacterial, antifungal, diabetic and pain fields of medicinal chemistry. Whilst not the focus of this review, boronic acid containing therapeutics pertaining to a range of other disease states are under investigation including dipeptidyl peptidase IV inhibitors for diabetes,98 antifungal treatments,99 Fatty Acid Amide Hydrolase inhibitors100 implicated in inflammation and pain relief and inhibitors of factor Xia101 implicated in thromboembolitic disease amongst an array of other disease targets.
Notes and references
- E. Frankland and B. F. Duppa, Justus Liebigs Ann. Chem., 1860, 115, 319 CrossRef.
- H. C. Brown and B. C. Subba Rao, J. Am. Chem. Soc., 1956, 78, 5694 CrossRef CAS.
- (a) N. Miyaura, K. Yamada and A. Suzuki, Tetrahedron Lett., 1979, 20, 3437 CrossRef; (b) N. Miyaura and A. Suzuki, Chem. Commun., 1979, 866 RSC.
- P. Y. S. Lam, C. G. Clark, S. Saubern, J. Adams, M. P. Winters, D. M. T. Chan and A. Combs, Tetrahedron Lett., 1998, 39, 2941 CrossRef CAS.
- D. G. Hall, Boronic Acids: Preparation, Applications in Organic Synthesis and Medicine, Wiley-VCH, Weinheim, 2005 Search PubMed.
- D. S. Matterson, Stereodirected Synthesis with Organoboranes, Springer, Berlin, 1995 Search PubMed.
- H. C. Brown, Organic Synthesis via Boranes, Wiley, New York, 1975 Search PubMed.
- (a) A. H. Soloway, W. Tjarks, B. A. Barnum, F.-G. Rong, R. F. Barth, I. M. Codogni and G. Wilson, Chem. Rev., 1998, 98, 1515 CrossRef CAS; (b) B. F. Spielvogel, A. Sood, B. R. Shaw and I. H. Hall, Pure Appl. Chem., 1991, 63, 415 CAS.
- J. Kohno, T. Kawahata, T. Otake, M. Morimoto, H. Mori, N. Ueba, M. Nishio, A. Kinumaki, S. Komatsubara and K. Kawashima, Biosci., Biotechnol., Biochem., 1996, 60, 1036 CrossRef CAS.
- T. Okazaki, T. Kitahara and Y. Okami, J. Antibiot., 1975, 28, 176 CAS.
- B. S. Moore and C. Hertweck, Nat. Prod. Rep., 2002, 19, 70 RSC.
- (a) E. J. Corey, B.-C. Pan, D. H. Hua and D. R. Deardorff, J. Am. Chem. Soc., 1982, 104, 6816 CrossRef CAS; (b) E. J. Corey, D. H. Hua, B.-C. Pan and S. P. Seitz, J. Am. Chem. Soc., 1982, 104, 6818 CrossRef CAS.
- H. E. Goldbach and M. A. Wimmer, J. Plant Nutr. Soil Sci., 2007, 170, 39 CrossRef CAS.
- (a) A. Michaelis and P. Becker, Ber. Dtsch. Chem. Ges., 1880, 13, 58 CrossRef; (b) A. Michaelis and P. Becker, Ber. Dtsch. Chem. Ges., 1882, 15, 180 CrossRef.
- M. Philipp and M. L. Bender, Proc. Natl. Acad. Sci. U. S. A., 1971, 68, 478 CAS.
- (a) E. Tsilikounas, C. A. Kettner and W. W. Bachovchin, Biochemistry, 1992, 31, 12839 CrossRef CAS; (b) E. Tsilikounas, C. A. Kettner and W. W. Bachovchin, Biochemistry, 1993, 32, 12651 CrossRef CAS; (c) D. W. Tanner and T. C. Brucie, J. Am. Chem. Soc., 1967, 89, 6954 CrossRef CAS; (d) H. Schafer, Z. Anorg. Allg. Chem., 1942, 250, 82 CrossRef CAS.
- T. D. James, M. D. Phillips, S. Shinkai, Boronic Acids in Saccharide Recognition, Royal Society of Chemistry, Cambridge, 2006 Search PubMed.
- R. N. Fedoak, M. D. Gershon and M. Field, Gastroenterology, 1989, 96, 37.
- J. Yan, H. Fang and B. Wang, Med. Res. Rev., 2005, 25, 490 CrossRef CAS.
- J. Balzarini, Lancet Infect. Dis., 2005, 5, 726 CrossRef CAS.
- S. Kitano, Y. Koyama, K. Kataoka, T. Okano and Y. Sakurai, J. Controlled Release, 1992, 19, 161 CrossRef CAS.
- C. Morin, Tetrahedron, 1994, 50, 12521 CrossRef CAS.
- W. Yang, X. Gao and B. Wang, Med. Res. Rev., 2003, 23, 346 CrossRef CAS.
- M. A. Jordan and L. Wilson, Nat. Rev. Cancer, 2004, 4, 253 CrossRef CAS and references therein.
- Y. Kong, J. Grembecka, M. C. Edler, E. Hamel, S. L. Mooberry, M. Sabat, J. Rieger and M. L. Brown, Chem. Biol., 2005, 12, 1007 CrossRef CAS.
- G. R. Pettit, G. M. Cragg, D. L. Herald, J. M. Schmidt and P. Lohavanijaya, Can. J. Chem., 1982, 60, 1374 CAS.
- Y. Kong, K. Wang, M. C. Edler, E. Hamel, S. L. Mooberry, M. A. Paige and M. L. Brown, Bioorg. Med. Chem., 2010, 18, 971 CrossRef CAS.
- A. Monks, A. Scudiero, P. Skehan, R. Shoemaker, K. Paull, D. Vistica, J. T. Warren, H. Bokesch, S. Kenney and M. R. Boyd, J. Natl. Cancer Inst., 1990, 82, 1107 CrossRef CAS.
- (a) X. Zi and A. R. Simoneau, Cancer Res., 2005, 65, 9762 CrossRef CAS; (b) S. Ducki, R. Forrest, J. A. Hadfield, A. Kendall, N. J. Lawrence, A. T. McGown and D. Rennish, Bioorg. Med. Chem. Lett., 1998, 8, 1051 CrossRef CAS.
- R. J. Autom, K. Sukumaran, G. Kuttan, M. N. A. Rao, V. Subbaraju and R. Kuttan, Cancer Lett., 1995, 97, 33 CrossRef CAS.
- H. Nakamura, H. Kuroda, H. Saito, R. Suzuki, T. Yamori, K. Maruyama and T. Haga, ChemMedChem, 2006, 1, 729 CrossRef CAS.
- (a) T. Hideshima, P. Richardson, D. Chauhan, V. J. Palombella, P. J. Elliott, J. Adams and K. C. Anderson, Cancer Res., 2001, 61, 3071 CAS; (b) A. Paramore and S. Frantz, Nat. Rev. Drug Discovery, 2003, 2, 611 CrossRef CAS.
- J. Adams, V. J. Palombella, E. A. Sausville, J. Johnson, A. Destree, D. D. Lazarus, J. Maas, C. S. Pien, S. Prakash and P. J. Elliott, Cancer Res., 1999, 59, 2615 CAS.
- D. S. Matteson, Med. Res. Rev., 2008, 28, 233 CrossRef CAS and references therein.
- P. G. Richardson, C. Mitsiades, T. Hideshima and K. C. Anderson, Annu. Rev. Med., 2006, 57, 33 CrossRef CAS.
- M. P. Curran and K. McKeage, Drugs, 2009, 69, 859 CrossRef CAS.
- S. K. Kumar, E. Hager, C. Pettit, H. Gurulingappa, N. E. Davidson and S. R. Khan, J. Med. Chem., 2003, 46, 2813 CrossRef CAS.
- C. Wasylyk, R. Salvi, M. Argentini, C. Dureuil, I. Delumeau, J. Abecassis, L. Debusche and B. Wasylyk, Oncogene, 1999, 18, 1921 CrossRef CAS.
- A. Modzelewska, C. Pettit, G. Achanta, N. E. Davidson, P. Huang and S. R. Khan, Bioorg. Med. Chem., 2006, 14, 3491 CrossRef CAS.
- (a) S. A. Williams and D. J. McConkey, Cancer Res., 2003, 63, 7338 CAS; (b) Y. H. Ling, L. Liebes, J. D. Jiang, J. F. Holland, P. J. Elliott, J. Adams, F. M. Muggia and R. Perez-Soler, Clin. Cancer Res., 2003, 9, 1145 CAS; (c) A. P. Maclaren, R. S. Chapman, A. H. Wyllie and C. J. Watson, Cell Death Differ., 2001, 8, 210 CrossRef CAS.
- Y. Zhu, S. Yao, B. Xu, Z. Ge, J. Cui, T. Cheng and R. Li, Bioorg. Med. Chem., 2009, 17, 6851 CrossRef CAS.
- (a) M. Groll, L. Ditzel, J. Lowe, D. Stock, M. Bochtler, H. D. Bartunik and R. Huber, Nature, 1997, 386, 463 CrossRef CAS; (b) M. Groll, C. R. Berkers, H. L. Ploegh and H. Ovaa, Structure, 2006, 14, 451 CrossRef CAS.
- M. Verdoes, B. I. Florea, V. enendez-Benito, C. J. Maynard, M. D. Witte, W. A. van der Linden, A. M. C. H. van der Nieuwendijk, T. Hofmann, C. R. Berkers, F. W. van Leeuwen, T. A. Groothuis, M. A. Leeuwenburgh, H. Ovaa, J. J. Neefjes, D. V. Filipov, G. A. van der Marel, N. P. Dantuma and H. S. Overkleeft, Chem. Biol., 2006, 13, 1217 CrossRef CAS.
- A. Asai, T. Tsujita, S. V. Sharma, Y. Yamashita, S. Akinaga, M. Funakoshi, H. Kobayashi and T. Mizukami, Biochem. Pharmacol., 2004, 67, 227 CrossRef CAS.
- H. Nakamura, M. Watanabe, H. S. Ban, W. Nabeyama and A. Asai, Bioorg. Med. Chem. Lett., 2009, 19, 3220 CrossRef CAS.
- T. Watanabe, I. Momose, M. Abe, H. Abe, R. Sawa, Y. Umezawa, D. Ikeda, Y. Takahashi and Y. Akamatsu, Bioorg. Med. Chem. Lett., 2009, 19, 2343 CrossRef CAS.
- J. Adams, M. Behnke, S. Chen, A. A. Cruickshank, L. R. Dick, L. Greiner, J. M. Klunder, Y.-T. Ma, L. Plamondon and R. L. Stein, Bioorg. Med. Chem. Lett., 1998, 8, 333 CrossRef CAS.
- Y. Zhu, X. Zhao, X. Zhu, G. Wu, Y. Li, Y. Ma, Y. Yuan, J. Yang, Y. Hu, L. Ai and Q. Gao, J. Med. Chem., 2009, 52, 4192 CrossRef CAS.
- Y. Q. Zhu, M. Lei, A. J. Lu, X. Zhao, X. J. Yin and Q. Z. Gao, Eur. J. Med. Chem., 2009, 44, 1486 CrossRef CAS.
- Y. Zhu, X. Zhu, G. Wu, Y. Ma, Y. Li, X. Zhao, Y. Yuan, J. Yang, S. Yu, F. Shao, R. Li, Y. Ke, A. Lu, Z. Liu and L. Zhang, J. Med. Chem., 2010, 53, 1990 CrossRef CAS.
- D. Seebach, S. Abele, J. V. Schrieber, B. Martinoni, A. K. Nussbaum, H. Schild, H. Schulz, H. Henneke, R. Woessner and F. Bitsch, Chimia, 1998, 52, 734 CAS.
- U. Langheinrich, BioEssays, 2003, 25, 904 CrossRef CAS.
- B. D. Dorsey, M. Iqbal, S. Chatterjee, E. Menta, R. Bernardini, A. Bernareggi, P. G. Cassara, G. D'Arasmo, E. Ferretti, S. De Munari, A. Oliva, G. Pezzoni, C. Allievi, I. Strepponi, B. Ruggeri, M. A. Ator, M. Williams and J. P. Mallamo, J. Med. Chem., 2008, 51, 1068 CrossRef CAS.
- R. Piva, B. Ruggeri, M. Williams, G. Costa, I. Tamagno, D. Ferrero, V. Giai, M. Coscia, S. Peola, M. Massaia, G. Pezzoni, C. Allievi, N. Pescalli, M. Cassin, S. di Giovine, P. Nicoli, P. de Feudis, I. Strepponi, I. Roato, R. Ferracini, B. Bussolati, G. Camussi, S. Jones-Bolin, K. Hunter, H. Zhao, A. Neri, A. Palumbo, C. Berkers, H. Ovaa, A. Bernareggi and G. Inghirami, Blood, 2008, 111, 2765 CrossRef CAS.
- L. R. Dick and P. E. Fleming, Drug Discovery Today, 2010, 15, 243 CrossRef CAS.
- B. Walker and J. F. Lynas, Cell. Mol. Life Sci., 2001, 58, 596 CAS.
- A. M. LeBeau, P. Singh, J. T. Isaacs and S. R. Denmeade, Chem. Biol., 2008, 15, 665 CrossRef CAS.
- S. R. Denmeade, C. Jakobsen, S. Janssen, S. R. Khan, H. Lilja, S. B. Christensen and J. T. Isaacs, J. Natl. Cancer Inst., 2003, 95, 990 CrossRef CAS.
- R. Bone, A. B. Shenvi, C. A. Kettner and D. A. Agard, Biochemistry, 1987, 26, 7609 CrossRef CAS.
- N. Suzuki, T. Suzuki, Y. Ota, T. Nakano, M. Kurihara, H. Okuda, T. Yamori, H. Tsumoto, H. Nakagawa and N. Miyata, J. Med. Chem., 2009, 52, 2909 CrossRef CAS.
- C. T. Supuran, Nat. Rev. Drug Discovery, 2008, 7, 168 CrossRef CAS and references therein.
- J.-Y. Winum, A. Innocenti, A. Scozzafava, J.-L. Montero and C. T. Supuran, Bioorg. Med. Chem., 2009, 17, 3649 CrossRef CAS.
- V. Ahmed, Y. Liu, C. Silvestro and S. D. Taylor, Bioorg. Med. Chem., 2006, 14, 8564 CrossRef CAS.
- J. D. Moyer, E. G. Barbacci, K. K. Iwata, L. Arnold, B. Boman, A. Cunningham, C. DiOrio, J. Doty, M. J. Morin, M. P. Moyer, M. Neveu, V. A. Pollack, L. R. Pustilnik, M. M. Reynolds, D. Sloan, A. Theleman and P. Miller, Cancer Res., 1997, 57, 4838 CAS.
- H. S. Ban, T. Usui, W. Nabeyama, H. Morita, K. Fukuzawa and H. Nakamura, Org. Biomol. Chem., 2009, 7, 4415 RSC.
- K. Shimizu, M. Maruyama, Y. Yasui, H. Minegishi, H. S. Ban and H. Nakamura, Bioorg. Med. Chem. Lett., 2010, 20, 1453 CrossRef CAS.
- R. L. Julius, O. K. Farha, J. Chiang, L. J. Perry and M. F. Hawthorne, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 4808 CrossRef CAS and references therein.
- R. M. Klevens, M. A. Morrison, J. Nadle, S. Petit, K. Gershman, S. Ray, L. H. Harrison, R. Lynfield, G. Dumyati, J. M. Townes, A. S. Craig, E. R. Zell, G. E. Fosheim, L. K. McDougal, R. B. Carey and S. K. Fridkin, JAMA, J. Am. Med. Assoc., 2007, 298, 1763 Search PubMed.
- (a) E. P. Abraham and E. Chain, Nature, 1940, 146, 837 CrossRef CAS; (b) R. Pongdee and H.-W. Liu, Bioorg. Chem., 2004, 32, 393 CrossRef CAS.
- S. Ness, R. Martin, A. M. Kindler, M. Paetzel, M. Gold, S. E. Jensen, J. B. Jones and N. J. C. Strynadka, Biochemistry, 2000, 39, 5312 CrossRef CAS.
- R. A. Powers, E. Caselli, P. J. Focia, F. Prati and B. K. Shoichet, Biochemistry, 2001, 40, 9014 CrossRef.
- (a) E. Caselli, R. A. Powers, L. C. Blasczcak, C. Y. Wu, F. Prati and B. K. Shoichet, Chem. Biol., 2001, 8, 17 CrossRef CAS; (b) F. Morandi, E. Caselli, S. Morandi, P. J. Focia, J. Blasquez, B. K. Shoichet and F. Prati, J. Am. Chem. Soc., 2003, 125, 685 CrossRef CAS.
- B. M. Beadle, I. Trehan, P. J. Focia and B. K. Shoichet, Structure, 2002, 10, 413 CrossRef CAS.
- S. Morandi, F. Morandi, E. Caselli, B. K. Shoichet and F. Prati, Bioorg. Med. Chem., 2008, 16, 1195 CrossRef CAS.
- J. M. Thomson, F. Prati, C. R. Bethel and R. A. Bonomo, Antimicrob. Agents Chemother., 2007, 51, 1577 CrossRef CAS.
- C. R. Bethel, A. M. Hujer, K. M. Hujer, J. M. Thomson, M. W. Ruszczycky, V. E. Anderson, M. Pusztai-Carey, M. Taracila, M. S. Helfrand and R. A. Bonomo, Antimicrob. Agents Chemother., 2006, 50, 4124 CrossRef CAS.
- E. Sauvage, F. Kerff, M. Terrak, J. A. Ayala and P. Charlier, FEMS Microbiol. Rev., 2008, 32, 234 CrossRef CAS.
- S. R. Inglis, A. Zervosen, E. C. Y. Woon, T. Gerards, N. Teller, D. S. Fischer, A. Luxen and C. J. Schofield, J. Med. Chem., 2009, 52, 6097 CrossRef CAS.
- A. Pechenov, M. E. Stefanova, R. A. Nicholas, S. Peddi and W. G. Gutheil, Biochemistry, 2003, 42, 579 CrossRef CAS.
- G. Nicola, S. Peddi, M. Stefanova, R. A. Nicholas, W. G. Gutheil and C. Davies, Biochemistry, 2005, 44, 8207 CrossRef CAS.
- M. E. Stefanova, J. Tomberg, C. Davies, R. A. Nicholas and W. G. Gutheil, Eur. J. Biochem., 2003, 271, 23 CrossRef.
- A. Venturelli, D. Tondi, L. Cancian, F. Morandi, G. Cannazza, B. Segatore, F. Prati, G. Amicosante, B. K. Shoichet and M. P. Costi, J. Med. Chem., 2007, 50, 5644 CrossRef CAS.
- N. Ni, H.-T. Chou, J. Wang, M. Li, C.-D. Lu, P. C. Tai and B. Wang, Biochem. Biophys. Res. Commun., 2008, 369, 590 CrossRef CAS.
- X. Chen, S. Schauder, N. Potier, A. V. Dorsselaer, I. Pelczer, B. L. Bassler and F. M. Hughson, Nature, 2002, 415, 545 CrossRef CAS.
- S. J. Benkovic, S. J. Baker, M. R. K. Alley, Y.-H. Woo, Y.-K. Zhang, T. Akama, W. Mao, J. Baboval, P. T. R. Rajagopalan, M. Wall, L. S. Kahng, A. Tavassoli and L. Shapiro, J. Med. Chem., 2005, 48, 7468 CrossRef CAS.
- S. J. Baker, T. Akama, Y.-K. Zhang, V. Sauro, C. Pandit, R. Singh, M. Kully, J. Khan, J. J. Plattner, S. J. Benkovic, V. Lee and K. R. Maples, Bioorg. Med. Chem. Lett., 2006, 16, 5963 CrossRef CAS.
- S. Venkatraman, W. Wu, A. Prongay, V. Girijavallabhan and F. G. Njoroge, Bioorg. Med. Chem. Lett., 2009, 19, 180 CrossRef CAS.
- A. Boloor, D. Hanway, M. Joshi, D. T. Winn, G. Mendez, M. Walls, P. Wei, F. Qian, X. Zhang, Y. Zhang, M. E. Hepperle, X. Li, D. A. Campbell and J. M. Betancroft, Bioorg. Med. Chem. Lett., 2009, 19, 5708 CrossRef CAS.
- K. Lohr, J. E. Knox, W. Y. Phong, N. L. Ma, Z. Yin, A. Sampath, S. J. Patel, W.-L. Wang, W.-L. Chan, K. R. R. Rao, G. Wang, S. G. Vasudevan, T. H. Keller and S. P. Lim, J. Gen. Virol., 2007, 88, 2223 Search PubMed.
- J. Balzarini, Nat. Rev. Microbiol., 2007, 5, 583 Search PubMed.
- G. Springsteen and B. Wang, Tetrahedron, 2002, 58, 5291 CrossRef CAS.
- J. Yan, G. Springsteen, S. Deeter and B. Wang, Tetrahedron, 2004, 60, 11205 CAS.
- R. P. Singhal, B. Ramamurhy, N. Govindraj and Y. Sarwar, J. Chromatogr., A, 1991, 543, 17 CrossRef CAS.
- J. I. Jay, B. E. Lai, D. G. Myszka, A. Mahalingam, K. Langheinrich, D. F. Katz and P. F. Kiser, Mol. Pharmaceutics, 2010, 7, 116 CrossRef CAS.
- M. Berube, M. Dowlut and D. G. Hall, J. Org. Chem., 2008, 73, 6471 CrossRef CAS.
- P. C. Trippier, C. McGuigan and J. Balzarini, Antiviral Chem. Chemother., 2010 DOI:10.3851/IMP1632.
- G. Kaur, H. Fang, X. Gao, H. Li and B. Wang, Tetrahedron, 2006, 62, 2583 CrossRef.
- B. A. Connolly, D. G. Sanford, A. K. Chiluwal, S. E. Healey, D. E. Peters, M. T. Dimare, W. Wu, Y. Liu, H. Maw, Y. Zhou, Y. Li, Z. Jin, J. L. Sudmeier, J. H. Lai and W. W. Bachovchin, J. Med. Chem., 2008, 51, 6005 CrossRef CAS.
- (a) J. W. Hicks, C. B. Kyle, C. M. Vogels, S. L. Wheaton, F. J. Baerlocher, A. Decken and S. A. Westcott, Chem. Biodiversity, 2008, 5, 2415 CrossRef CAS; (b) F. E. Appoh, S. L. Wheaton, C. M. Vogels, F. J. Baerlocher, A. Decken and S. A. Westcott, Heteroatom Chem., 2009, 20, 56 CrossRef CAS.
- A. Minkkila, S. M. Saario, H. Kasnanen, J. Leppanen, A. Poso and T. Nevalainen, J. Med. Chem., 2008, 51, 7057 CrossRef CAS.
- T. I. Lazarova, L. Jin, M. Rynkiewicz, J. C. Gorga, F. Bibbins, H. V. Meyers, R. Babine and J. Strickler, Bioorg. Med. Chem. Lett., 2006, 16, 5022 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |